
Ferroptosis in Gastrointestinal Diseases: A New Frontier in Pathogenesis and Therapy

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Molecular Mechanism of Ferroptosis
2.1. Iron Metabolism and Oxidative Stress
2.2. Regulatory Systems
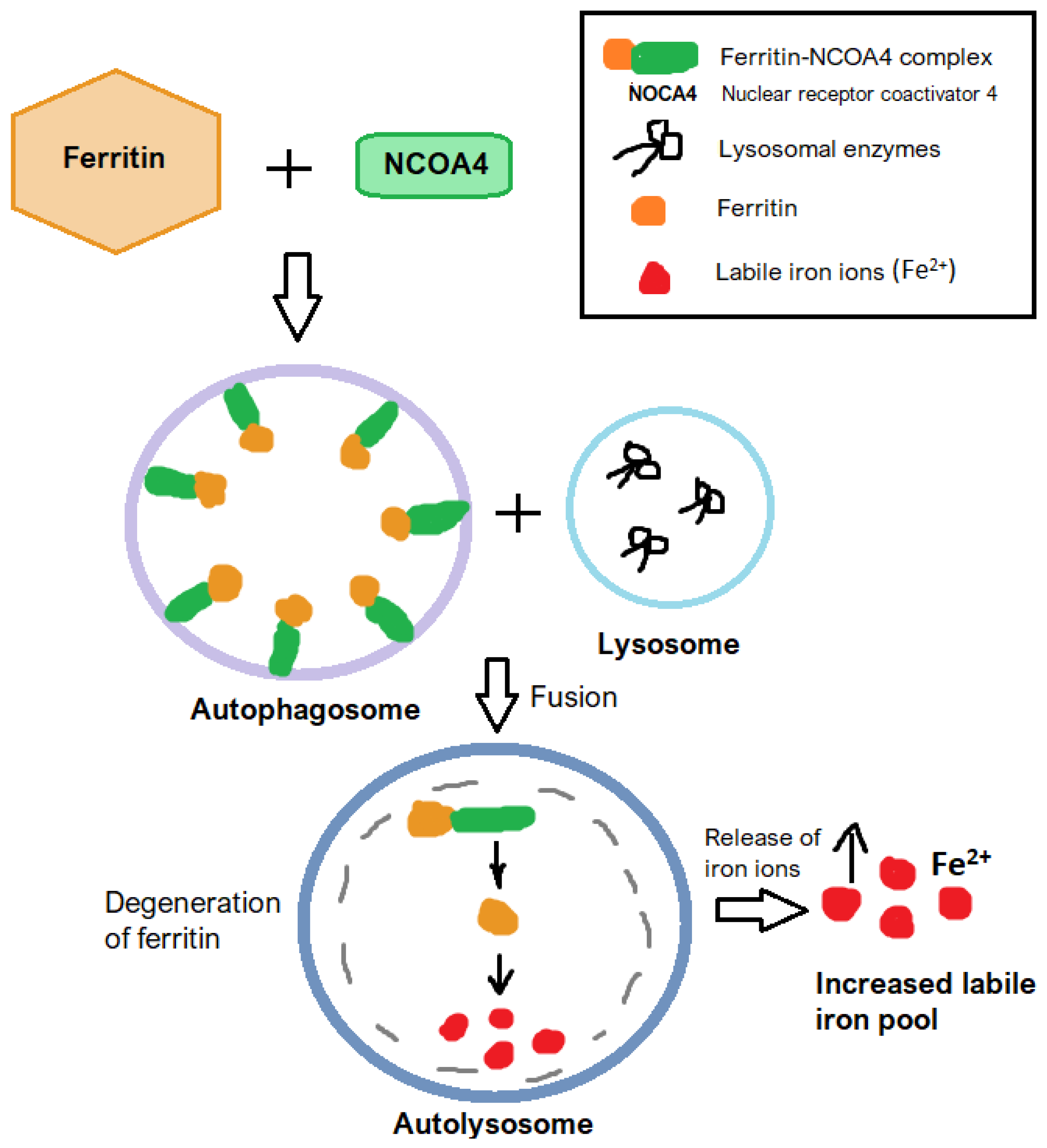
2.3. Ferritinophagy
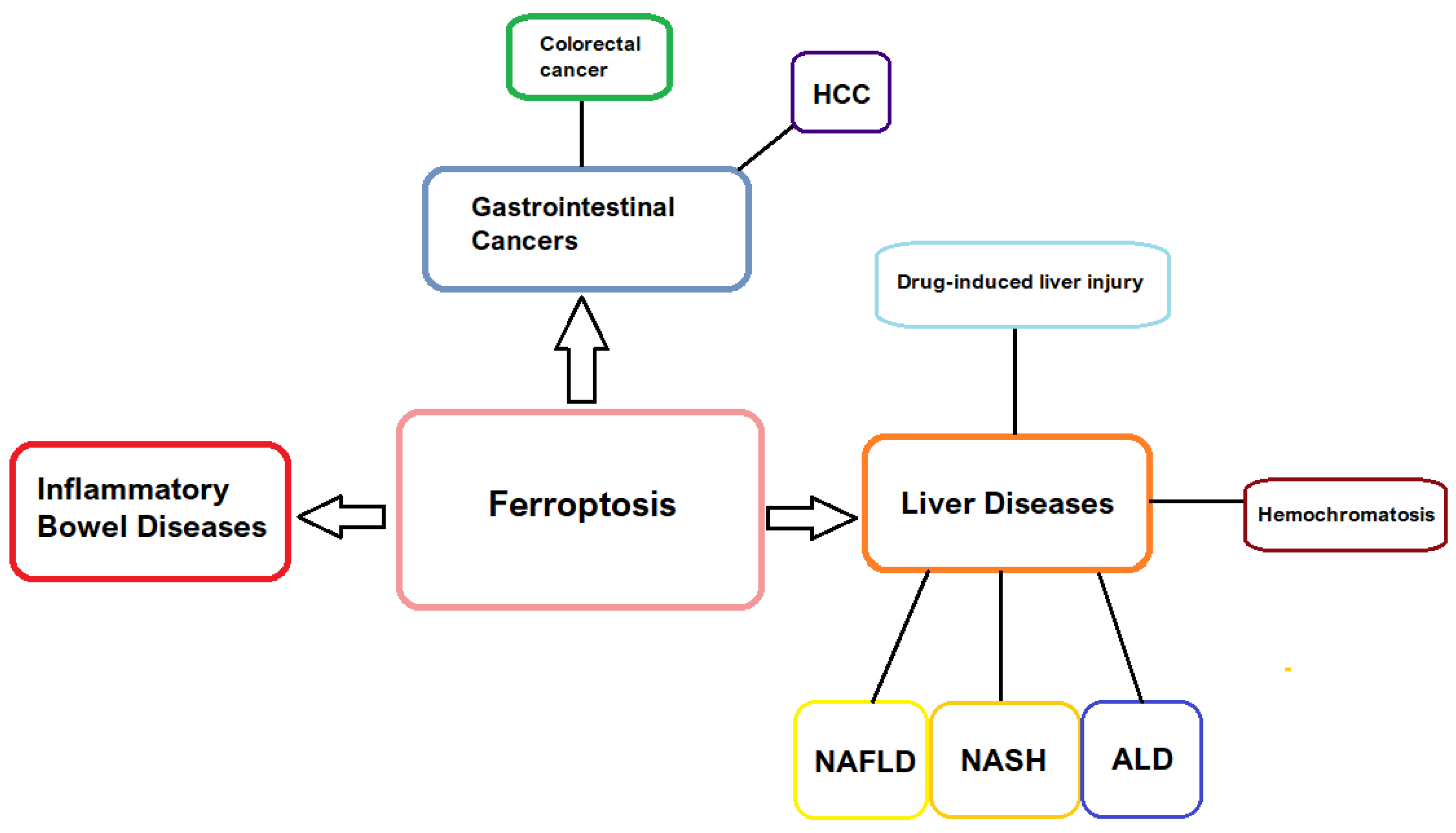
3. Ferroptosis in Gastrointestinal Pathology
3.1. Inflammatory Bowel Disease
3.2. Liver Disorders
4. Therapeutic Targets of Ferroptosis
5. Clinical Applications and Future Directions
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Newton, K.; Strasser, A.; Kayagaki, N.; Dixit, V.M. Cell death. Cell 2024, 187, 235–256. [Google Scholar] [CrossRef] [PubMed]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a new form of cell death: Opportunities and challenges in cancer. J. Hematol. Oncol. 2019, 12, 34. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wiredu Ocansey, D.K.; Yuan, J.; Wei, Z.; Mao, F.; Zhang, Z. Role of ferroptosis in the pathogenesis and as a therapeutic target of inflammatory bowel disease. Int. J. Mol. Med. 2023, 51, 53. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhu, J.Y.; Zang, X.; Zhai, Y.Z. The Emerging Role of Ferroptosis in Liver Diseases. Front. Cell Dev. Biol. 2021, 9, 801365. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ma, Y.; Lv, G.; Wang, H. Ferroptosis as a therapeutic target for inflammation-related intestinal diseases. Front. Pharmacol. 2023, 14, 1095366. [Google Scholar] [CrossRef]
- Shen, X.; Yu, Z.; Wei, C.; Hu, C.; Chen, J. Iron metabolism and ferroptosis in nonalcoholic fatty liver disease: What is our next step? Am. J. Physiol. Endocrinol. Metab. 2024, 326, E767–E775. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kajarabille, N.; Latunde-Dada, G.O. Programmed Cell-Death by Ferroptosis: Antioxidants as Mitigators. Int. J. Mol. Sci. 2019, 20, 4968. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Čepelak, I.; Dodig, S.; Dodig, D.Č. Ferroptosis: Regulated cell death. Arh. Hig. Rada Toksikol. 2020, 71, 99–109. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Henning, Y.; Blind, U.S.; Larafa, S.; Matschke, J.; Fandrey, J. Hypoxia aggravates ferroptosis in RPE cells by promoting the Fenton reaction. Cell Death Dis. 2022, 13, 662. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wen, R.J.; Dong, X.; Zhuang, H.W.; Pang, F.X.; Ding, S.C.; Li, N.; Mai, Y.X.; Zhou, S.T.; Wang, J.Y.; Zhang, J.F. Baicalin induces ferroptosis in osteosarcomas through a novel Nrf2/xCT/GPX4 regulatory axis. Phytomedicine 2023, 116, 154881. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.H.; Tseng, W.H.; Chi, J.T. The Intersection of DNA Damage Response and Ferroptosis-A Rationale for Combination Therapeutics. Biology 2020, 9, 187. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xu, C.; Liu, Z.; Xiao, J. Ferroptosis: A double-edged sword in gastrointestinal disease. Int. J. Mol. Sci. 2021, 22, 12403. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kang, R.; Kroemer, G. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef]
- Zeng, F.; Nijiati, S.; Tang, L.; Ye, J.; Zhou, Z.; Chen, X. Ferroptosis detection: From approaches to applications. Angew. Chem. Int. Ed. Engl. 2023, 62, e202300379. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Ma, W.; Wang, S.; Zhang, K.; Xiong, Q.; Li, Y.; Yu, H.; Du, H. Differentiation of intestinal stem cells toward goblet cells under systemic iron overload stress are associated with inhibition of Notch signaling pathway and ferroptosis. Redox Biol. 2024, 72, 103160. [Google Scholar] [CrossRef]
- Zhang, X.D.; Liu, Z.Y.; Wang, M.S.; Guo, Y.X.; Wang, X.K.; Luo, K.; Huang, S.; Li, R.F. Mechanisms and regulations of ferroptosis. Front Immunol. 2023, 14, 1269451. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, M.; Kong, X.Y.; Yao, Y.; Wang, X.A.; Yang, W.; Wu, H.; Li, S.; Ding, J.W.; Yang, J. The critical role and molecular mechanisms of ferroptosis in antioxidant systems. Ann. Transl. Med. 2022, 10, 368. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Nemeth, E.; Ganz, T. Hepcidin-Ferroportin Interaction Controls Systemic Iron Homeostasis. Int. J. Mol. Sci. 2021, 22, 6493. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, J.; Kang, R.; Tang, D. Signaling pathways and defense mechanisms of ferroptosis. FEBS J. 2022, 289, 7038–7050. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Chu, H.; Zhu, Q.; Yang, L. Ferroptosis in non-alcoholic liver disease: Molecular mechanisms and therapeutic implications. Front. Nutr. 2023, 10, 1090338. [Google Scholar] [CrossRef]
- Zhou, P.; Zhang, S.; Wang, M.; Zhou, J. The induction mechanism of ferroptosis, necroptosis, and pyroptosis in inflammatory bowel disease, colorectal cancer, and intestinal injury. Biomolecules 2023, 13, 820. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, J.; Yu, Y.; Liu, X.; Shang, X.; Du, Z.; Xu, M.L.; Zhang, T. Recent Advances in the Inhibition of Membrane Lipid Peroxidation by Food-Borne Plant Polyphenols via the Nrf2/GPx4 Pathway. J. Agric. Food Chem. 2024, 72, 12340–12355. [Google Scholar] [CrossRef] [PubMed]
- Imai, H.; Matsuoka, M.; Kumagai, T.; Sakamoto, T.; Koumura, T. Lipid Peroxidation-Dependent Cell Death Regulated by GPx4 and Ferroptosis. Curr. Top. Microbiol. Immunol. 2017, 403, 143–170. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Seiler, A.; Perisic, T.; Kölle, P.; Banjac Canak, A.; Förster, H.; Weiss, N.; Kremmer, E.; Lieberman, M.W.; Bannai, S.; et al. System x(c)- and thioredoxin reductase 1 cooperatively rescue glutathione deficiency. J. Biol. Chem. 2010, 285, 22244–22253. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bridges, R.J.; Natale, N.R.; Patel, S.A. System xc⁻ cystine/glutamate antiporter: An update on molecular pharmacology and roles within the CNS. Br. J. Pharmacol. 2012, 165, 20–34. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Averill-Bates, D.A. The antioxidant glutathione. Vitam. Horm. 2023, 121, 109–141. [Google Scholar] [CrossRef] [PubMed]
- Pei, W.; Jiang, M.; Liu, H.; Song, J.; Hu, J. The prognostic and antitumor roles of key genes of ferroptosis in liver hepatocellular cancer and stomach adenocarcinoma. Cancer Biomark. 2024, 39, 335–347. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cao, Z.; Tian, K.; Ran, Y.; Zhou, H.; Zhou, L.; Ding, Y.; Tang, X. Beclin-1: A therapeutic target at the intersection of autophagy, immunotherapy, and cancer treatment. Front. Immunol. 2024, 15, 1506426. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Song, Y.; Yang, H.; Lin, R.; Jiang, K.; Wang, B. The role of ferroptosis in digestive system cancer. Oncol. Lett. 2019, 18, 2159–2164. [Google Scholar] [CrossRef]
- Shojaie, L.; Iorga, A.; Dara, L. Cell death in liver diseases: A review. Int. J. Mol. Sci. 2020, 21, 9682. [Google Scholar] [CrossRef] [PubMed]
- Macías-Rodríguez, R.U.; Inzaugarat, M.E.; Ruiz-Margáin, A.; Nelson, L.J.; Trautwein, C.; Cubero, F.J. Reclassifying hepatic cell death during liver damage: Ferroptosis—A novel form of non-apoptotic cell death? Int. J. Mol. Sci. 2020, 21, 1651. [Google Scholar] [CrossRef]
- Santana-Codina, N.; Gikandi, A.; Mancias, J.D. The Role of NCOA4-Mediated Ferritinophagy in Ferroptosis. Adv. Exp. Med. Biol. 2021, 1301, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Zhang, H.; Qin, Q.; Hou, Y.; Zhang, X.; Chen, D.; Zhang, H.; Chen, Y. Ferroptosis as a new therapeutic opportunity for nonviral liver disease. Eur. J. Pharmacol. 2021, 908, 174319. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Hu, X.; Ai, X.; Zhang, Y. Research progress of ferroptosis and inflammatory bowel disease. BioMetals 2024, 37, 1039–1062. [Google Scholar] [CrossRef]
- Lam, I.-H.; Chan, C.-I.; Han, M.; Li, L.; Yu, H.-H. ACSL4 mediates inflammatory bowel disease and contributes to LPS-induced intestinal epithelial cell dysfunction by activating ferroptosis and inflammation. Open Med. 2024, 19, 1–12. [Google Scholar] [CrossRef]
- Zhao, Y.; Ren, P.; Luo, Q.; Li, X.; Cheng, X.; Wen, Y.; Wu, X.; Zhou, J. Ferroptosis, pathogenesis and therapy in AS co-depression disease. Front. Pharmacol. 2025, 16, 1516601. [Google Scholar] [CrossRef]
- Muro, P.; Zhang, L.; Li, S.; Zhao, Z.; Jin, T.; Mao, F.; Mao, Z. The emerging role of oxidative stress in inflammatory bowel disease. Front. Endocrinol. 2024, 15, 1390351. [Google Scholar] [CrossRef]
- Wang, Y.; Chu, T.; Meng, C.; Bian, Y.; Li, J. Piezo1-specific Deletion in Macrophage Protects the Progression of Chronic Inflammatory Bowel Disease in Mice. Cell Mol. Gastroenterol. Hepatol. 2025, 19, 101495. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Wu, Y.; Zhang, L.; Bai, B.; Han, W.; Wang, H.; Mei, Q. Epithelial Piezo1 deletion ameliorates intestinal barrier damage by regulating ferroptosis in ulcerative colitis. Free Radic. Biol. Med. 2024, 224, 272–286. [Google Scholar] [CrossRef]
- Lin, S.; Zhang, X.; Zhu, X.; Jiao, J.; Wu, Y.; Li, Y.; Zhao, L. Fusobacterium nucleatum aggravates ulcerative colitis through promoting gut microbiota dysbiosis and dysmetabolism. J. Periodontol. 2023, 94, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Wang, N.; Yao, Y.; Wang, Y.; An, X.; Wang, H.; Liu, H.; Jiang, Y.; Li, H.; Cheng, X.; et al. NEDD4L mediates intestinal epithelial cell ferroptosis to restrict inflammatory bowel diseases and colorectal tumorigenesis. J. Clin. Investig. 2025, 135, e173994. [Google Scholar] [CrossRef]
- Sun, S.P.; Lu, Y.F.; Li, H.; Weng, C.Y.; Chen, J.J.; Lou, Y.J.; Lyu, D.; Lyu, B. AMPK activation alleviated dextran sulfate sodium-induced colitis by inhibiting ferroptosis. J. Dig. Dis. 2023, 24, 213–223. [Google Scholar] [CrossRef]
- Niu, R.; Lan, J.; Liang, D.; Xiang, L.; Wu, J.; Zhang, X.; Li, Z.; Chen, H.; Geng, L.; Xu, W.; et al. GZMA suppressed GPX4-mediated ferroptosis to improve intestinal mucosal barrier function in inflammatory bowel disease. Cell Commun. Signal. 2024, 22, 474. [Google Scholar] [CrossRef] [PubMed]
- Han, S.K.; Baik, S.K.; Kim, M.Y. Non-alcoholic fatty liver disease: Definition and subtypes. Clin Mol Hepatol. 2023, 29, S5–S16. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sheka, A.C.; Adeyi, O.; Thompson, J.; Hameed, B.; Crawford, P.A.; Ikramuddin, S. Nonalcoholic Steatohepatitis: A Review. JAMA 2020, 323, 1175–1183. [Google Scholar] [CrossRef]
- Zhu, X.; Li, S. Ferroptosis, necroptosis, and pyroptosis in gastrointestinal cancers: The chief culprits of tumor progression and drug resistance. Adv. Sci. 2023, 10, e2300824. [Google Scholar] [CrossRef]
- Martín-Fernández, M.; Arroyo, V.; Carnicero, C.; Sigüenza, R.; Busta, R.; Mora, N.; Antolín, B.; Tamayo, E.; Aspichueta, P.; Carnicero-Frutos, I.; et al. Role of Oxidative Stress and Lipid Peroxidation in the Pathophysiology of NAFLD. Antioxidants 2022, 11, 2217. [Google Scholar] [CrossRef]
- Kouroumalis, E.; Tsomidis, I.; Voumvouraki, A. Iron as a therapeutic target in chronic liver disease. World J. Gastroenterol. 2023, 29, 616–655. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bo, J.Q.; Guo, Z.P.; Han, Y.H.; Liu, L.X. Advancements in ferroptosis research and therapeutic strategies for alcoholic liver disease: A narrative review. Eur. Rev. Med. Pharmacol. Sci. 2023, 27, 9296–9308. [Google Scholar] [CrossRef] [PubMed]
- Safari, M.H.; Rahimzadeh, P.; Alaei, E.; Alimohammadi, M.; Esfandiari, N.; Daneshi, S.; Malgard, N.; Farahani, N.; Taheriazam, A.; Hashemi, M. Targeting ferroptosis in gastrointestinal tumors: Interplay of iron-dependent cell death and autophagy. Mol. Cell. Probes 2025, 79, 102013. [Google Scholar] [CrossRef]
- Miotto, G.; Rossetto, M.; Di Paolo, M.L.; Orian, L.; Venerando, R.; Roveri, A.; Vučković, A.M.; Bosello Travain, V.; Zaccarin, M.; Zennaro, L.; et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 2020, 28, 101328. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, D.; Zhang, M.; Liu, J.; Li, Z.; Ni, B. Potential therapies for HCC involving targeting the ferroptosis pathway. Am. J. Cancer Res. 2024, 14, 1446–1465. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, F.; Huang, H.; Wei, X.; Tan, P.; Wang, Z.; Hu, Z. Targeting cell death pathways in intestinal ischemia-reperfusion injury: A comprehensive review. Cell Death Discov. 2024, 10, 112. [Google Scholar] [CrossRef]
- Si, C.; Zhou, X.; Deng, J.; Ye, S.; Kong, L.; Zhang, B.; Wang, W. Role of ferroptosis in gastrointestinal tumors: From mechanisms to therapies. Cell Biol. Int. 2022, 46, 997–1008. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell. 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fang, Y.; Chen, X.; Tan, Q.; Zhou, H.; Xu, J.; Gu, Q. Inhibiting Ferroptosis through Disrupting the NCOA4-FTH1 Interaction: A New Mechanism of Action. ACS Cent. Sci. 2021, 7, 980–989. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, P.; Feng, Y.; Li, H.; Chen, X.; Wang, G.; Xu, S.; Li, Y.; Zhao, L. Ferrostatin-1 alleviates lipopolysaccharide-induced acute lung injury via inhibiting ferroptosis. Cell Mol. Biol. Lett. 2020, 25, 10. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rabitha, R.; Shivani, S.; Showket, Y.; Sudhandiran, G. Ferroptosis regulates key signaling pathways in gastrointestinal tumors: Underlying mechanisms and therapeutic strategies. World J. Gastroenterol. 2023, 29, 2433–2451. [Google Scholar] [CrossRef]
- Yan, H.; Talty, R.; Johnson, C.H. Targeting ferroptosis to treat colorectal cancer. Trends Cell Biol. 2023, 33, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Lv, Y.; Zou, Y.; Kang, Z.; Li, Z.; Tian, J.; Zhou, H.; Su, W.; Zhong, J. The role of ferroptosis in colorectal cancer and its potential synergy with immunotherapy. Front. Immunol. 2025, 15, 1526749. [Google Scholar] [CrossRef]
- Sun, X.; Zhang, Q.; Lin, X.; Shu, P.; Gao, X.; Shen, K. Imatinib induces ferroptosis in gastrointestinal stromal tumors by promoting STUB1-mediated GPX4 ubiquitination. Cell Death Dis. 2023, 14, 839. [Google Scholar] [CrossRef]
- Tang, K.; Chen, Q.; Liu, Y.; Wang, L.; Lu, W. Combination of Metformin and Sorafenib Induces Ferroptosis of Hepatocellular Carcinoma Through p62-Keap1-Nrf2 Pathway. J. Cancer 2022, 13, 3234–3243. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ji, J.; Wu, L.; Wei, J.; Wu, J.; Guo, C. The Gut Microbiome and Ferroptosis in MAFLD. J. Clin. Transl. Hepatol. 2023, 11, 174–187. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cui, W.; Guo, M.; Liu, D.; Xiao, P.; Yang, C.; Huang, H.; Liang, C.; Yang, Y.; Fu, X.; Zhang, Y.; et al. Gut microbial metabolite facilitates colorectal cancer development via ferroptosis inhibition. Nat. Cell Biol. 2024, 26, 124–137. [Google Scholar] [CrossRef]
- Yao, T.; Li, L. The influence of microbiota on ferroptosis in intestinal diseases. Gut Microbes 2023, 15, 2263210. [Google Scholar] [CrossRef]
- Li, C.; Zhang, Y.; Liu, J.; Kang, R.; Klionsky, D.J.; Tang, D. Mitochondrial DNA stress triggers autophagy-dependent ferroptotic death. Autophagy 2021, 17, 948–960. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yan, H.; Talty, R.; Aladelokun, O.; Bosenberg, M.; Johnson, C.H. Ferroptosis in colorectal cancer: A future target? Br. J. Cancer. 2023, 128, 1439–1451. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Balihodzic, A.; Prinz, F.; Dengler, M.A.; Calin, G.A.; Jost, P.J.; Pichler, M. Non-coding RNAs and ferroptosis: Potential implications for cancer therapy. Cell Death Differ. 2022, 29, 1094–1106. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wawrzeńczyk, A.; Napiórkowska-Baran, K.; Alska, E.; Gruszka-Koselska, A.; Szynkiewicz, E.; Sławatycki, J.; Klemenska, P.; Bartuzi, Z. Ferroptosis in Gastrointestinal Diseases: A New Frontier in Pathogenesis and Therapy. J. Clin. Med. 2025, 14, 4035. https://doi.org/10.3390/jcm14124035
Wawrzeńczyk A, Napiórkowska-Baran K, Alska E, Gruszka-Koselska A, Szynkiewicz E, Sławatycki J, Klemenska P, Bartuzi Z. Ferroptosis in Gastrointestinal Diseases: A New Frontier in Pathogenesis and Therapy. Journal of Clinical Medicine. 2025; 14(12):4035. https://doi.org/10.3390/jcm14124035
Chicago/Turabian StyleWawrzeńczyk, Adam, Katarzyna Napiórkowska-Baran, Ewa Alska, Alicja Gruszka-Koselska, Ewa Szynkiewicz, Józef Sławatycki, Paula Klemenska, and Zbigniew Bartuzi. 2025. "Ferroptosis in Gastrointestinal Diseases: A New Frontier in Pathogenesis and Therapy" Journal of Clinical Medicine 14, no. 12: 4035. https://doi.org/10.3390/jcm14124035
APA StyleWawrzeńczyk, A., Napiórkowska-Baran, K., Alska, E., Gruszka-Koselska, A., Szynkiewicz, E., Sławatycki, J., Klemenska, P., & Bartuzi, Z. (2025). Ferroptosis in Gastrointestinal Diseases: A New Frontier in Pathogenesis and Therapy. Journal of Clinical Medicine, 14(12), 4035. https://doi.org/10.3390/jcm14124035