
Integrative Analysis of Drug Co-Prescriptions in Peritoneal Dialysis Reveals Molecular Targets and Novel Strategies for Intervention
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
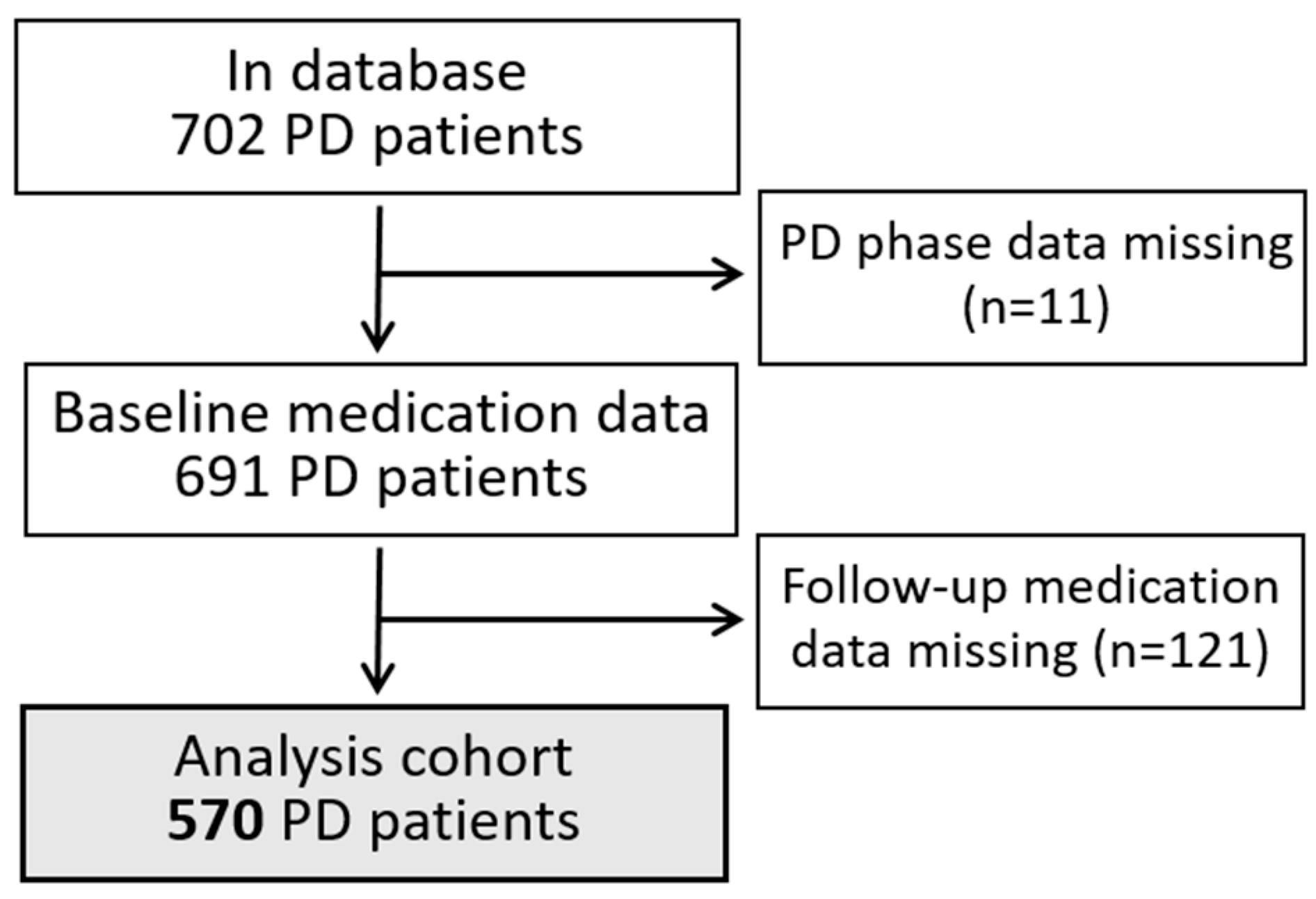
2.1. PD Patient Database
2.2. Drug Data Harmonization and Co-Prescription Analysis in PD Patients
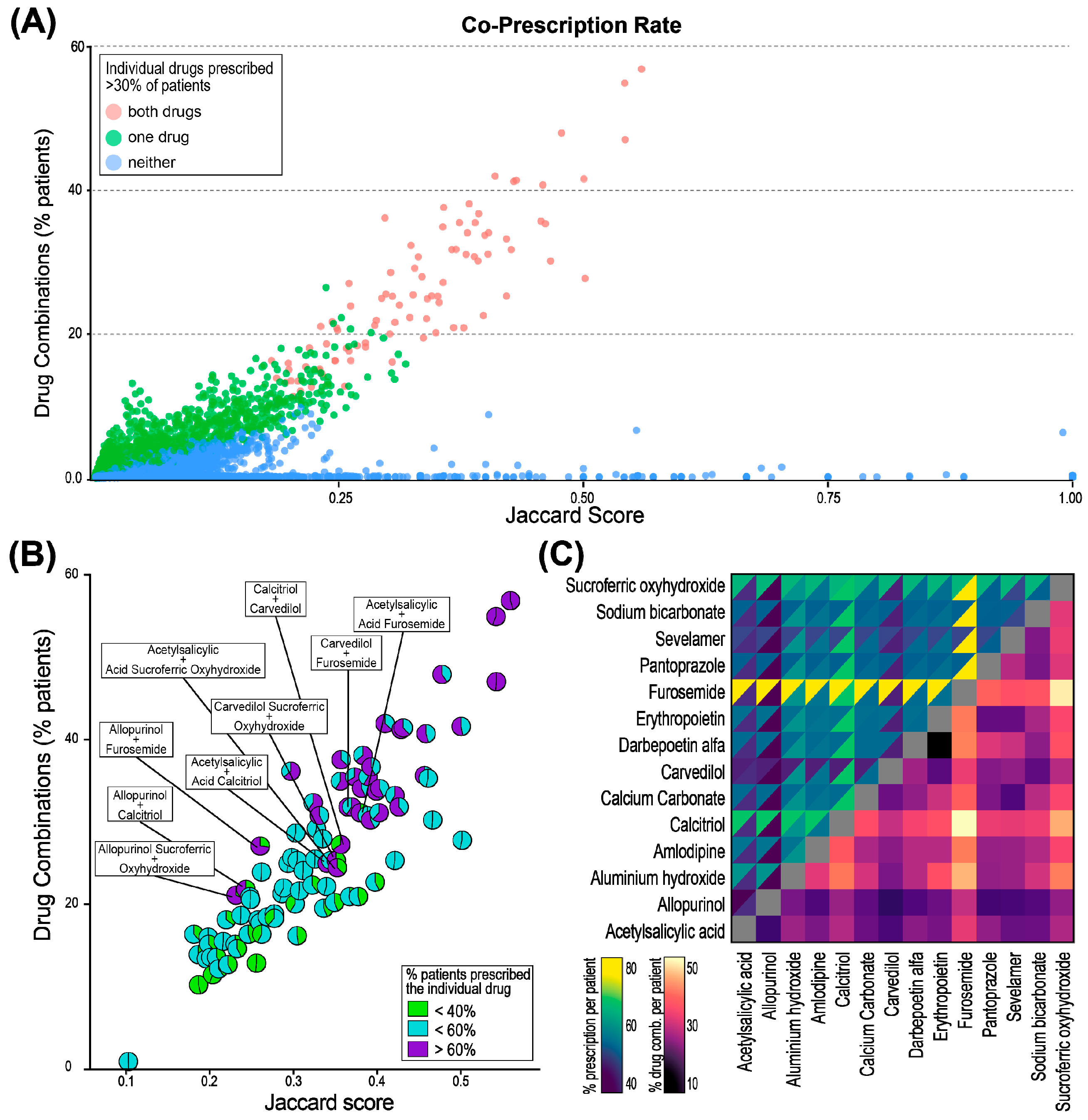
2.3. Co-Prescription Rate and Similarity of Co-Prescribed Drugs
2.4. Peritoneal Dialysis Molecular Model (“PD Disease Network”)
2.5. Drug–Target Interactions from the Public Domain and Network Clustering
2.6. Identification and Prioritization of Novel Drug Combinations Interfering with PD-Associated Molecular Processes
2.7. Statistics
3. Results
3.1. Co-Prescribed Drugs During PD Therapy Show Three Distinct Patterns
3.2. Network Analysis for Drugs Targeting Biological Processes in the “PD Disease Network”
3.3. Drug Shortlisting in PD: Targeting Angiogenesis and Inflammation
3.4. Synergistic Potential of Drug Combinations in PD: Structural and Pharmacological Similarity Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marques, L.; Costa, B.; Pereira, M.; Silva, A.; Santos, J.; Saldanha, L.; Silva, I.; Magalhaes, P.; Schmidt, S.; Vale, N. Advancing Precision Medicine: A Review of Innovative In Silico Approaches for Drug Development, Clinical Pharmacology and Personalized Healthcare. Pharmaceutics 2024, 16, 332. [Google Scholar] [CrossRef] [PubMed]
- Lopes, M.B.; Coletti, R.; Duranton, F.; Glorieux, G.; Jaimes Campos, M.A.; Klein, J.; Ley, M.; Perco, P.; Sampri, A.; Tur-Sinai, A. The Omics-Driven Machine Learning Path to Cost-Effective Precision Medicine in Chronic Kidney Disease. Proteomics 2025, e202400108. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Kar, S. Application of artificial intelligence and machine learning in early detection of adverse drug reactions (ADRs) and drug-induced toxicity. Artif. Intell. Chem. 2023, 1, 100011. [Google Scholar] [CrossRef]
- Checa-Ros, A.; Locascio, A.; Steib, N.; Okojie, O.J.; Malte-Weier, T.; Bermudez, V.; D’Marco, L. In silico medicine and -omics strategies in nephrology: Contributions and relevance to the diagnosis and prevention of chronic kidney disease. Kidney Res. Clin. Pract. 2025, 44, 49–57. [Google Scholar] [CrossRef]
- Loftus, T.J.; Shickel, B.; Ozrazgat-Baslanti, T.; Ren, Y.; Glicksberg, B.S.; Cao, J.; Singh, K.; Chan, L.; Nadkarni, G.N.; Bihorac, A. Artificial intelligence-enabled decision support in nephrology. Nat. Rev. Nephrol. 2022, 18, 452–465. [Google Scholar] [CrossRef] [PubMed]
- Karalis, V.D. The Integration of Artificial Intelligence into Clinical Practice. Appl. Biosci. 2024, 3, 14–44. [Google Scholar] [CrossRef]
- Perco, P.; Ley, M.; Keska-Izworska, K.; Wojenska, D.; Bono, E.; Walter, S.M.; Fillinger, L.; Kratochwill, K. Computational Drug Repositioning in Cardiorenal Disease: Opportunities, Challenges, and Approaches. Proteomics 2025, e202400109. [Google Scholar] [CrossRef]
- Mehrotra, R.; Devuyst, O.; Davies, S.J.; Johnson, D.W. The Current State of Peritoneal Dialysis. J. Am. Soc. Nephrol. 2016, 27, 3238–3252. [Google Scholar] [CrossRef]
- Witowski, J.; Lopez-Cabrera, M. Peritoneal Dialysis and Its Local and Systemic Complications: From the Bench to the Clinic. Front. Physiol. 2020, 11, 188. [Google Scholar] [CrossRef]
- Teitelbaum, I. Peritoneal Dialysis. N. Engl. J. Med. 2021, 385, 1786–1795. [Google Scholar] [CrossRef]
- McCormick, B.B.; Bargman, J.M. Noninfectious complications of peritoneal dialysis: Implications for patient and technique survival. J. Am. Soc. Nephrol. 2007, 18, 3023–3025. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Ronco, C.; Rosner, M.H. Computerized decision support systems: Improving patient safety in nephrology. Nat. Rev. Nephrol. 2011, 7, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Damgov, I.; Bartosova, M.; Marinovic, I.; Istanbuly, O.; Kieser, M.; Lambie, M.; Davies, S.J.; Schmitt, C.P.; Consortium, I.-P. IMPROVE-PD Finder: A Web-Based Platform to Search and Share Peritoneal Dialysis Biobank, Registry, and Clinical Trial Metadata. Kidney Int. Rep. 2023, 8, 912–915. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, P.; Sun, Z.; Hu, J. Data-Driven Prediction of Beneficial Drug Combinations in Spontaneous Reporting Systems. AMIA Annu. Symp. Proc. 2016, 2016, 808–817. [Google Scholar]
- Macalino, S.J.; Gosu, V.; Hong, S.; Choi, S. Role of computer-aided drug design in modern drug discovery. Arch. Pharm. Res. 2015, 38, 1686–1701. [Google Scholar] [CrossRef]
- Seidel, T.; Schuetz, D.A.; Garon, A.; Langer, T. The Pharmacophore Concept and Its Applications in Computer-Aided Drug Design. In Progress in the Chemistry of Organic Natural Products 110; Springer: Cham, Switzerland, 2019; Volume 110, pp. 99–141. [Google Scholar] [CrossRef]
- Gini, G. QSAR Methods. In In Silico Methods for Predicting Drug Toxicity; Methods in Molecular Biology; Humana: New York, NY, USA, 2022; Volume 2425, pp. 1–26. [Google Scholar] [CrossRef]
- Barabasi, A.L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2011, 12, 56–68. [Google Scholar] [CrossRef]
- Boezio, B.; Audouze, K.; Ducrot, P.; Taboureau, O. Network-based Approaches in Pharmacology. Mol. Inform. 2017, 36, 1700048. [Google Scholar] [CrossRef]
- Guney, E.; Menche, J.; Vidal, M.; Barabasi, A.L. Network-based in silico drug efficacy screening. Nat. Commun. 2016, 7, 10331. [Google Scholar] [CrossRef]
- Hopkins, A.L. Network pharmacology: The next paradigm in drug discovery. Nat. Chem. Biol. 2008, 4, 682–690. [Google Scholar] [CrossRef]
- Nogales, C.; Mamdouh, Z.M.; List, M.; Kiel, C.; Casas, A.I.; Schmidt, H. Network pharmacology: Curing causal mechanisms instead of treating symptoms. Trends Pharmacol. Sci. 2022, 43, 136–150. [Google Scholar] [CrossRef]
- Cheng, F.; Kovacs, I.A.; Barabasi, A.L. Network-based prediction of drug combinations. Nat. Commun. 2019, 10, 1197. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Vilar, S.; Tatonetti, N.P. High-throughput methods for combinatorial drug discovery. Sci. Transl. Med. 2013, 5, 205rv201. [Google Scholar] [CrossRef] [PubMed]
- Fillinger, L.; Walter, S.; Ley, M.; Keska-Izworska, K.; Dehkordi, L.G.; Kratochwill, K.; Perco, P. Computational modeling approaches and regulatory pathways for drug combinations. Drug Discov. Today 2025, 30, 104345. [Google Scholar] [CrossRef] [PubMed]
- Jaro, M.A. Advances in Record-Linkage Methodology as Applied to Matching the 1985 Census of Tampa, Florida. J. Am. Stat. Assoc. 1989, 84, 414–420. [Google Scholar] [CrossRef]
- Widoyo, R.; Djafri, D.; Putri, A.S.E.; Yani, F.F.; Kusumawati, R.L.; Wongsirichot, T.; Chongsuvivatwong, V. Missing Cases of Bacteriologically Confirmed TB/DR-TB from the National Treatment Registers in West and North Sumatra Provinces, Indonesia. Trop. Med. Infect. Dis. 2023, 8, 31. [Google Scholar] [CrossRef]
- Jaccard, P. The distribution of the flora in the alpine zone. New Phytol. 1912, 11, 37–50. [Google Scholar] [CrossRef]
- Gebeshuber, C.A.; Daniel-Fischer, L.; Regele, H.; Schachner, H.; Aufricht, C.; Kornauth, C.; Ley, M.; Alper, S.L.; Herzog, R.; Kratochwill, K.; et al. Computational drug repositioning of clopidogrel as a novel therapeutic option for focal segmental glomerulosclerosis. Transl. Res. 2023, 259, 28–34. [Google Scholar] [CrossRef]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef]
- Gillespie, M.; Jassal, B.; Stephan, R.; Milacic, M.; Rothfels, K.; Senff-Ribeiro, A.; Griss, J.; Sevilla, C.; Matthews, L.; Gong, C.; et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res. 2022, 50, D687–D692. [Google Scholar] [CrossRef]
- Rogers, D.; Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 update. Nucleic Acids Res. 2023, 51, D1373–D1380. [Google Scholar] [CrossRef] [PubMed]
- Evgeniou, M.; Sacnun, J.M.; Kratochwill, K.; Perco, P. A Meta-Analysis of Human Transcriptomics Data in the Context of Peritoneal Dialysis Identifies Novel Receptor-Ligand Interactions as Potential Therapeutic Targets. Int. J. Mol. Sci. 2021, 22, 3277. [Google Scholar] [CrossRef] [PubMed]
- Boehm, M.; Bukosza, E.N.; Huttary, N.; Herzog, R.; Aufricht, C.; Kratochwill, K.; Gebeshuber, C.A. A systems pharmacology workflow with experimental validation to assess the potential of anakinra for treatment of focal and segmental glomerulosclerosis. PLoS ONE 2019, 14, e0214332. [Google Scholar] [CrossRef] [PubMed]
- Fechete, R.; Heinzel, A.; Söllner, J.; Perco, P.; Lukas, A.; Mayer, B. Using Information Content for Expanding Human Protein Coding Gene Interaction Networks. J. Comput. Sci. Syst. Biol. 2013, 6, 73–82. [Google Scholar] [CrossRef]
- Fiscon, G.; Paci, P. SAveRUNNER: An R-based tool for drug repurposing. BMC Bioinform. 2021, 22, 150. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021; Available online: http://www.R-project.org (accessed on 15 May 2025).
- Balzer, M.S. Molecular pathways in peritoneal fibrosis. Cell Signal 2020, 75, 109778. [Google Scholar] [CrossRef]
- Li, J.; Liu, Y.; Liu, J. A review of research progress on mechanisms of peritoneal fibrosis related to peritoneal dialysis. Front. Physiol. 2023, 14, 1220450. [Google Scholar] [CrossRef]
- Wang, L.; Wang, R.; Liu, L.; Qiao, D.; Baldwin, D.S.; Hou, R. Effects of SSRIs on peripheral inflammatory markers in patients with major depressive disorder: A systematic review and meta-analysis. Brain Behav. Immun. 2019, 79, 24–38. [Google Scholar] [CrossRef]
- Roumestan, C.; Michel, A.; Bichon, F.; Portet, K.; Detoc, M.; Henriquet, C.; Jaffuel, D.; Mathieu, M. Anti-inflammatory properties of desipramine and fluoxetine. Respir. Res. 2007, 8, 35. [Google Scholar] [CrossRef]
- Waiskopf, N.; Ofek, K.; Gilboa-Geffen, A.; Bekenstein, U.; Bahat, A.; Bennett, E.R.; Podoly, E.; Livnah, O.; Hartmann, G.; Soreq, H. AChE and RACK1 promote the anti-inflammatory properties of fluoxetine. J. Mol. Neurosci. 2014, 53, 306–315. [Google Scholar] [CrossRef]
- Walker, F.R. A critical review of the mechanism of action for the selective serotonin reuptake inhibitors: Do these drugs possess anti-inflammatory properties and how relevant is this in the treatment of depression? Neuropharmacology 2013, 67, 304–317. [Google Scholar] [CrossRef]
- Kumar, A.; Zhang, K.Y.J. Advances in the Development of Shape Similarity Methods and Their Application in Drug Discovery. Front. Chem. 2018, 6, 315. [Google Scholar] [CrossRef] [PubMed]
- Jagirdar, R.M.; Bozikas, A.; Zarogiannis, S.G.; Bartosova, M.; Schmitt, C.P.; Liakopoulos, V. Encapsulating Peritoneal Sclerosis: Pathophysiology and Current Treatment Options. Int. J. Mol. Sci. 2019, 20, 5765. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]
- van Hasselt, J.G.C.; Iyengar, R. Systems Pharmacology: Defining the Interactions of Drug Combinations. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 21–40. [Google Scholar] [CrossRef]
- Lampe, D.; Grosser, J.; Gensorowsky, D.; Witte, J.; Muth, C.; van den Akker, M.; Dinh, T.S.; Greiner, W. The Relationship of Continuity of Care, Polypharmacy and Medication Appropriateness: A Systematic Review of Observational Studies. Drugs Aging 2023, 40, 473–497. [Google Scholar] [CrossRef]
- Hanlon, P.; Nicholl, B.I.; Jani, B.D.; McQueenie, R.; Lee, D.; Gallacher, K.I.; Mair, F.S. Examining patterns of multimorbidity, polypharmacy and risk of adverse drug reactions in chronic obstructive pulmonary disease: A cross-sectional UK Biobank study. BMJ Open 2018, 8, e018404. [Google Scholar] [CrossRef]
- Colombijn, J.M.T.; Bonenkamp, A.A.; van Eck van der Sluijs, A.; Bijlsma, J.A.; Boonstra, A.H.; Ozyilmaz, A.; Abrahams, A.C.; van Jaarsveld, B.C.; Domestico Study Group. Impact of Polypharmacy on Health-Related Quality of Life in Dialysis Patients. Am. J. Nephrol. 2021, 52, 735–744. [Google Scholar] [CrossRef]
- Marin, J.G.; Beresford, L.; Lo, C.; Pai, A.; Espino-Hernandez, G.; Beaulieu, M. Prescription Patterns in Dialysis Patients: Differences Between Hemodialysis and Peritoneal Dialysis Patients and Opportunities for Deprescription. Can. J. Kidney Health Dis. 2020, 7, 2054358120912652. [Google Scholar] [CrossRef]
- Paik, J.M.; Zhuo, M.; York, C.; Tsacogianis, T.; Kim, S.C.; Desai, R.J. Medication Burden and Prescribing Patterns in Patients on Hemodialysis in the USA, 2013-2017. Am. J. Nephrol. 2021, 52, 919–928. [Google Scholar] [CrossRef]
- Tatonetti, N.P.; Ye, P.P.; Daneshjou, R.; Altman, R.B. Data-driven prediction of drug effects and interactions. Sci. Transl. Med. 2012, 4, 125ra131. [Google Scholar] [CrossRef] [PubMed]
- Heinzel, A.; Perco, P.; Mayer, G.; Oberbauer, R.; Lukas, A.; Mayer, B. From molecular signatures to predictive biomarkers: Modeling disease pathophysiology and drug mechanism of action. Front. Cell Dev. Biol. 2014, 2, 37. [Google Scholar] [CrossRef]
- Masola, V.; Bonomini, M.; Borrelli, S.; Di Liberato, L.; Vecchi, L.; Onisto, M.; Gambaro, G.; Palumbo, R.; Arduini, A. Fibrosis of Peritoneal Membrane as Target of New Therapies in Peritoneal Dialysis. Int. J. Mol. Sci. 2022, 23, 4831. [Google Scholar] [CrossRef]
- Tomino, Y. Mechanisms and interventions in peritoneal fibrosis. Clin. Exp. Nephrol. 2012, 16, 109–114. [Google Scholar] [CrossRef]
- Ada, S.; Ersan, S.; Sifil, A.; Unlu, M.; Kolatan, E.; Sert, M.; Sarioglu, S.; Yilmaz, O.; Camsari, T. Effect of bevacizumab, a vascular endothelial growth factor inhibitor, on a rat model of peritoneal sclerosis. Int. Urol. Nephrol. 2015, 47, 2047–2051. [Google Scholar] [CrossRef] [PubMed]
- Acikgoz Mert, G.S.; Ceri, M.; Calli Demirkan, N.; Sahin, B.; Mert, M.; Dursun, B. Effect of bevacizumab and everolimus combination treatment on peritoneal sclerosis in an experimental rat model. Ther. Apher. Dial. 2021, 25, 323–330. [Google Scholar] [CrossRef] [PubMed]
- ElSheikh, R.H.; Chauhan, M.Z.; Sallam, A.B. Current and Novel Therapeutic Approaches for Treatment of Neovascular Age-Related Macular Degeneration. Biomolecules 2022, 12, 1629. [Google Scholar] [CrossRef]
- Ding, X.; Patel, M.; Chan, C.C. Molecular pathology of age-related macular degeneration. Prog. Retin. Eye Res. 2009, 28, 1–18. [Google Scholar] [CrossRef]
- McMahon, C.G. Efficacy of dapoxetine in the treatment of premature ejaculation. Clin. Med. Insights Reprod. Health 2011, 5, 25–39. [Google Scholar] [CrossRef]
- Sangkum, P.; Badr, R.; Serefoglu, E.C.; Hellstrom, W.J. Dapoxetine and the treatment of premature ejaculation. Transl. Androl. Urol. 2013, 2, 301–311. [Google Scholar] [CrossRef]
- Gul, M.; Bocu, K.; Serefoglu, E.C. Current and emerging treatment options for premature ejaculation. Nat. Rev. Urol. 2022, 19, 659–680. [Google Scholar] [CrossRef] [PubMed]
- Cilan, H.; Sipahioglu, M.H.; Oguzhan, N.; Unal, A.; Turan, T.; Koc, A.N.; Tokgoz, B.; Utas, C.; Oymak, O. Association between depression, nutritional status, and inflammatory markers in peritoneal dialysis patients. Ren. Fail. 2013, 35, 17–22. [Google Scholar] [CrossRef] [PubMed]
- King-Wing Ma, T.; Kam-Tao Li, P. Depression in dialysis patients. Nephrology 2016, 21, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, P.; Zhang, L.; Zhang, Y.; Xie, L.; Niu, J. Prevalence and predisposing factors of depressive symptoms in continuous ambulatory peritoneal dialysis patients: A cross-sectional single center study. BMC Nephrol. 2023, 24, 104. [Google Scholar] [CrossRef]
- Yirmiya, R. The inflammatory underpinning of depression: An historical perspective. Brain Behav. Immun. 2024, 122, 433–443. [Google Scholar] [CrossRef]
- Fattahian, E.; Hajhashemi, V.; Rabbani, M.; Minaiyan, M.; Mahzouni, P. Anti-inflammatory Effect of Amitriptyline on Ulcerative Colitis in Normal and Reserpine-Induced Depressed Rats. Iran. J. Pharm. Res. 2016, 15, 125–137. [Google Scholar]
- Takenaka, Y.; Tanaka, R.; Kitabatake, K.; Kuramochi, K.; Aoki, S.; Tsukimoto, M. Profiling Differential Effects of 5 Selective Serotonin Reuptake Inhibitors on TLRs-Dependent and -Independent IL-6 Production in Immune Cells Identifies Fluoxetine as Preferred Anti-Inflammatory Drug Candidate. Front. Pharmacol. 2022, 13, 874375. [Google Scholar] [CrossRef]
- Hatamnejad, M.R.; Baradaran Ghavami, S.; Shirvani, M.; Asghari Ahmadabad, M.; Shahrokh, S.; Farmani, M.; Sherkat, G.; Asadzadeh Aghdaei, H.; Zali, M.R. Selective serotonin reuptake inhibitors and inflammatory bowel disease; Beneficial or malpractice. Front. Immunol. 2022, 13, 980189. [Google Scholar] [CrossRef]
- Meikle, C.K.S.; Creeden, J.F.; McCullumsmith, C.; Worth, R.G. SSRIs: Applications in inflammatory lung disease and implications for COVID-19. Neuropsychopharmacol. Rep. 2021, 41, 325–335. [Google Scholar] [CrossRef]
- Sacre, S.; Medghalchi, M.; Gregory, B.; Brennan, F.; Williams, R. Fluoxetine and citalopram exhibit potent antiinflammatory activity in human and murine models of rheumatoid arthritis and inhibit toll-like receptors. Arthritis Rheum. 2010, 62, 683–693. [Google Scholar] [CrossRef]
- Hishida, E.; Ito, H.; Komada, T.; Karasawa, T.; Kimura, H.; Watanabe, S.; Kamata, R.; Aizawa, E.; Kasahara, T.; Morishita, Y.; et al. Crucial Role of NLRP3 Inflammasome in the Development of Peritoneal Dialysis-related Peritoneal Fibrosis. Sci. Rep. 2019, 9, 10363. [Google Scholar] [CrossRef] [PubMed]
- Stavenuiter, A.W.; Schilte, M.N.; Ter Wee, P.M.; Beelen, R.H. Angiogenesis in peritoneal dialysis. Kidney Blood Press. Res. 2011, 34, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, P.; Schilte, M.N.; Zareie, M.; ter Wee, P.M.; Keuning, E.D.; Beelen, R.H.; van den Born, J. Celecoxib treatment reduces peritoneal fibrosis and angiogenesis and prevents ultrafiltration failure in experimental peritoneal dialysis. Nephrol. Dial. Transplant. 2009, 24, 3669–3676. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Hahn, H.J.; Oh, S.R.; Lee, H.J. Theophylline Attenuates BLM-Induced Pulmonary Fibrosis by Inhibiting Th17 Differentiation. Int. J. Mol. Sci. 2023, 24, 1019. [Google Scholar] [CrossRef]
- Bozkurt, D.; Sarsik, B.; Hur, E.; Ertilav, M.; Karaca, B.; Timur, O.; Bicak, S.; Akcicek, F.; Duman, S. A novel angiogenesis inhibitor, sunitinib malate, in encapsulating peritoneal sclerosis. J. Nephrol. 2011, 24, 359–365. [Google Scholar] [CrossRef]
- Tapiawala, S.N.; Bargman, J.M.; Oreopoulos, D.G.; Simons, M. Prolonged use of the tyrosine kinase inhibitor in a peritoneal dialysis patient with metastatic renal cell carcinoma: Possible beneficial effects on peritoneal membrane and peritonitis rates. Int. Urol. Nephrol. 2009, 41, 431–434. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mean ± SD/n(%) | |
|---|---|
| Age (years) a | 54 ± 14 |
| Sex (n, male/female) | 330 (58%)/240 (42%) |
| Diabetes mellitus (n (%)) b | 132 (23%) |
| Etiology of ESKD (n (%)) | |
| Chronic glomerulonephritis | 108 (19%) |
| Diabetic nephropathy | 72 (13%) |
| Polycystic kidney disease | 64 (11%) |
| Vascular nephropathy | 26 (5%) |
| Other or unknown | 300 (53%) |
| Time on PD (months) a | 29 ± 22 |
| PD modality (n, APD/CAPD/IPD) c | 224 (39%)/292 (51%)/54 (10%) |
| Endpoint at time of study end (n (%)) d | |
| Kidney transplantation | 219 (38%) |
| Transfer to HD | 141 (25%) |
| Death | 182 (32%) |
| Recovery of kidney function | 8 (1%) |
| Transfer to another PD center | 19 (3%) |
| Drug prescription timepoints | 5835 |
| Individual drugs e | 547 |
| ATC code 4th level (drug category) | 323 |
| ATC code 5th level | 764 |
| Patients in decade 1/2/3 (n (%)) f | 137 (24%)/262 (46%)/171 (30%) |
| Drug Class (ATC 2nd Level) | Drug | Drug Target (s) | Proximity Score * | p-Value | z-Score |
|---|---|---|---|---|---|
| Analgesics | Dihydrocodeine | OPRM1 | 0 | 0.0305 | −1.916 |
| Hydromorphone | OPRM1 | 0 | 0.0305 | −1.916 | |
| Morphine | OPRM1 | 0 | 0.0305 | −1.916 | |
| Tramadol | OPRM1 | 0 | 0.0305 | −1.916 | |
| Antibacterials for systemic use | Doxycycline # | MMP7, MMP8, MMP13, MMP1, rpsB, […] | 0.500 | 0.0174 | −0.673 |
| Antidiarrheals | Loperamide | OPRM1 | 0 | 0.0305 | −1.916 |
| Antiemetics | Ondansetron | HTR3A | 0.600 | 0.0249 | −0.425 |
| Antihistamines for systematic use | Cetirizine # | HRH1 | 0 | 0.0041 | −1.916 |
| Desloratadine | HRH1 | 0 | 0.0041 | −1.916 | |
| Levocetirizine | HRH1 | 0 | 0.0041 | −1.916 | |
| Loratadine | HRH1 | 0 | 0.0041 | −1.916 | |
| Diphenhydramine # | HRH1 | 0 | 0.0219 | −1.916 | |
| Antihypertensives | Clonidine | ADRA2A, ADRA2C, ADRA2B | 0.556 | 0.0077 | −0.535 |
| Anti-inflammatory and antirheumatic products | Celecoxib # | PTGS2 | 0 | 0.0383 | −1.916 |
| Meloxicam | PTGS2 | 0 | 0.0383 | −1.916 | |
| Rofecoxib | PTGS2 | 0 | 0.0383 | −1.916 | |
| Antineoplastic agents | Pazopanib | CSF1R, KIT, LCK, FGFR3, FGFR1, PDGFRB, PDGFRA, FLT1, FLT4, KDR, ITK | 0.273 | 0.0002 | −1.238 |
| Capecitabine | TYMS | 0 | 0.0050 | −1.916 | |
| Cardiac therapy | Flecainide | SCN5A | 0 | 0.0343 | −1.916 |
| Drugs for acid-related disorders | Sucralfate | PGA5 | 0 | 0.0021 | −1.916 |
| Drugs for constipation | Naloxegol | OPRM1 | 0 | 0.0305 | −1.916 |
| Drugs for obstructive airway diseases | Theophylline | PDE4A, PDE4D, PDE4B, PDE4C, PDE3A, PDE3B, ADORA1, ADORA2B, ADORA3, ADORA2A | 0.600 | 0.0148 | −0.425 |
| Drugs for treatment of bone diseases | Denosumab | TNFSF11 | 0 | 0.0113 | −1.916 |
| Immunostimulants | Filgrastim | CSF3R | 0 | 0.0298 | −1.916 |
| Immunosuppressants | Mycophenolic acid | IMPDH1, IMPDH2 | 0.600 | 0.0303 | −0.425 |
| Other nervous system drugs | Methadone | OPRM1 | 0 | 0.0305 | −1.916 |
| Psychoanaleptics | Citalopram | SLC6A4 | 0 | 0.0068 | −1.916 |
| Escitalopram | SLC6A4 | 0 | 0.0068 | −1.916 | |
| Fluoxetine | SLC6A4 | 0 | 0.0068 | −1.916 | |
| Sertraline | SLC6A4 | 0 | 0.0068 | −1.916 | |
| Hydroxyzine | HRH1 | 0 | 0.0219 | −1.916 | |
| Paroxetine | SLC6A4 | 0 | 0.0343 | −1.916 | |
| Amitriptyline | SLC6A4, SLC6A2 | 0.500 | 0.0466 | −0.673 | |
| Duloxetine | SLC6A4, SLC6A2 | 0.500 | 0.0466 | −0.673 | |
| Milnacipran | SLC6A4, SLC6A2 | 0.500 | 0.0466 | −0.673 | |
| Venlafaxine | SLC6A4, SLC6A2 | 0.500 | 0.0466 | −0.673 | |
| Psycholeptics | Melatonin | MTNR1A, MTNR1B | 0 | 0.0080 | −1.916 |
| Urologicals | Finasteride | SRD5A2 | 0 | 0.0124 | −1.916 |
| Sildenafil | PDE5A | 0 | 0.0168 | −1.916 | |
| Tadalafil | PDE5A | 0 | 0.0168 | −1.916 | |
| Vardenafil | PDE5A | 0 | 0.0168 | −1.916 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evgeniou, M.; Perco, P.; Eibensteiner, F.; Unterwurzacher, M.; Vychytil, A.; Herzog, R.; Kratochwill, K. Integrative Analysis of Drug Co-Prescriptions in Peritoneal Dialysis Reveals Molecular Targets and Novel Strategies for Intervention. J. Clin. Med. 2025, 14, 3733. https://doi.org/10.3390/jcm14113733
Evgeniou M, Perco P, Eibensteiner F, Unterwurzacher M, Vychytil A, Herzog R, Kratochwill K. Integrative Analysis of Drug Co-Prescriptions in Peritoneal Dialysis Reveals Molecular Targets and Novel Strategies for Intervention. Journal of Clinical Medicine. 2025; 14(11):3733. https://doi.org/10.3390/jcm14113733
Chicago/Turabian StyleEvgeniou, Michail, Paul Perco, Fabian Eibensteiner, Markus Unterwurzacher, Andreas Vychytil, Rebecca Herzog, and Klaus Kratochwill. 2025. "Integrative Analysis of Drug Co-Prescriptions in Peritoneal Dialysis Reveals Molecular Targets and Novel Strategies for Intervention" Journal of Clinical Medicine 14, no. 11: 3733. https://doi.org/10.3390/jcm14113733
APA StyleEvgeniou, M., Perco, P., Eibensteiner, F., Unterwurzacher, M., Vychytil, A., Herzog, R., & Kratochwill, K. (2025). Integrative Analysis of Drug Co-Prescriptions in Peritoneal Dialysis Reveals Molecular Targets and Novel Strategies for Intervention. Journal of Clinical Medicine, 14(11), 3733. https://doi.org/10.3390/jcm14113733