
Bilateral Sensorineural Hearing Loss in a Patient with Primary Ciliary Dyskinesia and Concomitant SH3TC2 Gene Mutation

 , , and
, , and {kind=link}
{kind=link}
Abstract
1. Introduction
2. Case Study
2.1. Patient Background
2.2. Respiratory and Nasal Assessment
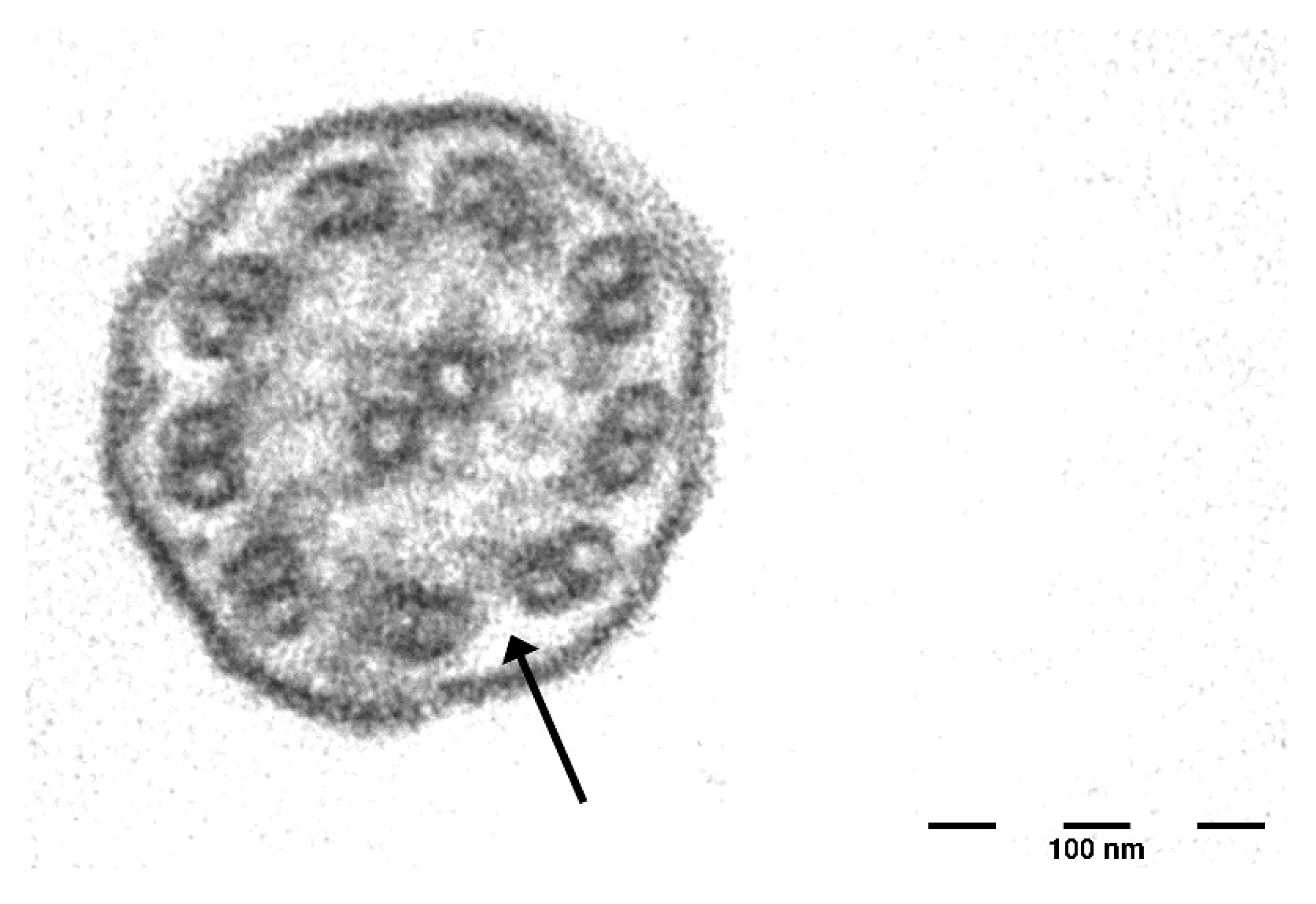
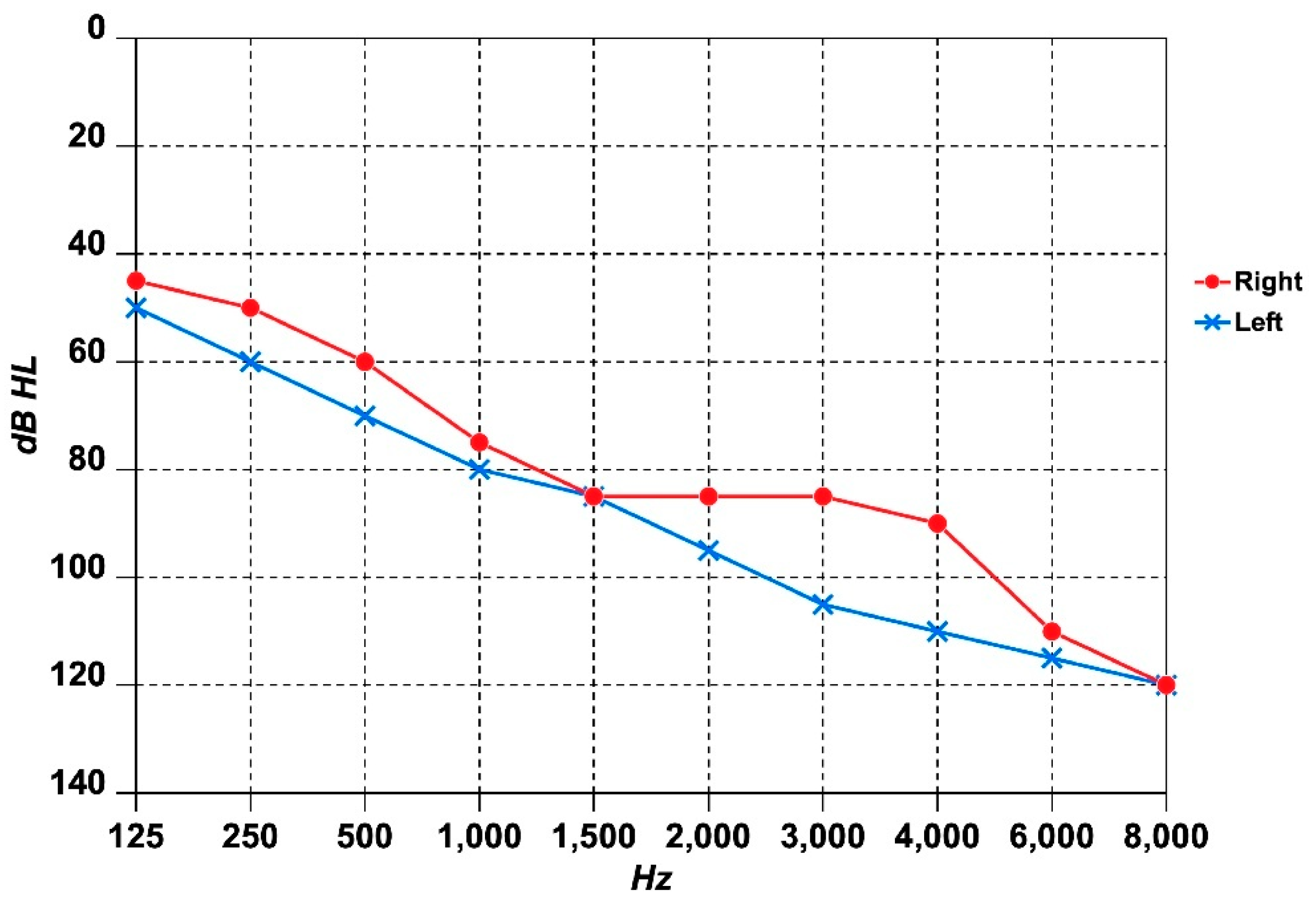
2.3. Audiological and Vestibular Assessment
2.4. Genetic Tests
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mirra, V.; Werner, C.; Santamaria, F. Primary Ciliary Dyskinesia: An Update on Clinical Aspects, Genetics, Diagnosis, and Future Treatment Strategies. Front. Pediatr. 2017, 5, 135. [Google Scholar] [CrossRef] [PubMed]
- Zariwala, M.A.; Knowles, M.R.; Leigh, M.W. Primary Ciliary Dyskinesia. In GeneReviews® [Internet]; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2007. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1122/ (accessed on 20 March 2025).
- Piatti, G.; Girotto, G.; Concas, M.P.; Braga, L.; Ambrosetti, U.; Aldè, M. TAS2R38 Genotype Does Not Affect SARS-CoV-2 Infection in Primary Ciliary Dyskinesia. Int. J. Mol. Sci. 2024, 25, 8635. [Google Scholar] [CrossRef]
- Despotes, K.A.; Zariwala, M.A.; Davis, S.D.; Ferkol, T.W. Primary Ciliary Dyskinesia: A Clinical Review. Cells 2024, 13, 974. [Google Scholar] [CrossRef] [PubMed]
- Tinoco, E.M.; Gigante, A.R.; Ferreira, E.; Sanches, I.; Pereira, R.; Sá, R.; Monteiro, R.; Sousa, M.; Pascoal, I. Primary Ciliary Dyskinesia in a Portuguese Bronchiectasis Outpatient Clinic. Genes 2023, 14, 541. [Google Scholar] [CrossRef]
- Alsamri, M.T.; Alabdouli, A.; Iram, D.; Alkalbani, A.M.; Almarzooqi, A.S.; Souid, A.-K.; Vijayan, R. A Study on the Genetics of Primary Ciliary Dyskinesia. J. Clin. Med. 2021, 10, 5102. [Google Scholar] [CrossRef] [PubMed]
- Nagappa, M.; Sharma, S.; Taly, A.B. Charcot-Marie-Tooth Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK562163/ (accessed on 20 March 2025).
- Timmerman, V.; Strickland, A.V.; Züchner, S. Genetics of Charcot-Marie-Tooth (CMT) Disease within the Frame of the Human Genome Project Success. Genes 2014, 5, 13–32. [Google Scholar] [CrossRef]
- Stavrou, M.; Sargiannidou, I.; Georgiou, E.; Kagiava, A.; Kleopa, K.A. Emerging Therapies for Charcot-Marie-Tooth Inherited Neuropathies. Int. J. Mol. Sci. 2021, 22, 6048. [Google Scholar] [CrossRef]
- Cortese, A.; Wilcox, J.E.; Polke, J.M.; Poh, R.; Skorupinska, M.; Rossor, A.M.; Laura, M.; Tomaselli, P.J.; Houlden, H.; Shy, M.E.; et al. Targeted next-generation sequencing panels in the diagnosis of Charcot-Marie-Tooth disease. Neurology 2020, 94, e51–e61. [Google Scholar] [CrossRef]
- Jerath, N.U.; Mankodi, A.; Crawford, T.O.; Grunseich, C.; Baloui, H.; Nnamdi-Emeratom, C.; Schindler, A.B.; Heiman-Patterson, T.; Chrast, R.; Shy, M.E. Charcot-Marie-Tooth Disease type 4C: Novel mutations, clinical presentations, and diagnostic challenges. Muscle Nerve 2018, 57, 749–755. [Google Scholar] [CrossRef]
- Avgeri, C.; Sideris, G.; Moriki, D.; Douros, K.; Delides, A.; Nikolopoulos, T. Bilateral Hearing Loss in Primary Ciliary Dyskinesia: A Study of Conductive and Sensorineural Mechanisms from Pediatric and Adult Cases. J. Int. Adv. Otol. 2025, 21, 1–4. [Google Scholar] [CrossRef]
- Sivera, R.; Cavalle, L.; Vílchez, J.J.; Espinós, C.; Pérez Garrigues, H.; Sevilla, T. Audiological Findings in Charcot-Marie-Tooth Disease Type 4C. J. Int. Adv. Otol. 2017, 13, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Aldè, M.; Ambrosetti, U.; Barozzi, S.; Aldè, S. The Ongoing Challenges of Hearing Loss: Stigma, Socio-Cultural Differences, and Accessibility Barriers. Audiol. Res. 2025, 15, 46. [Google Scholar] [CrossRef] [PubMed]
- Aldè, M.; Bosi, P.; Muck, S.; Mayr, T.; Di Mauro, P.; Berto, V.; Aleandri, B.G.; Folino, F.; Barozzi, S.; Zanetti, D.; et al. Long-Term Impact of Recurrent Acute Otitis Media on Balance and Vestibular Function in Children. Brain Sci. 2024, 14, 1246. [Google Scholar] [CrossRef]
- Aldè, M.; Ambrosetti, U.; Piatti, G.; Romanini, C.; Filipponi, E.; Di Berardino, F.; Zanetti, D.; Pignataro, L.; Cantarella, G.; Barozzi, S. Sudden Sensorineural Hearing Loss in Patients Aged from 15 to 40 Years. J. Clin. Med. 2024, 13, 3303. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Alexandru, M.; de Boissieu, P.; Benoudiba, F.; Moustarhfir, M.; Kim, S.; Bequignon, É.; Honoré, I.; Garcia, G.; Mitri-Frangieh, R.; Legendre, M.; et al. Otological Manifestations in Adults with Primary Ciliary Dyskinesia: A Controlled Radio-Clinical Study. J. Clin. Med. 2022, 11, 5163. [Google Scholar] [CrossRef]
- Bequignon, E.; Dupuy, L.; Zerah-Lancner, F.; Bassinet, L.; Honoré, I.; Legendre, M.; Devars du Mayne, M.; Escabasse, V.; Crestani, B.; Maître, B.; et al. Critical Evaluation of Sinonasal Disease in 64 Adults with Primary Ciliary Dyskinesia. J. Clin. Med. 2019, 8, 619. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.J.; Zariwala, M.A.; Ferkol, T.; Davis, S.D.; Sagel, S.D.; Dell, S.D.; Rosenfeld, M.; Olivier, K.N.; Milla, C.; Daniel, S.J.; et al. Diagnosis, monitoring, and treatment of primary ciliary dyskinesia: PCD foundation consensus recommendations based on state of the art review. Pediatr. Pulmonol. 2016, 51, 115–132. [Google Scholar] [CrossRef]
- Gooding, R.; Colomer, J.; King, R.; Angelicheva, D.; Marns, L.; Parman, Y.; Chandler, D.; Bertranpetit, J.; Kalaydjieva, L. A novel Gypsy founder mutation, p.Arg1109X in the CMT4C gene, causes variable peripheral neuropathy phenotypes. J. Med. Genet. 2005, 42, e69. [Google Scholar] [CrossRef]
- Claramunt, R.; Sevilla, T.; Lupo, V.; Cuesta, A.; Millán, J.M.; Vílchez, J.J.; Palau, F.; Espinós, C. The p.R1109X mutation in SH3TC2 gene is predominant in Spanish Gypsies with Charcot-Marie-Tooth disease type 4. Clin. Genet. 2007, 71, 343–349. [Google Scholar] [CrossRef]
- Lerat, J.; Magdelaine, C.; Lunati, A.; Dzugan, H.; Dejoie, C.; Rego, M.; Beze Beyrie, P.; Bieth, E.; Calvas, P.; Cintas, P.; et al. Implication of the SH3TC2 gene in Charcot-Marie-Tooth disease associated with deafness and/or scoliosis: Illustration with four new pathogenic variants. J. Neurol. Sci. 2019, 406, 116376. [Google Scholar] [CrossRef] [PubMed]
- Andersen, T.N.; Alanin, M.C.; von Buchwald, C.; Nielsen, L.H. A longitudinal evaluation of hearing and ventilation tube insertion in patients with primary ciliary dyskinesia. Int. J. Pediatr. Otorhinolaryngol. 2016, 89, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Goutaki, M.; Lam, Y.T.; Alexandru, M.; Anagiotos, A.; Armengot, M.; Boon, M.; Burgess, A.; Caversaccio, N.; Crowley, S.; Dheyauldeen, S.A.D.; et al. Characteristics of Otologic Disease Among Patients with Primary Ciliary Dyskinesia. JAMA Otolaryngol. Head Neck Surg. 2023, 149, 587–596. [Google Scholar] [CrossRef]
- Krawczyński, M.R.; Dmeńska, H.; Witt, M. Apparent X-linked primary ciliary dyskinesia associated with retinitis pigmentosa and a hearing loss. J. Appl. Genet. 2004, 45, 107–110. [Google Scholar]
- Zawawi, F.; Shapiro, A.J.; Dell, S.; Wolter, N.E.; Marchica, C.L.; Knowles, M.R.; Zariwala, M.A.; Leigh, M.W.; Smith, M.; Gajardo, P.; et al. Otolaryngology Manifestations of Primary Ciliary Dyskinesia: A Multicenter Study. Otolaryngol. Head Neck Surg. 2022, 166, 540–547. [Google Scholar] [CrossRef]
- Piatti, G.; De Santi, M.M.; Torretta, S.; Pignataro, L.; Soi, D.; Ambrosetti, U. Cilia and Ear. Ann. Otol. Rhinol. Laryngol. 2017, 126, 322–327. [Google Scholar] [CrossRef]
- Jones, C.; Roper, V.C.; Foucher, I.; Qian, D.; Banizs, B.; Petit, C.; Yoder, B.K.; Chen, P. Ciliary proteins link basal body polarization to planar cell polarity regulation. Nat. Genet. 2008, 40, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Ezan, J.; Lasvaux, L.; Gezer, A.; Novakovic, A.; May-Simera, H.; Belotti, E.; Lhoumeau, A.C.; Birnbaumer, L.; Beer-Hammer, S.; Borg, J.P.; et al. Primary cilium migration depends on G-protein signalling control of subapical cytoskeleton. Nat. Cell. Biol. 2013, 15, 1107–1115. [Google Scholar] [CrossRef]
- Senderek, J.; Bergmann, C.; Stendel, C.; Kirfel, J.; Verpoorten, N.; De Jonghe, P.; Timmerman, V.; Chrast, R.; Verheijen, M.H.; Lemke, G.; et al. Mutations in a gene encoding a novel SH3/TPR domain protein cause autosomal recessive Charcot-Marie-Tooth type 4C neuropathy. Am. J. Hum. Genet. 2003, 73, 1106–1119. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aldè, M.; Ambrosetti, U.; Guazzo, R.; Rocca, M.S.; Piatti, G. Bilateral Sensorineural Hearing Loss in a Patient with Primary Ciliary Dyskinesia and Concomitant SH3TC2 Gene Mutation. J. Clin. Med. 2025, 14, 3692. https://doi.org/10.3390/jcm14113692
Aldè M, Ambrosetti U, Guazzo R, Rocca MS, Piatti G. Bilateral Sensorineural Hearing Loss in a Patient with Primary Ciliary Dyskinesia and Concomitant SH3TC2 Gene Mutation. Journal of Clinical Medicine. 2025; 14(11):3692. https://doi.org/10.3390/jcm14113692
Chicago/Turabian StyleAldè, Mirko, Umberto Ambrosetti, Raffaella Guazzo, Maria Santa Rocca, and Gioia Piatti. 2025. "Bilateral Sensorineural Hearing Loss in a Patient with Primary Ciliary Dyskinesia and Concomitant SH3TC2 Gene Mutation" Journal of Clinical Medicine 14, no. 11: 3692. https://doi.org/10.3390/jcm14113692
APA StyleAldè, M., Ambrosetti, U., Guazzo, R., Rocca, M. S., & Piatti, G. (2025). Bilateral Sensorineural Hearing Loss in a Patient with Primary Ciliary Dyskinesia and Concomitant SH3TC2 Gene Mutation. Journal of Clinical Medicine, 14(11), 3692. https://doi.org/10.3390/jcm14113692