
Early Neonatal Fosdenopterin Treatment for Molybdenum Cofactor Deficiency Type A: New Insights into Its Natural History and Potential Role for Fetal Therapy

 , , and
, , and
Abstract

1. Introduction
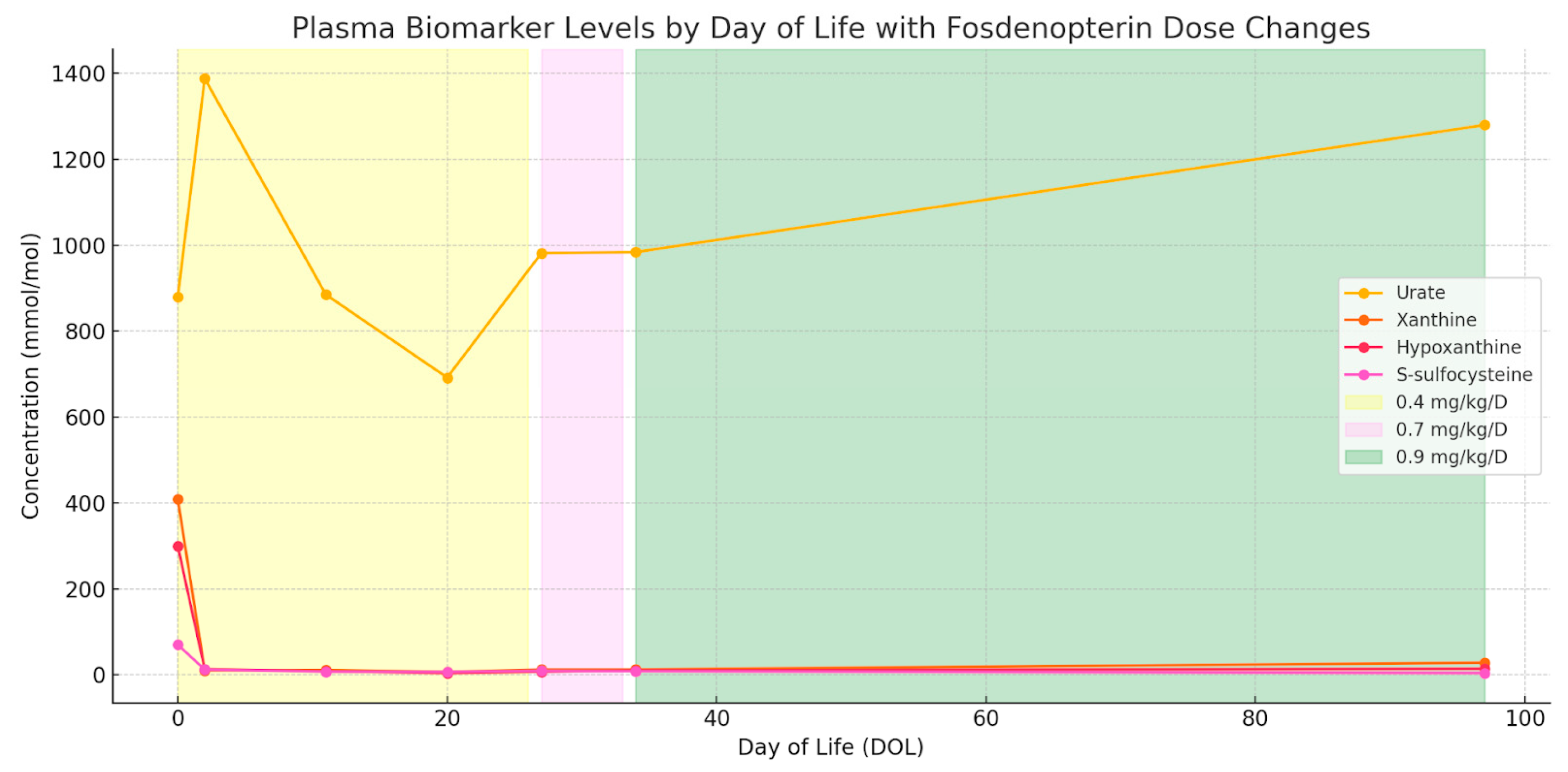
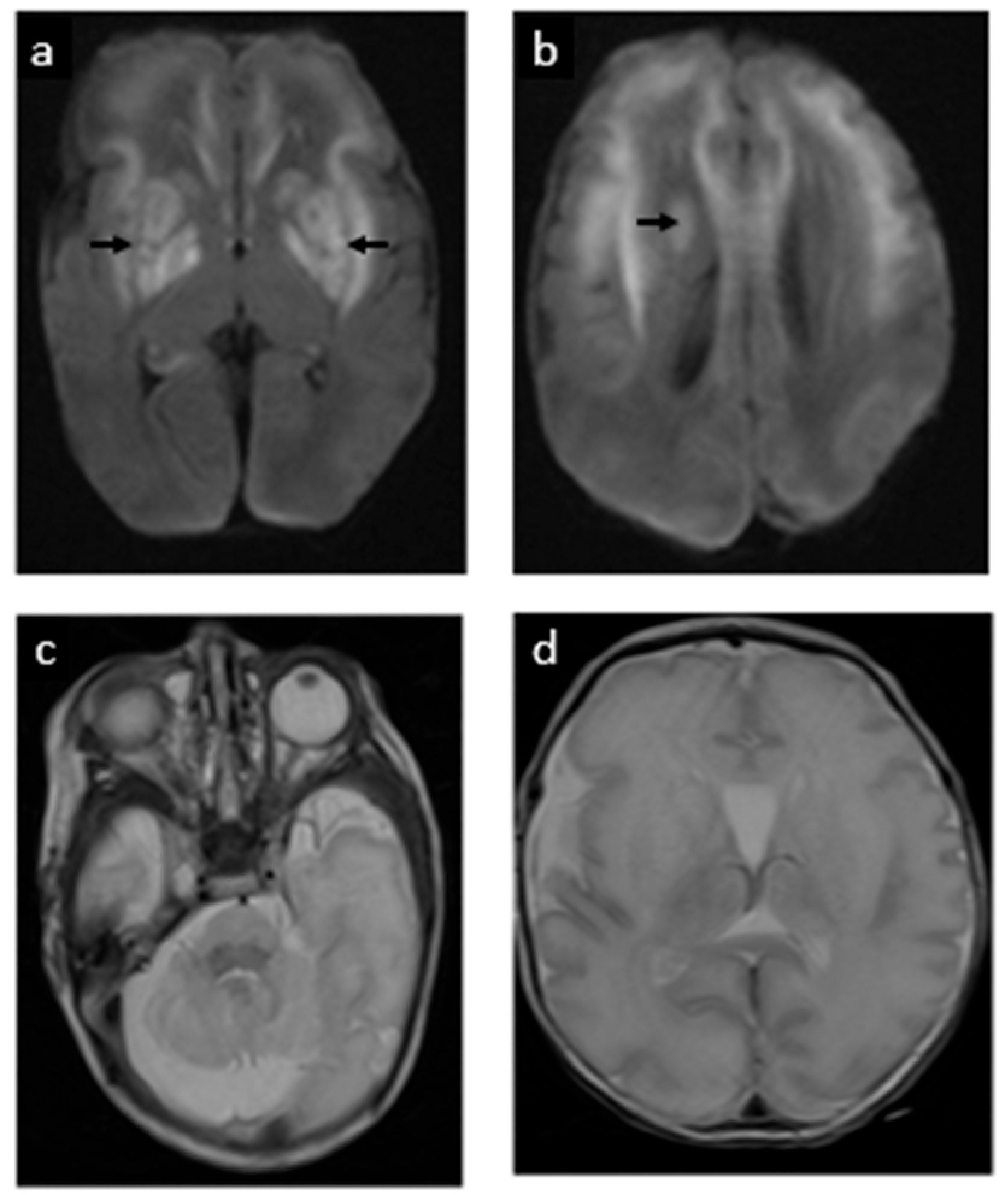
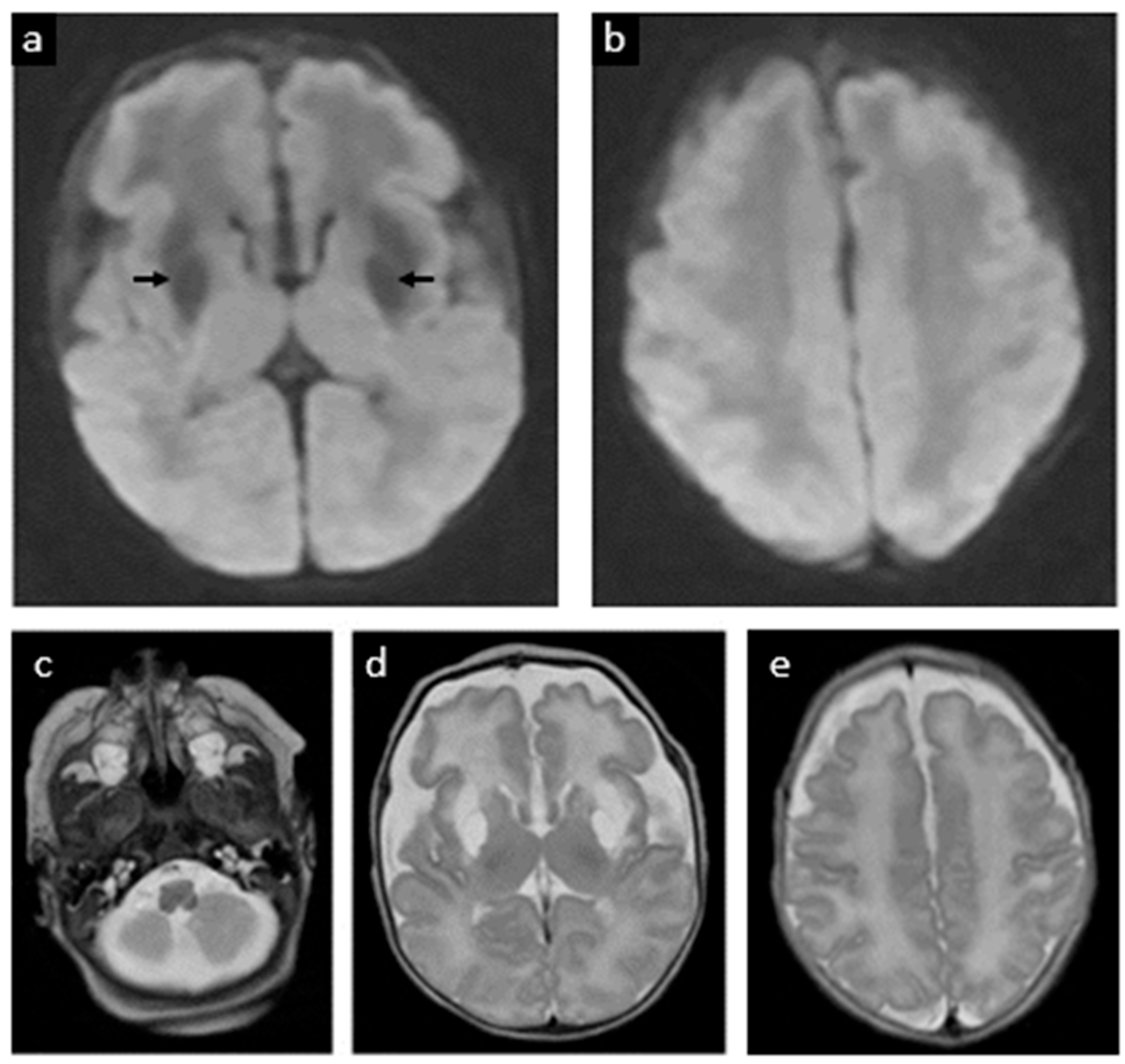
2. Case Report
3. Discussion
3.1. Diagnosis and Natural History
3.2. Treatment Approaches
3.3. Review of the Literature: Neonatal Treatment with Fosdenopterin
3.4. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Spiegel, R.; Schwahn, B.C.; Squires, L.; Confer, N. Molybdenum cofactor deficiency: A natural history. J. Inherit. Metab. Dis. 2022, 45, 456–469. [Google Scholar] [CrossRef] [PubMed]
- Per, H.; Gümüş, H.; Ichida, K.; Cağlayan, O.; Kumandaş, S. Molybdenum cofactor deficiency: Clinical features in a Turkish patient. Brain Dev. 2007, 29, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Chandran, S.; Muthanandam, D.; Ponmudi, N.; Kumar, M. Expanding the Phenotype of Molybdenum Cofactor Deficiency in Neonates: Report of Two Cases. J. Pediatr. Neurol. 2022, 20, 277–282. [Google Scholar] [CrossRef]
- Misko, A.; Mahtani, K.; Abbott, J.; Schwarz, G.; Atwal, P. Molybdenum Cofactor Deficiency. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Parini, R.; Briscioli, V.; Caruso, U.; Dorche, C.; Fortuna, R.; Minniti, G.; Selicorni, A.; Vismara, E.; Mancini, G. Spherophakia associated with molybdenum cofactor deficiency. Am. J. Med. Genet. 1997, 73, 272–275. [Google Scholar] [CrossRef]
- Atwal, P.S.; Scaglia, F. Molybdenum cofactor deficiency. Mol. Genet. Metab. 2016, 117, 1–4. [Google Scholar] [CrossRef]
- Waring, W.S.; Maxwell, S. Diagnosis of molybdenum cofactor deficiency. Lancet 1999, 353, 675–676. [Google Scholar] [CrossRef] [PubMed]
- Johannes, L.; Fu, C.-Y.; Schwarz, G. Molybdenum cofactor deficiency in humans. Molecules 2022, 27, 6896. [Google Scholar] [CrossRef]
- Carmi-Nawi, N.; Malinger, G.; Mandel, H.; Ichida, K.; Lerman-Sagie, T.; Lev, D. Prenatal brain disruption in molybdenum cofactor deficiency. J. Child Neurol. 2011, 26, 460–464. [Google Scholar] [CrossRef]
- Lubout, C.M.; Derks, T.G.; Meiners, L.; Erwich, J.J.; Bergman, K.A.; Lunsing, R.J.; Schwarz, G.; Veldman, A.; van Spronsen, F.J. Molybdenum cofactor deficiency type A: Prenatal monitoring using MRI. Eur. J. Paediatr. Neurol. 2018, 22, 536–540. [Google Scholar] [CrossRef]
- Alonzo Martínez, M.; Cazorla, E.; Cánovas, E.; Anniuk, K.; Cores, A.; Serrano, A. Molybdenum cofactor deficiency: Mega cisterna magna in two consecutive pregnancies and review of the literature. Appl. Clin. Genet. 2020, 13, 49–55. [Google Scholar] [CrossRef]
- Wyse, A.T.; Grings, M.; Wajner, M.; Leipnitz, G. The role of oxidative stress and bioenergetic dysfunction in sulfite oxidase deficiency: Insights from animal models. Neurotox. Res. 2019, 35, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Farrell, S.; Karp, J.; Hager, R.; Wang, Y.; Adeniyi, O.; Wang, J.; Li, L.; Ma, L.; Peretz, J.; Summan, M. Regulatory news: Nulibry (fosdenopterin) approved to reduce the risk of mortality in patients with molybdenum cofactor deficiency type A: FDA approval summary. J. Inherit. Metab. Dis. 2021, 44, 1085–1087. [Google Scholar] [CrossRef] [PubMed]
- Salman, M.S.; Ackerley, C.; Senger, C.; Becker, L. New insights into the neuropathogenesis of molybdenum cofactor deficiency. Can. J. Neurol. Sci. 2002, 29, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Veldman, A.; Santamaria-Araujo, J.A.; Sollazzo, S.; Pitt, J.; Gianello, R.; Yaplito-Lee, J.; Wong, F.; Ramsden, C.A.; Reiss, J.; Cook, I. Successful treatment of molybdenum cofactor deficiency type A with cPMP. Pediatrics 2010, 125, e1249–e1254. [Google Scholar] [CrossRef]
- Schwahn, B.C.; Van Spronsen, F.J.; Belaidi, A.A.; Bowhay, S.; Christodoulou, J.; Derks, T.G.; Hennermann, J.B.; Jameson, E.; König, K.; McGregor, T.L. Efficacy and safety of cyclic pyranopterin monophosphate substitution in severe molybdenum cofactor deficiency type A: A prospective cohort study. Lancet 2015, 386, 1955–1963. [Google Scholar] [CrossRef]
- Abe, Y.; Aihara, Y.; Endo, W.; Hasegawa, H.; Ichida, K.; Uematsu, M.; Kure, S. The effect of dietary protein restriction in a case of molybdenum cofactor deficiency with MOCS1 mutation. Mol. Genet. Metab. Rep. 2021, 26, 100716. [Google Scholar] [CrossRef]
- Kumar, A.; Dejanovic, B.; Hetsch, F.; Semtner, M.; Fusca, D.; Arjune, S.; Santamaria-Araujo, J.A.; Winkelmann, A.; Ayton, S.; Bush, A.I. S-sulfocysteine/NMDA receptor–dependent signaling underlies neurodegeneration in molybdenum cofactor deficiency. J. Clin. Investig. 2017, 127, 4365–4378. [Google Scholar] [CrossRef]
- Schwahn, B.C.; Hart, C.; Smith, L.A.; Hart, A.; Fairbanks, L.; Arenas-Hernandez, M.; Turner, C.; Horman, A.; Rust, S.; Santamaria-Araujo, J.A.; et al. cPMP rescue of a neonate with severe molybdenum cofactor deficiency after serendipitous early diagnosis, and characterisation of a novel MOCS1 variant. Mol. Genet. Metab. 2024, 143, 108598. [Google Scholar] [CrossRef]
- Hitzert, M.M.; Bos, A.F.; Bergman, K.A.; Veldman, A.; Schwarz, G.; Santamaria-Araujo, J.A.; Heiner-Fokkema, R.; Sival, D.A.; Lunsing, R.J.; Arjune, S. Favorable outcome in a newborn with molybdenum cofactor type A deficiency treated with cPMP. Pediatrics 2012, 130, e1005–e1010. [Google Scholar] [CrossRef]
- Schwahn, B.C.; van Spronsen, F.; Misko, A.; Pavaine, J.; Holmes, V.; Spiegel, R.; Schwarz, G.; Wong, F.; Horman, A.; Pitt, J. Consensus guidelines for the diagnosis and management of isolated sulfite oxidase deficiency and molybdenum cofactor deficiencies. J. Inherit. Metab. Dis. 2024, 47, 598–623. [Google Scholar] [CrossRef]
- Schwahn, B.; Barvíková, K.; Wu, H.; Horman, A.; Emmett, E.; Kožich, V. Pharmacodynamic profiling in three patients with molybdenum cofactor deficiency type A reveals prolonged biological effects after withdrawal of cyclic pyranopterin monophosphate. Mol. Genet. Metab. 2024, 143, 108563. [Google Scholar] [CrossRef]
- Kuegler, S.; Hahnewald, R.; Garrido, M.; Reiss, J. Long-term rescue of a lethal inherited disease by adeno-associated virus–mediated gene transfer in a mouse model of molybdenum-cofactor deficiency. Am. J. Hum. Genet. 2007, 80, 291–297. [Google Scholar] [CrossRef] [PubMed]
- de Coppi, P.; Loukogeorgakis, S.; Götherström, C.; David, A.L.; Almeida-Porada, G.; Chan, J.K.; Deprest, J.; Wong, K.K.Y.; Tam, P.K.H. Regenerative medicine: Prenatal approaches. Lancet Child Adolesc. Health 2022, 6, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Derderian, S.C.; Jeanty, C.; Walters, M.C.; Vichinsky, E.; MacKenzie, T.C. In utero hematopoietic cell transplantation for hemoglobinopathies. Front. Pharmacol. 2015, 5, 278. [Google Scholar] [CrossRef] [PubMed]
- Götherström, C.; David, A.L.; Walther-Jallow, L.; Åström, E.; Westgren, M. Mesenchymal stem cell therapy for osteogenesis imperfecta. Clin. Obstet. Gynecol. 2021, 64, 898–903. [Google Scholar] [CrossRef]
- Spencer, R.; Ambler, G.; Brodszki, J.; Diemert, A.; Figueras, F.; Gratacós, E.; Hansson, S.R.; Hecher, K.; Huertas-Ceballos, A.; Marlow, N. EVERREST prospective study: A 6-year prospective study to define the clinical and biological characteristics of pregnancies affected by severe early onset fetal growth restriction. BMC Pregnancy Childbirth 2017, 17, 43. [Google Scholar] [CrossRef]
- Sudrié-Arnaud, B.; Marguet, F.; Patrier, S.; Martinovic, J.; Louillet, F.; Broux, F.; Charbonnier, F.; Dranguet, H.; Coutant, S.; Vezain, M. Metabolic causes of nonimmune hydrops fetalis: A next-generation sequencing panel as a first-line investigation. Clin. Chim. Acta 2018, 481, 1–8. [Google Scholar] [CrossRef]
- Al-Kouatly, H.B.; Shivashankar, K.; Mossayebi, M.H.; Makhamreh, M.; Critchlow, E.; Gao, Z.; Fasehun, L.K.; Alkuraya, F.S.; Ryan, E.E.; Hegde, M. Diagnostic yield from prenatal exome sequencing for non-immune hydrops fetalis: A systematic review and meta-analysis. Clin. Genet. 2023, 103, 503–512. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | Author (Year) | Prenatal Diagnosis | Prenatal Findings | Day of First Fosdenopterin Dose | Corrected GA at Start | Clinical Outcome |
|---|---|---|---|---|---|---|
| 1 | Hitzert et al., 2012 [20] | Yes | None reported | Day 0 (4 h after birth) | 36 w 3 d | Brief neonatal seizures; mildly delayed cognitive, rest normal neurodevelopment at follow-up (21 months) |
| 2 | Schwahn et al., 2015 (Patient 3) [16] | No (family history; not prenatal) | None reported (dysmorphic features at birth) | Day 7 | ~40 w | Seizure-free, near-normal development long-term (21 months) |
| 3 | Schwahn et al., 2015 (Patient 4) [16] | No | None reported (dysmorphic features at birth) | Day 5 | ~42 w | Marked improvement, but residual neurologic deficits (not fully seizure-free) |
| 4 | Schwahn et al., 2015 (Patient 5) [16] | Yes (older sib; prenatal) | None reported (dysmorphic features at birth) | Day 0 | ~36 w | Seizure-free, normal development by age 3 years |
| 5 | Schwahn et al., 2015 (Patient 6) [16] | No (family history; not prenatal) | None reported | Day 4 | ~39 w | Initial improvement, parents decided to abandon treatment after 111 days and the patient was lost to follow-up |
| 6 | Schwahn et al., 2015 (Patient 7) [16] | Yes (older sib; prenatal) | None reported (dysmorphic features at birth) | Day 0 | 40 w | Seizure-free, near-normal development long-term by age 2 years |
| 7 | Schwahn et al., 2015 (Patient 8) [16] | No | None reported | Day 11 | ~40 w | Transient stabilization, but died in neonatal period (~3 weeks) |
| 8 | Schwahn et al., 2015 (Patient 10) [16] | No | None reported | Day 8 | ~41 w | Rapid clinical decline; died in neonatal period (~11 days old) |
| 9 | Schwahn et al., 2015 (Patient 11) [16] | No | None reported | Day 6 | ~42 w | Survived; treatment stopped after 5 days (irreversible damage)– Severe CP, intractable impairment |
| 10 | Lubout et al., 2018 (Patient A) [10] | Yes (via chorionic villus sampling) | Fetal MRI normal until ~36 wk; then mild ventriculomegaly and subtle white-matter T2 hyperintensities observed | Day 0 (~4 h after birth) | 36 + 4 w | Neonatal seizures controlled; at 41 mo ~30–35 mo developmental level (Bayley ~16th percentile) with persistent macrocephaly (HC > P98; familial trait) |
| 11 | Lubout et al., 2018 (Patient B) [10] | Yes (via amniocentesis at 32 weeks) | Fetal MRI from 32 wk: mega cisterna magna with slightly small cerebellum; by 36 wk mild ventriculomegaly and subtle white-matter T2 hyperintensities | Day 0 (~5 h after birth) | ~39–40 wk | Only brief subclinical neonatal seizures (on aEEG) at 41 mo~18–26 mo developmental level (Bayley; cognitive ~5th, motor ~0.5th percentile), with clumsy gait |
| 12 | Schwahn et al., 2024 (Patient A) [21] | No | None reported | Day 7 | Term | No seizure control. Severe dystonic and spastic quadriplegia. Received cPMP for 7 days before brain MRI results showed widespread severe diffusion restriction and signs of brain necrosis |
| 13 | Schwahn et al., 2024 (Patient B) [21] | No | None reported | Day 3 | Term | No seizure control. Severe dystonic and spastic quadriplegia. Received cPMP for 8 days before stopping due to lack of benefit |
| 14 | Schwahn et al., 2024 (Patient C) [21] | No | None reported | Day 4 | Term | No seizure control. Severe dystonic and spastic quadriplegia. Received cPMP for 22 days before stopping due to lack of benefit |
| 15 | Schwahn et al., 2024 [19] | No | None reported | Day 4 | 39 w 2 d | Early brain MRI at 5 days revealed transient edema and signal changes in globi pallidi, resolving by week 6. Long-term neurodevelopment showed mild muscular hypotonia and delayed cognitive milestones |
| 16 | Etchegaray et al., 2025 (this case) | Yes (16 w via amniocentesis) | Fetal MRI normal at 22 w; mega cisterna magna at 28 w | Day 0 (within 10 min of birth) | 32 w 6 d | Neonatal seizures (resolved by 60 h); discharged D37. Developed dystonic quadriplegic CP by 6 mo (cognition relatively spared); remains seizure-free at 24 mo |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Etchegaray, A.; Haffner, D.; Cruz, S.M.; Ogunleye, O.; Xia, J.; Schlegel, A.; Olutoye, O.O.; Chaudhari, B.P. Early Neonatal Fosdenopterin Treatment for Molybdenum Cofactor Deficiency Type A: New Insights into Its Natural History and Potential Role for Fetal Therapy. J. Clin. Med. 2025, 14, 3561. https://doi.org/10.3390/jcm14103561
Etchegaray A, Haffner D, Cruz SM, Ogunleye O, Xia J, Schlegel A, Olutoye OO, Chaudhari BP. Early Neonatal Fosdenopterin Treatment for Molybdenum Cofactor Deficiency Type A: New Insights into Its Natural History and Potential Role for Fetal Therapy. Journal of Clinical Medicine. 2025; 14(10):3561. https://doi.org/10.3390/jcm14103561
Chicago/Turabian StyleEtchegaray, Adolfo, Darrah Haffner, Stephanie M. Cruz, Oluseyi Ogunleye, Jason Xia, Amy Schlegel, Oluyinka O. Olutoye, and Bimal P. Chaudhari. 2025. "Early Neonatal Fosdenopterin Treatment for Molybdenum Cofactor Deficiency Type A: New Insights into Its Natural History and Potential Role for Fetal Therapy" Journal of Clinical Medicine 14, no. 10: 3561. https://doi.org/10.3390/jcm14103561
APA StyleEtchegaray, A., Haffner, D., Cruz, S. M., Ogunleye, O., Xia, J., Schlegel, A., Olutoye, O. O., & Chaudhari, B. P. (2025). Early Neonatal Fosdenopterin Treatment for Molybdenum Cofactor Deficiency Type A: New Insights into Its Natural History and Potential Role for Fetal Therapy. Journal of Clinical Medicine, 14(10), 3561. https://doi.org/10.3390/jcm14103561