
Behavior of Complement System Effectors in Chronic and Acute Coronary Artery Disease

 , ,
, ,  , , ,
, , ,  ,
,
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. The Main Clinical, Electrocardiographic, Cardiac Ultrasound, Coronary Angiography, Blood Biochemical Parameters and Therapeutic Data for the Three Groups of Patients (NSTEMI, APS, and Control Groups)
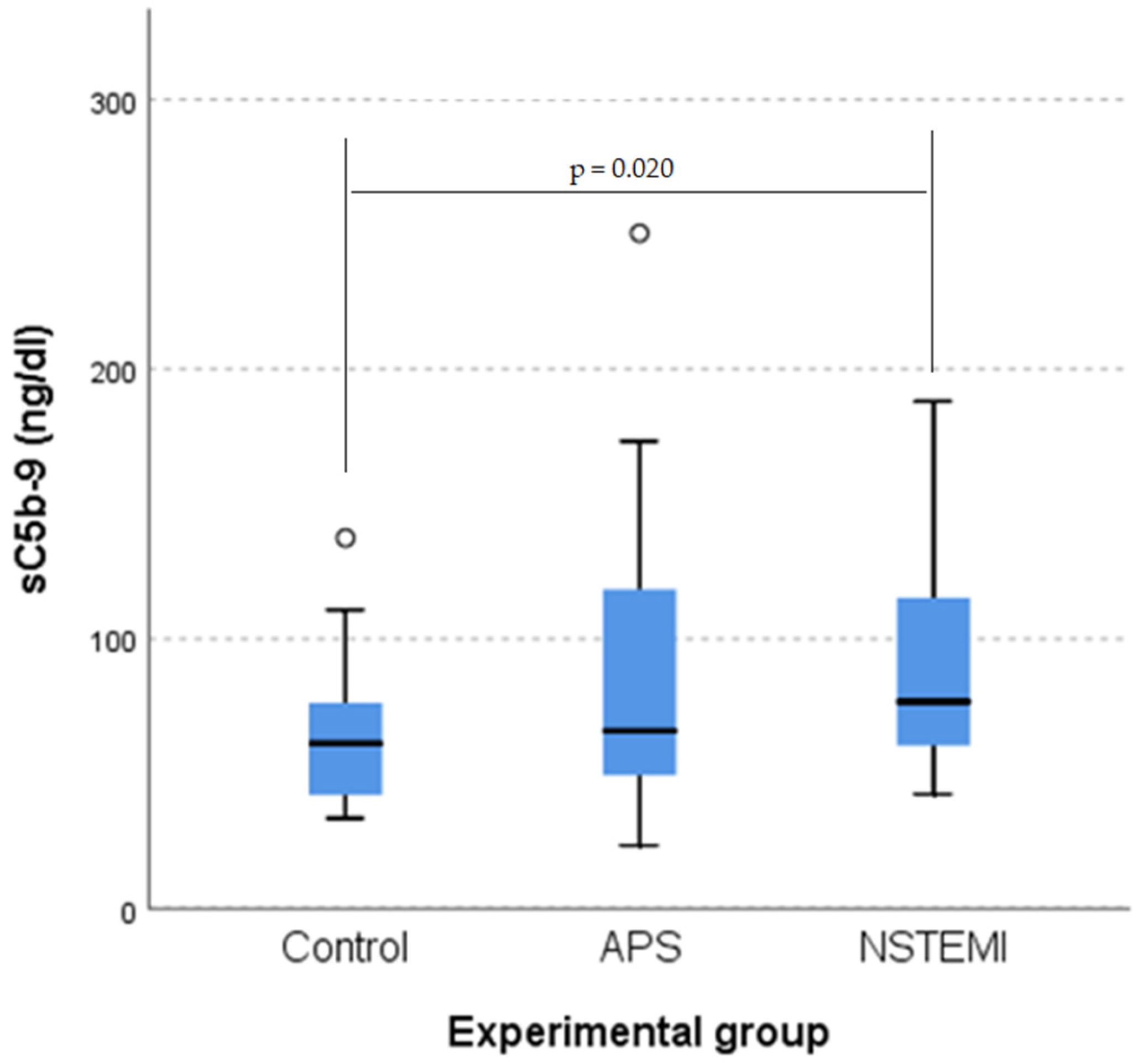
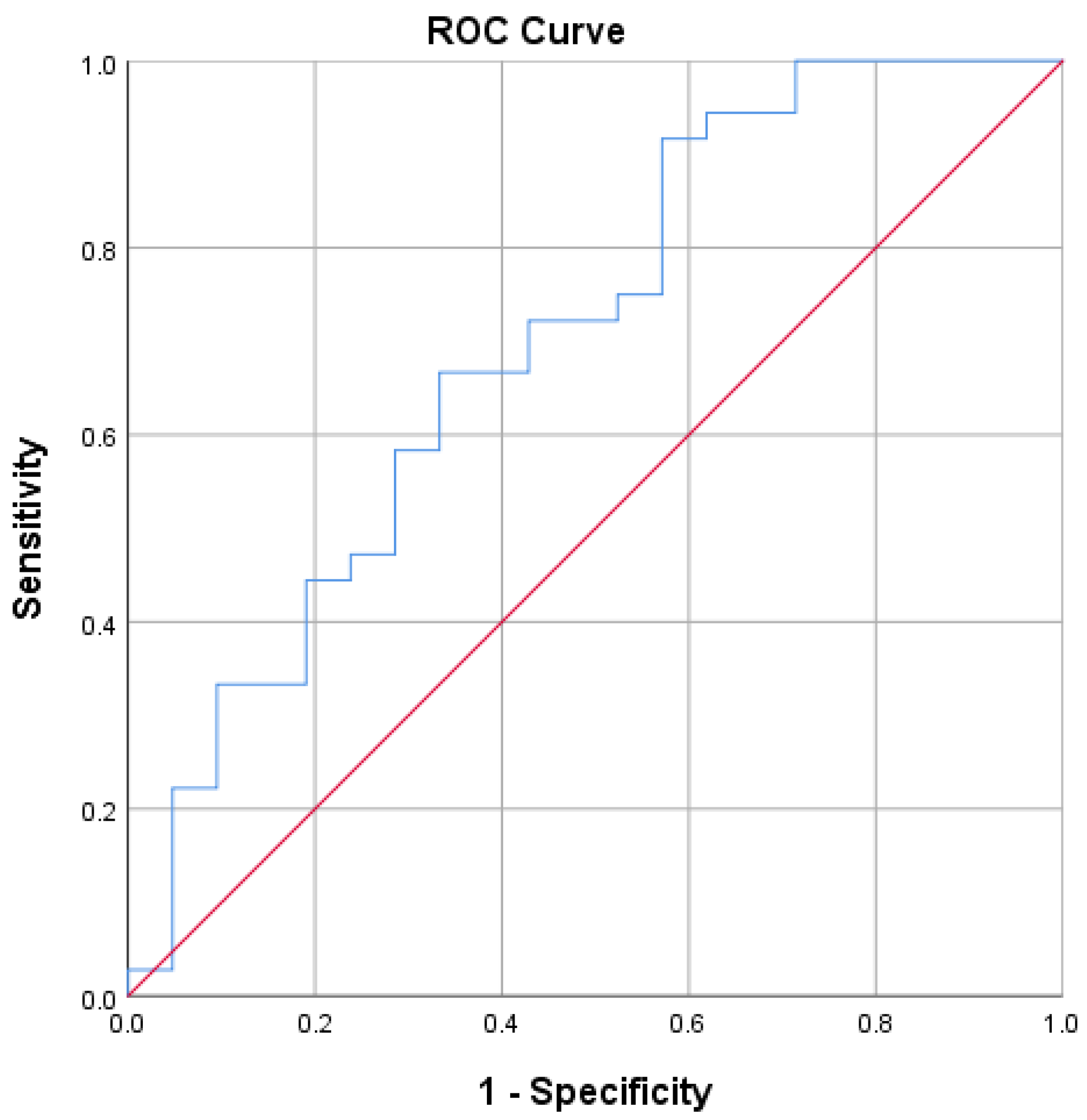
3.2. sC5b-9 as a Biomarker of Coronary Plaque Vulnerability
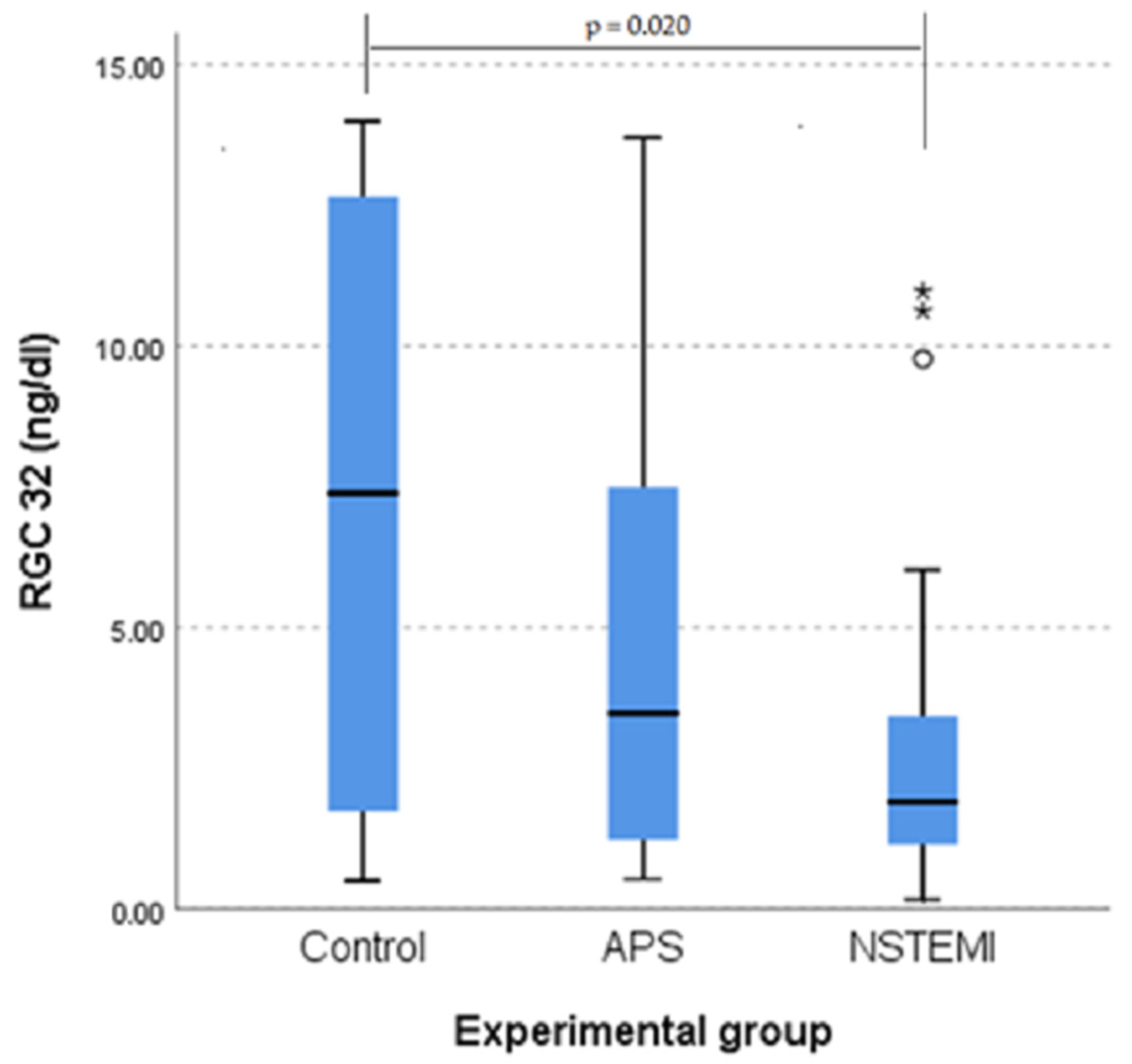
3.3. RGC-32 Is Significantly Lower in Patients with NSTEMI
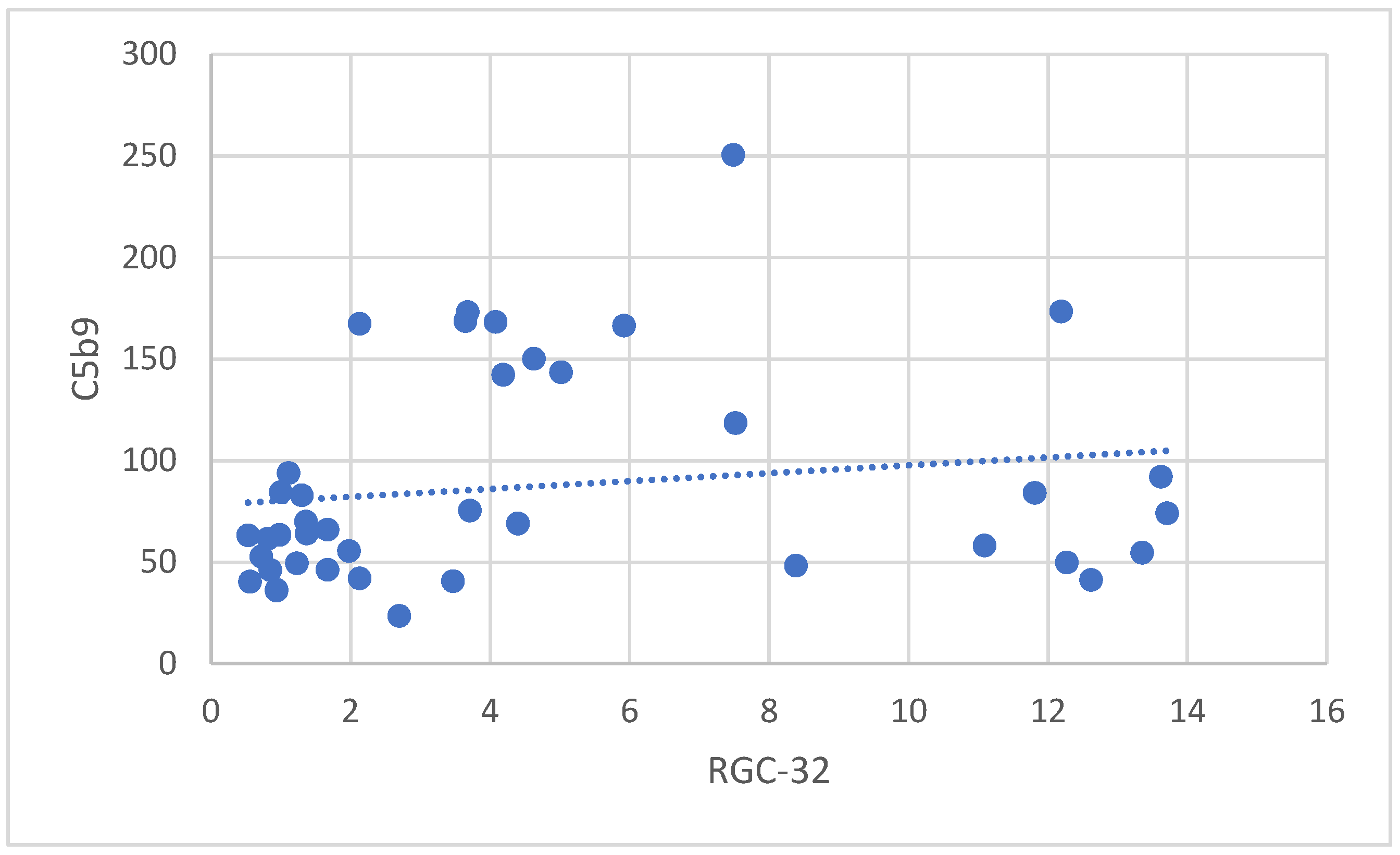
3.3.1. Correlation Between RGC-32 and sC5b-9 in Patients with APS
3.3.2. Correlation Between RGC-32 and the Extension of Atherosclerosis in Patients with APS
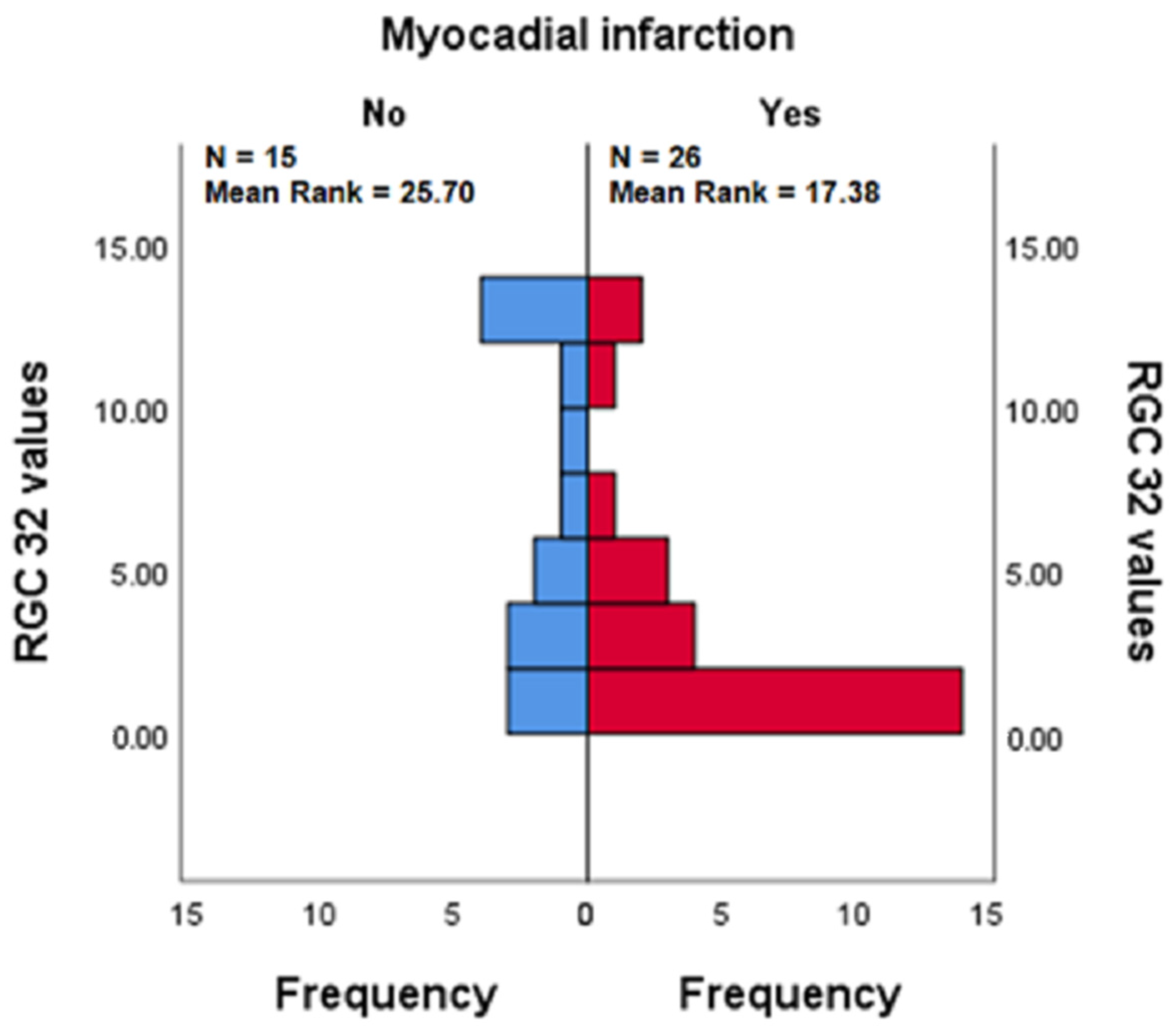
3.3.3. RGC-32 in the Presence vs. Absence of Myocardial Infarction in Patients with APS
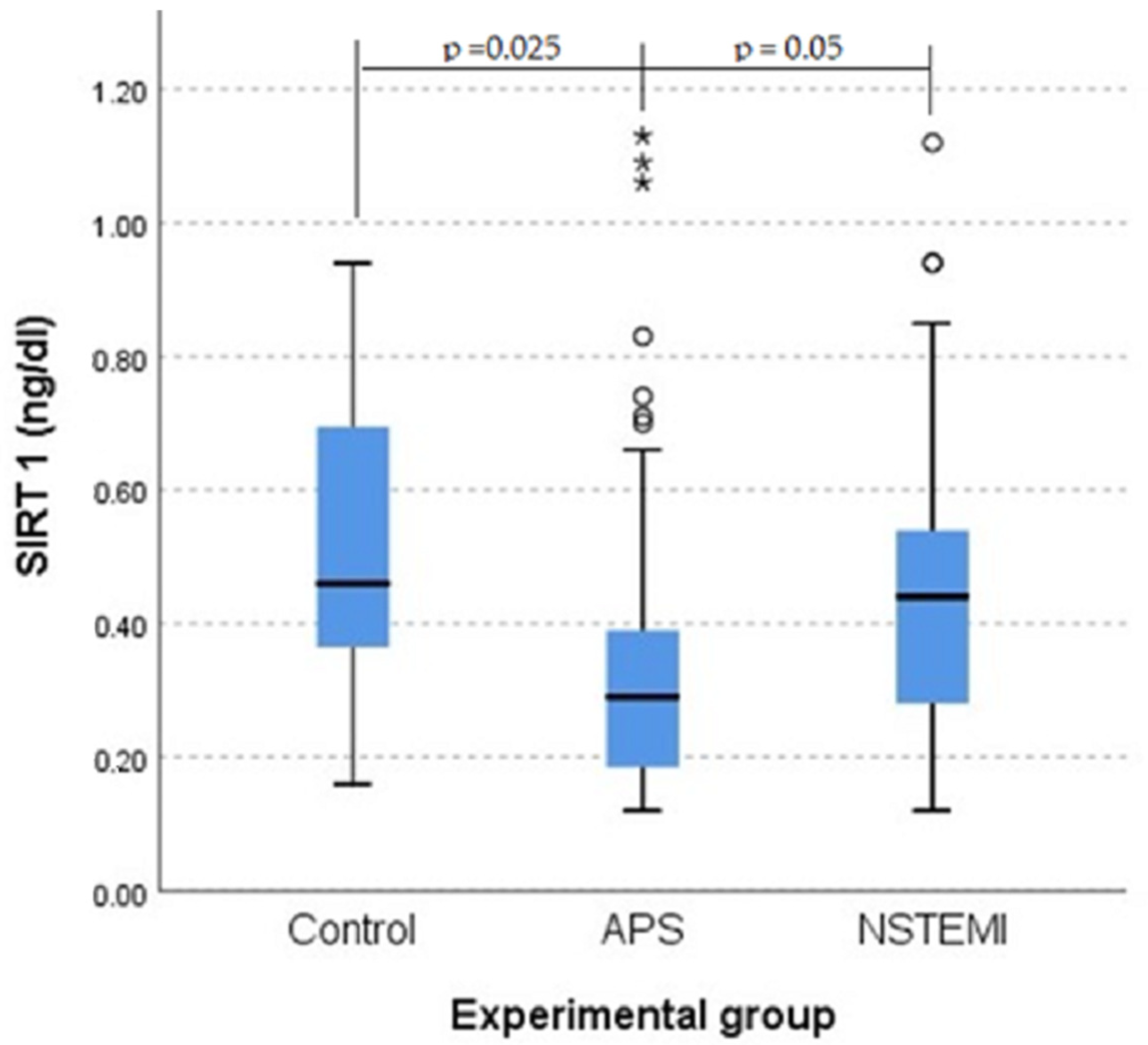
3.4. SIRT1 Plays a Role in Protection Against Atherosclerosis
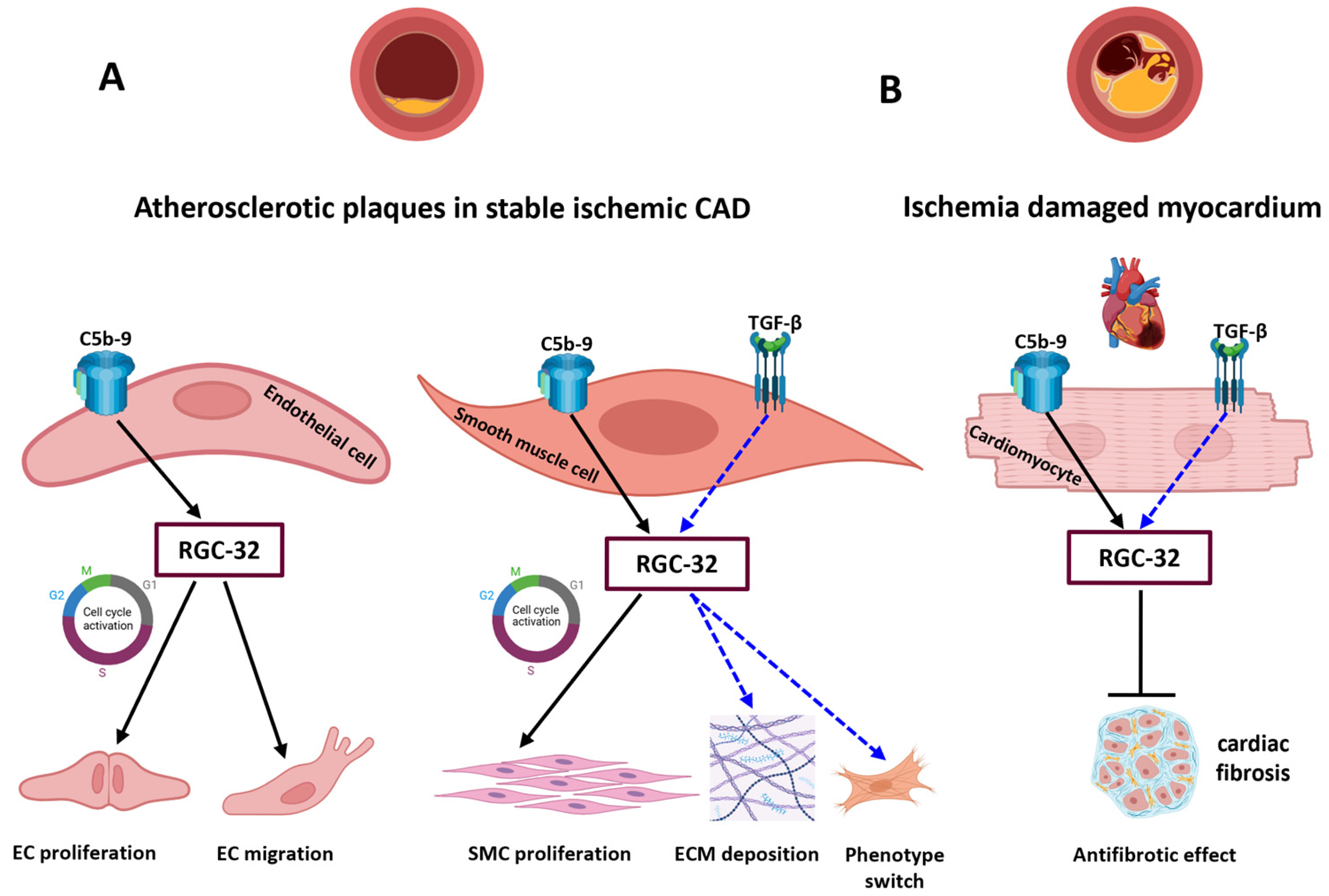
4. Discussion
Study Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACE-I | Angiotensin-converting enzyme inhibitor |
| ACS | Acute coronary syndrome |
| AMI | Acute myocardial infarction |
| ANOVA | Analysis of variance |
| APS | Angina pectoris stable |
| AUC | Area under the curve |
| CAD | Coronary artery disease |
| CO | Control group |
| Cx | Circumflex artery |
| DOAJ | Directory of open access journals |
| ECG | Electrocardiography |
| ECM | Extracellular matrix |
| EDD | End-diastolic diameters |
| EF | Ejection fraction of left ventricle |
| EKG | Electrocardiogram |
| ELISAs | Enzyme-linked immunosorbent assays |
| ESD | End-systolic diameters |
| GA | Glatiramer acetate |
| HTN | Hypertension |
| I/R | Ischemia/reperfusion |
| IMT | Intima-media thickness |
| IQR | Interquartile range |
| LAD | Left Anterior Descending artery |
| LD | Linear dichroism |
| LV | Left ventricular |
| MDPI | Multidisciplinary Digital Publishing Institute |
| MS | Multiple sclerosis |
| NO | Nitric oxide |
| NSTEMI | Non-ST-segment elevation myocardial infarction |
| PBMCs | Peripheral blood monocytes |
| RCA | Right Coronary Artery |
| RGC-32 | Response Gene to Complement-32 |
| ROC | Receiver operating characteristic curve |
| SD | Standard deviation |
| SIRT1 | Sirtuin 1 |
| TLA | Three-letter acronym |
| UA | Unstable angina |
| YI | Youden Index |
| 1VD | Single-vessel disease |
| 2VD | Two-vessel disease |
| 3VD | Three-vessel disease |
References
- Nedkoff, L.; Briffa, T.; Zemedikun, D.; Herrington, S.; Wright, F.L. Global Trends in Atherosclerotic Cardiovascular Disease. Clin. Ther. 2023, 45, 1087–1091. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Libby, P.; Tabas, I. Inflammation and Plaque Vulnerability. J. Intern. Med. 2015, 278, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Mangge, H.; Almer, G. Immune-Mediated Inflammation in Vulnerable Atherosclerotic Plaques. Molecules 2019, 24, 3072. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.J. Eradicating Atherosclerotic Events by Targeting Early Subclinical Disease: It Is Time to Retire the Therapeutic Paradigm of Too Much, Too Late. Arter. Thromb. Vasc. Biol. 2024, 44, 48–64. [Google Scholar] [CrossRef]
- Aday, A.W.; Ridker, P.M. Targeting Residual Inflammatory Risk: A Shifting Paradigm for Atherosclerotic Disease. Front. Cardiovasc. Med. 2019, 6, 16. [Google Scholar] [CrossRef]
- Gistera, A.; Hansson, G.K. The Immunology of Atherosclerosis. Nat. Rev. Nephrol. 2017, 13, 368–380. [Google Scholar] [CrossRef]
- Ajala, O.N.; Everett, B.M. Targeting Inflammation to Reduce Residual Cardiovascular Risk. Curr. Atheroscler. Rep. 2020, 22, 66. [Google Scholar] [CrossRef]
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgozoglu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef]
- Medina-Leyte, D.J.; Zepeda-García, O.; Domínguez-Pérez, M.; González-Garrido, A.; Villarreal-Molina, T.; Jacobo-Albavera, L. Endothelial Dysfunction, Inflammation and Coronary Artery Disease: Potential Biomarkers and Promising Therapeutical Approaches. Int. J. Mol. Sci. 2021, 22, 3850. [Google Scholar] [CrossRef]
- Li, J.; Song, X.; Liao, X.; Shi, Y.; Chen, H.; Xiao, Q.; Liu, F.; Zhan, J.; Cai, Y. Adaptive Enzyme-Responsive Self-Assembling Multivalent Apelin Ligands for Targeted Myocardial Infarction Therapy. J. Control. Release Off. J. Control. Release Soc. 2024, 372, 571–586. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, Z.; Ma, M.; He, Y. Soluble St2 in Coronary Artery Disease: Clinical Biomarkers and Treatment Guidance. Front. Cardiovasc. Med. 2022, 9, 924461. [Google Scholar] [CrossRef] [PubMed]
- Carter, A.M. Complement Activation: An Emerging Player in the Pathogenesis of Cardiovascular Disease. Scientifica 2012, 2012, 402783. [Google Scholar] [CrossRef] [PubMed]
- Kiss, M.G.; Binder, C.J. The Multifaceted Impact of Complement on Atherosclerosis. Atherosclerosis 2022, 351, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Vlaicu, S.I.; Tatomir, A.; Rus, V.; Mekala, A.P.; Mircea, P.A.; Niculescu, F.; Rus, H. The Role of Complement Activation in Atherogenesis: The First 40 Years. Immunol. Res. 2015, 64, 1–13. [Google Scholar] [CrossRef]
- Maffia, P.; Mauro, C.; Case, A.; Kemper, C. Canonical and Non-Canonical Roles of Complement in Atherosclerosis. Nat. Rev. Cardiol. 2024, 21, 743–761. [Google Scholar] [CrossRef]
- Badea, T.; Niculescu, F.; Soane, L.; Fosbrink, M.; Sorana, H.; Rus, V.; Shin, M.L.; Rus, H. Rgc-32 Increases P34cdc2 Kinase Activity and Entry of Aortic Smooth Muscle Cells into S-Phase. J. Biol. Chem. 2002, 277, 502–508. [Google Scholar] [CrossRef]
- Vlaicu, S.I.; Tatomir, A.; Anselmo, F.; Boodhoo, D.; Chira, R.; Rus, V.; Rus, H. Rgc-32 and Diseases: The First 20 Years. Immunol. Res. 2019, 67, 267–279. [Google Scholar] [CrossRef]
- Cui, X.B.; Chen, S.Y. Response Gene to Complement 32 in Vascular Diseases. Front. Cardiovasc. Med. 2018, 5, 128. [Google Scholar] [CrossRef]
- Cui, X.B.; Luan, J.N.; Dong, K.; Chen, S.; Wang, Y.; Watford, W.T.; Chen, S.Y. Rgc-32 (Response Gene to Complement 32) Deficiency Protects Endothelial Cells from Inflammation and Attenuates Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2018, 38, e36–e47. [Google Scholar] [CrossRef]
- Wang, J.N.; Shi, N.; Xie, W.B.; Guo, X.; Chen, S.Y. Response Gene to Complement 32 Promotes Vascular Lesion Formation through Stimulation of Smooth Muscle Cell Proliferation and Migration. Arterioscler. Thromb. Vasc. Biol. 2011, 31, e19–e26. [Google Scholar] [CrossRef]
- Vlaicu, S.I.; Tatomir, A.; Fosbrink, M.; Nguyen, V.; Boodhoo, D.; Cudrici, C.; Badea, T.C.; Rus, V.; Rus, H. Rgc-32’ Dual Role in Smooth Muscle Cells and Atherogenesis. Clin. Immunol. 2022, 238, 109020. [Google Scholar] [CrossRef] [PubMed]
- Martínez-López, D.; Roldan-Montero, R.; García-Marqués, F.; Nuñez, E.; Jorge, I.; Camafeita, E.; Minguez, P.; Rodriguez de Cordoba, S.; López-Melgar, B.; Lara-Pezzi, E.; et al. Complement C5 Protein as a Marker of Subclinical Atherosclerosis. J. Am. Coll. Cardiol. 2020, 75, 1926–1941. [Google Scholar] [CrossRef] [PubMed]
- Si, W.; He, P.; Wang, Y.; Fu, Y.; Li, X.; Lin, X.; Chen, F.; Cao, G.; Zhang, H. Complement Complex C5b-9 Levels Are Associated with the Clinical Outcomes of Acute Ischemic Stroke and Carotid Plaque Stability. Transl. Stroke Res. 2019, 10, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Kitada, M.; Ogura, Y.; Koya, D. The Protective Role of Sirt1 in Vascular Tissue: Its Relationship to Vascular Aging and Atherosclerosis. Aging 2016, 8, 2290–2307. [Google Scholar] [CrossRef]
- Breitenstein, A.; Wyss, C.A.; Spescha, R.D.; Franzeck, F.C.; Hof, D.; Riwanto, M.; Hasun, M.; Akhmedov, A.; von Eckardstein, A.; Maier, W.; et al. Peripheral Blood Monocyte Sirt1 Expression Is Reduced in Patients with Coronary Artery Disease. PLoS ONE 2013, 8, e53106. [Google Scholar] [CrossRef]
- Ionut, V.-S. Civil Liability of the Doctor; Hamangiu: Bucharest, Romania, 2013; 408p. [Google Scholar]
- Byrne, R.A.; Rossello, X.; Coughlan, J.J.; Barbato, E.; Berry, C.; Chieffo, A.; Claeys, M.J.; Dan, G.A.; Dweck, M.R.; Galbraith, M.; et al. 2023 Esc Guidelines for the Management of Acute Coronary Syndromes. Eur. Heart. J. 2023, 44, 3720–3826. [Google Scholar] [CrossRef]
- Knuuti, J.; Wijns, W.; Saraste, A.; Capodanno, D.; Barbato, E.; Funck-Brentano, C.; Prescott, E.; Storey, R.F.; Deaton, C.; Cuisset, T.; et al. 2019 Esc Guidelines for the Diagnosis and Management of Chronic Coronary Syndromes. Eur. Heart. J. 2020, 41, 407–477. [Google Scholar] [CrossRef]
- Fluss, R.; Faraggi, D.; Reiser, B. Estimation of the Youden Index and Its Associated Cutoff Point. Biom. J. Biom. Z. 2005, 47, 458–472. [Google Scholar] [CrossRef]
- Luzina, I.G.; Rus, V.; Lockatell, V.; Courneya, J.P.; Hampton, B.S.; Fishelevich, R.; Misharin, A.V.; Todd, N.W.; Badea, T.C.; Rus, H.; et al. Regulator of Cell Cycle Protein (Rgcc/Rgc-32) Protects against Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2022, 66, 146–157. [Google Scholar] [CrossRef]
- Vlaicu, S.I.; Tatomir, A.; Boodhoo, D.; Ito, T.; Fosbrink, M.; Cudrici, C.; Mekala, A.P.; Ciriello, J.; Crisan, D.; Botan, E.; et al. Rgc-32 Is Expressed in the Human Atherosclerotic Arterial Wall: Role in C5b-9-Induced Cell Proliferation and Migration. Exp. Mol. Pathol. 2016, 101, 221–230. [Google Scholar] [CrossRef]
- Chiorescu, R.M.; Mocan, M.; Inceu, A.I.; Buda, A.P.; Blendea, D.; Vlaicu, S.I. Vulnerable Atherosclerotic Plaque: Is There a Molecular Signature? Int. J. Mol. Sci. 2022, 23, 13638. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Jin, Y.; Guo, H.; Foo, S.Y.; Cully, B.L.; Wu, J.; Zeng, H.; Rosenzweig, A.; Li, J. Response Gene to Complement 32, a Novel Hypoxia-Regulated Angiogenic Inhibitor. Circulation 2009, 120, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Talman, V.; Ruskoaho, H. Cardiac Fibrosis in Myocardial Infarction-from Repair and Remodeling to Regeneration. Cell Tissue Res. 2016, 365, 563–581. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Chen, S.Y. Rgc32 Promotes Bleomycin-Induced Systemic Sclerosis in a Murine Disease Model by Modulating Classically Activated Macrophage Function. J. Immunol. 2018, 200, 2777–2785. [Google Scholar] [CrossRef]
- Huang, W.Y.; Li, Z.G.; Rus, H.; Wang, X.; Jose, P.A.; Chen, S.Y. Rgc-32 Mediates Transforming Growth Factor-Beta-Induced Epithelial-Mesenchymal Transition in Human Renal Proximal Tubular Cells. J. Biol. Chem. 2009, 284, 9426–9432. [Google Scholar] [CrossRef]
- Hashmi, K.A.; Adnan, F.; Ahmed, O.; Yaqeen, S.R.; Ali, J.; Irfan, M.; Edhi, M.M.; Hashmi, A.A. Risk Assessment of Patients after St-Segment Elevation Myocardial Infarction by Killip Classification: An Institutional Experience. Cureus 2020, 12, e12209. [Google Scholar] [CrossRef]
- Langlois, P.F.; Gawryl, M.S. Detection of the Terminal Complement Complex in Patient Plasma Following Acute Myocardial Infarction. Atherosclerosis 1988, 70, 95–105. [Google Scholar] [CrossRef]
- Liu, D.; Qi, X.; Li, Q.; Jia, W.; Wei, L.; Huang, A.; Liu, K.; Li, Z. Increased Complements and High-Sensitivity C-Reactive Protein Predict Heart Failure in Acute Myocardial Infarction. Biomed. Rep. 2016, 5, 761–765. [Google Scholar] [CrossRef]
- Shao, A.; Qi, X.; Li, Q.; Jia, W.; Wei, L.; Hou, W.; Qi, Y.; Liu, Y. Dynamic Changes of Complement Level in Patients with Acute Coronary Syndrome and Its Relationships with Myocardial Injury. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 2017, 29, 515–519. [Google Scholar] [CrossRef]
- Bavia, L.; Lidani, K.C.F.; Andrade, F.A.; Sobrinho, M.; Nisihara, R.M.; de Messias-Reason, I.J. Complement Activation in Acute Myocardial Infarction: An Early Marker of Inflammation and Tissue Injury? Immunol. Lett. 2018, 200, 18–25. [Google Scholar] [CrossRef]
- Vlaicu, R.; Rus, H.G.; Niculescu, F.; Cristea, A. Immunoglobulins and Complement Components in Human Aortic Atherosclerotic Intima. Atherosclerosis 1985, 55, 35–50. [Google Scholar] [CrossRef] [PubMed]
- Rus, H.G.; Niculescu, F.; Vlaicu, R. Presence of C5b-9 Complement Complex and S-Protein in Human Myocardial Areas with Necrosis and Sclerosis. Immunol. Lett. 1987, 16, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Väkevä, A.; Laurila, P.; Meri, S. Regulation of Complement Membrane Attack Complex Formation in Myocardial Infarction. Am. J. Pathol. 1993, 143, 65–75. [Google Scholar]
- Jia, D.; Ping, W.; Wang, M.; Wang, D.; Zhang, L.; Cao, Y. Sirt1 Mediates the Inflammatory Response of Macrophages and Regulates the Timp3/Adam17 Pathway in Atherosclerosis. Exp. Cell Res. 2024, 442, 114253. [Google Scholar] [CrossRef] [PubMed]
- Miranda, M.X.; van Tits, L.J.; Lohmann, C.; Arsiwala, T.; Winnik, S.; Tailleux, A.; Stein, S.; Gomes, A.P.; Suri, V.; Ellis, J.L.; et al. The Sirt1 Activator Srt3025 Provides Atheroprotection in Apoe−/− Mice by Reducing Hepatic Pcsk9 Secretion and Enhancing Ldlr Expression. Eur. Heart J. 2015, 36, 51–59. [Google Scholar] [CrossRef]
- Hu, Y.; Wang, L.; Chen, S.; Liu, X.; Li, H.; Lu, X.; Yang, X.; Huang, J.; Gu, D. Association between the Sirt1 Mrna Expression and Acute Coronary Syndrome. J. Atheroscler. Thromb. 2015, 22, 165–182. [Google Scholar] [CrossRef]
- Kilic, U.; Gok, O.; Bacaksiz, A.; Izmirli, M.; Elibol-Can, B.; Uysal, O. Sirt1 Gene Polymorphisms Affect the Protein Expression in Cardiovascular Diseases. PLoS ONE 2014, 9, e90428. [Google Scholar] [CrossRef]
- Hsu, C.P.; Zhai, P.; Yamamoto, T.; Maejima, Y.; Matsushima, S.; Hariharan, N.; Shao, D.; Takagi, H.; Oka, S.; Sadoshima, J. Silent Information Regulator 1 Protects the Heart from Ischemia/Reperfusion. Circulation 2010, 122, 2170–2182. [Google Scholar] [CrossRef]
- Xu, J.-J.; Cui, J.; Lin, Q.; Chen, X.-Y.; Zhang, J.; Gao, E.-H.; Wei, B.; Zhao, W. Protection of the Enhanced Nrf2 Deacetylation and Its Downstream Transcriptional Activity by Sirt1 in Myocardial Ischemia/Reperfusion Injury. Int. J. Cardiol. 2021, 342, 82–93. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Control Group (n = 21) | APS (n = 41) | NSTEMI (n = 36) | p |
|---|---|---|---|---|
| Age (years) | 59.8 ± 13.48 | 68.43 ± 12.4 | 68.85 ± 12.9 | 0.02 |
| Men (%) | 28.5 | 71.79 | 72.22 | 0.002 |
| Cardiovascular risk factors: | ||||
| HTN (%) | 71.42 | 79.48 | 91.66 | 0.49 |
| Diabetes (%) | 14.28 | 30.76 | 30.55 | 0.27 |
| Obesity (%) | 57.14 | 35.89 | 25 | 0.05 |
| Smoking (%) | 19.5 | 15.38 | 22.22 | 0.45 |
| AMI History (%) | 0 | 58.97 | 16.66% | 0 |
| Dyslipidemia (%) | 28.57 | 38.46 | 47.22 | 0.30 |
| Sedentary lifestyle | 38.09 | 15.79 | 59.38 | 0.008 |
| ECG changes | ||||
| ST segment depression > 1 mm (%) and/or negative T wave | 14.28 | 46.15 | 72.22 | <0.001 |
| Rhythm or conduction disorders (%) | 9.52 | 25.64 | 16.66 | 0.14 |
| Echocardiographic parameters: | ||||
| EF (%) | 54 | 52.12 ± 4.18 | 46.46 ± 5.12 | 0.38 |
| Ventricular kinetics disorders (n%) | 9.52 | 51.28 | 68.33 | <0.001 |
| Diastolic dysfunction (n%) | 51 | 52 | 60 | 0.45 |
| Coronary angiography (n%) | - | 87.8% | 88.88% | 0.48 |
| Univascular coronary involvement (%) | - | 55.55% | 22.22% | <0.001 |
| Serum biochemical parameters: | ||||
| Triglycerides (mg/dL) | 124.86 ± 60.18 | 148.1 ± 82.76 | 126.57 ± 67.44 | 0.28 |
| Cholesterol (mg/dL) | 171.53 ± 10.11 | 167.9 ± 51 | 165.57 ± 46.89 | 0.39 |
| LDL cholesterol (mg/dL) | 98.12 ± 42.51 | 106.4 ± 43.44 | 96 ± 37.09 | 0.74 |
| HDL cholesterol (mg/dL) | 53.13 ± 13.67 | 48 ± 11.36 | 47.40 ± 22.5 | 0.03 |
| Creatinine (mg/dL) | 0.8 ± 0.4 | 0.86 ± 0.6 | 0.98 ± 0.1 | 0.24 |
| Uric acid (mg/dL) | 5 ± 1.03 | 5.02 ± 1.84 | 7.19 ± 5.3 | 0.25 |
| Pre-study medication | ||||
| ACE-I (n%) | 44.33 | 56.14 | 51.43 | 0.23 |
| Beta-blockers (n %) | 33.33 | 74.35 | 47.22 | 0.007 |
| Aspirin (n%) | 30 | 75 | 100 | <0.001 |
| Statin (n%) | 20 | 95 | 70 | <0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiorescu, R.M.; Mocan, M.; Iacobescu, M.; Iuga, C.A.; Blendea, D.; Roșian, H.S.; Tat, R.M.; Mate, E.; Rus, H.; Vlaicu, S.I. Behavior of Complement System Effectors in Chronic and Acute Coronary Artery Disease. J. Clin. Med. 2025, 14, 3947. https://doi.org/10.3390/jcm14113947
Chiorescu RM, Mocan M, Iacobescu M, Iuga CA, Blendea D, Roșian HS, Tat RM, Mate E, Rus H, Vlaicu SI. Behavior of Complement System Effectors in Chronic and Acute Coronary Artery Disease. Journal of Clinical Medicine. 2025; 14(11):3947. https://doi.org/10.3390/jcm14113947
Chicago/Turabian StyleChiorescu, Roxana Mihaela, Mihaela Mocan, Maria Iacobescu, Cristina Adela Iuga, Dan Blendea, Horia Stefan Roșian, Raluca Mihaela Tat, Edina Mate, Horea Rus, and Sonia Irina Vlaicu. 2025. "Behavior of Complement System Effectors in Chronic and Acute Coronary Artery Disease" Journal of Clinical Medicine 14, no. 11: 3947. https://doi.org/10.3390/jcm14113947
APA StyleChiorescu, R. M., Mocan, M., Iacobescu, M., Iuga, C. A., Blendea, D., Roșian, H. S., Tat, R. M., Mate, E., Rus, H., & Vlaicu, S. I. (2025). Behavior of Complement System Effectors in Chronic and Acute Coronary Artery Disease. Journal of Clinical Medicine, 14(11), 3947. https://doi.org/10.3390/jcm14113947