
The Impact of Anti-Amyloid Immunotherapies on Stroke Care

Abstract
1. Introduction
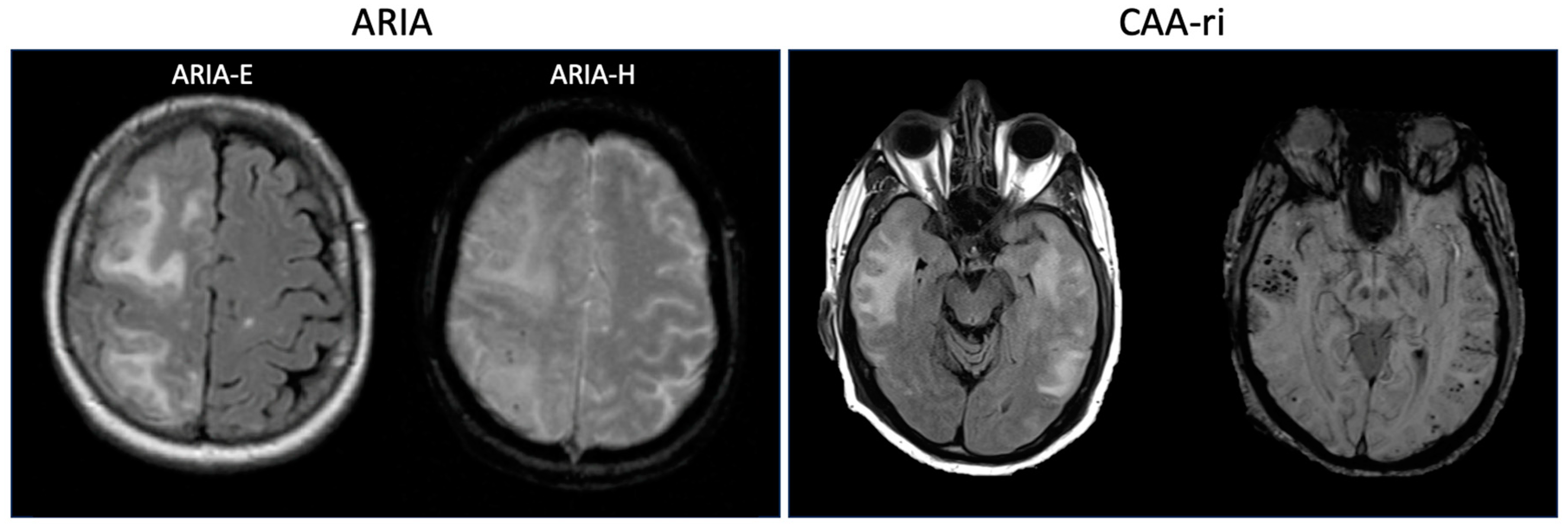
2. Defining Amyloid-Related Imaging Abnormalities: Insights from Clinical Trials
3. Risk Factors for ARIA
4. Cerebral Amyloid Angiopathy-Related Inflammation: Spontaneous ARIA?
5. Secondary Ischemic Stroke Prevention in Patients Receiving Anti-Amyloid Immunotherapy
5.1. Antiplatelets
5.2. Anticoagulation
5.3. Hypertension and Hyperlipidemia Management
6. Acute Stroke Therapy and Emergent Anticoagulation in Patients Receiving Anti-Amyloid Immunotherapy
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Swanson, C.J.; Zhang, Y.; Dhadda, S.; Wang, J.; Kaplow, J.; Lai, R.Y.K.; Lannfelt, L.; Bradley, H.; Rabe, M.; Koyama, A.; et al. A Randomized, Double-Blind, Phase 2b Proof-of-Concept Clinical Trial in Early Alzheimer’s Disease with Lecanemab, an Anti-Aβ Protofibril Antibody. Alzheimer’s Res. Ther. 2021, 13, 80. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The Antibody Aducanumab Reduces Aβ Plaques in Alzheimer’s Disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Budd Haeberlein, S.; Aisen, P.S.; Barkhof, F.; Chalkias, S.; Chen, T.; Cohen, S.; Dent, G.; Hansson, O.; Harrison, K.; von Hehn, C.; et al. Two Randomized Phase 3 Studies of Aducanumab in Early Alzheimer’s Disease. J. Prev. Alzheimer’s Dis. 2022, 9, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Sims, J.R.; Zimmer, J.A.; Evans, C.D.; Lu, M.; Ardayfio, P.; Sparks, J.; Wessels, A.M.; Shcherbinin, S.; Wang, H.; Monkul Nery, E.S.; et al. Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. JAMA 2023, 330, 512–527. [Google Scholar] [CrossRef]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Salloway, S.; Sperling, R.; Fox, N.C.; Blennow, K.; Klunk, W.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Porsteinsson, A.P.; Ferris, S.; et al. Two Phase 3 Trials of Bapineuzumab in Mild-to-Moderate Alzheimer’s Disease. N. Engl. J. Med. 2014, 370, 322–333. [Google Scholar] [CrossRef]
- Doody, R.S.; Thomas, R.G.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; Raman, R.; Sun, X.; Aisen, P.S.; et al. Phase 3 Trials of Solanezumab for Mild-to-Moderate Alzheimer’s Disease. N. Engl. J. Med. 2014, 370, 311–321. [Google Scholar] [CrossRef]
- Honig, L.S.; Vellas, B.; Woodward, M.; Boada, M.; Bullock, R.; Borrie, M.; Hager, K.; Andreasen, N.; Scarpini, E.; Liu-Seifert, H.; et al. Trial of Solanezumab for Mild Dementia Due to Alzheimer’s Disease. N. Engl. J. Med. 2018, 378, 321–330. [Google Scholar] [CrossRef]
- Salloway, S.; Farlow, M.; McDade, E.; Clifford, D.B.; Wang, G.; Llibre-Guerra, J.J.; Hitchcock, J.M.; Mills, S.L.; Santacruz, A.M.; Aschenbrenner, A.J.; et al. A Trial of Gantenerumab or Solanezumab in Dominantly Inherited Alzheimer’s Disease. Nat. Med. 2021, 27, 1187–1196. [Google Scholar] [CrossRef]
- Ostrowitzki, S.; Bittner, T.; Sink, K.M.; Mackey, H.; Rabe, C.; Honig, L.S.; Cassetta, E.; Woodward, M.; Boada, M.; van Dyck, C.H.; et al. Evaluating the Safety and Efficacy of Crenezumab vs Placebo in Adults with Early Alzheimer Disease: Two Phase 3 Randomized Placebo-Controlled Trials. JAMA Neurol. 2022, 79, 1113–1121. [Google Scholar] [CrossRef]
- Bateman, R.J.; Smith, J.; Donohue, M.C.; Delmar, P.; Abbas, R.; Salloway, S.; Wojtowicz, J.; Blennow, K.; Bittner, T.; Black, S.E.; et al. Two Phase 3 Trials of Gantenerumab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 389, 1862–1876. [Google Scholar] [CrossRef]
- Jäkel, L.; De Kort, A.M.; Klijn, C.J.M.; Schreuder, F.H.B.M.; Verbeek, M.M. Prevalence of Cerebral Amyloid Angiopathy: A Systematic Review and Meta-Analysis. Alzheimer’s Dement. 2022, 18, 10–28. [Google Scholar] [CrossRef]
- Piazza, F.; Greenberg, S.M.; Savoiardo, M.; Gardinetti, M.; Chiapparini, L.; Raicher, I.; Nitrini, R.; Sakaguchi, H.; Brioschi, M.; Billo, G.; et al. Anti–Amyloid β Autoantibodies in Cerebral Amyloid Angiopathy–Related Inflammation: Implications for Amyloid-Modifying Therapies. Ann. Neurol. 2013, 73, 449–458. [Google Scholar] [CrossRef]
- Boche, D.; Zotova, E.; Weller, R.O.; Love, S.; Neal, J.W.; Pickering, R.M.; Wilkinson, D.; Holmes, C.; Nicoll, J.A.R. Consequence of Abeta Immunization on the Vasculature of Human Alzheimer’s Disease Brain. Brain 2008, 131, 3299–3310. [Google Scholar] [CrossRef]
- Sveikata, L.; Charidimou, A.; Viswanathan, A. Vessels Sing Their ARIAs: The Role of Vascular Amyloid in the Age of Aducanumab. Stroke 2022, 53, 298–302. [Google Scholar] [CrossRef]
- Greenberg, S.M.; Bacskai, B.J.; Hernandez-Guillamon, M.; Pruzin, J.; Sperling, R.; van Veluw, S.J. Cerebral Amyloid Angiopathy and Alzheimer Disease—One Peptide, Two Pathways. Nat. Rev. Neurol. 2020, 16, 30–42. [Google Scholar] [CrossRef]
- Greenberg, S.M.; Cordonnier, C.; Schneider, J.A.; Smith, E.E.; van Buchem, M.A.; van Veluw, S.J.; Verbeek, M.M.; Viswanathan, A.; Werring, D.J. Off-Label Use of Aducanumab for Cerebral Amyloid Angiopathy. Lancet Neurol. 2021, 20, 596–597. [Google Scholar] [CrossRef]
- Orgogozo, J.-M.; Gilman, S.; Dartigues, J.-F.; Laurent, B.; Puel, M.; Kirby, L.C.; Jouanny, P.; Dubois, B.; Eisner, L.; Flitman, S.; et al. Subacute Meningoencephalitis in a Subset of Patients with AD after Abeta42 Immunization. Neurology 2003, 61, 46–54. [Google Scholar] [CrossRef]
- Black, R.S.; Sperling, R.A.; Safirstein, B.; Motter, R.N.; Pallay, A.; Nichols, A.; Grundman, M. A Single Ascending Dose Study of Bapineuzumab in Patients with Alzheimer Disease. Alzheimer Dis. Assoc. Disord. 2010, 24, 198–203. [Google Scholar] [CrossRef]
- Salloway, S.; Sperling, R.; Gilman, S.; Fox, N.C.; Blennow, K.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Doody, R.; van Dyck, C.H.; et al. A Phase 2 Multiple Ascending Dose Trial of Bapineuzumab in Mild to Moderate Alzheimer Disease. Neurology 2009, 73, 2061–2070. [Google Scholar] [CrossRef]
- Sperling, R.A.; Jack, C.R.; Black, S.E.; Frosch, M.P.; Greenberg, S.M.; Hyman, B.T.; Scheltens, P.; Carrillo, M.C.; Thies, W.; Bednar, M.M.; et al. Amyloid-Related Imaging Abnormalities in Amyloid-Modifying Therapeutic Trials: Recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimer’s Dement. 2011, 7, 367–385. [Google Scholar] [CrossRef]
- Joseph-Mathurin, N.; Llibre-Guerra, J.J.; Li, Y.; McCullough, A.A.; Hofmann, C.; Wojtowicz, J.; Park, E.; Wang, G.; Preboske, G.M.; Wang, Q.; et al. Amyloid-Related Imaging Abnormalities in the DIAN-TU-001 Trial of Gantenerumab and Solanezumab: Lessons from a Trial in Dominantly Inherited Alzheimer Disease. Ann. Neurol. 2022, 92, 729–744. [Google Scholar] [CrossRef]
- Reish, N.J.; Jamshidi, P.; Stamm, B.; Flanagan, M.E.; Sugg, E.; Tang, M.; Donohue, K.L.; McCord, M.; Krumpelman, C.; Mesulam, M.-M.; et al. Multiple Cerebral Hemorrhages in a Patient Receiving Lecanemab and Treated with T-PA for Stroke. N. Engl. J. Med. 2023, 388, 478–479. [Google Scholar] [CrossRef]
- FDA’s Decision to Approve New Treatment for Alzheimer’s Disease. Available online: https://www.fda.gov/drugs/news-events-human-drugs/fdas-decision-approve-new-treatment-Alzheimer’s-disease (accessed on 8 August 2023).
- FDA Converts Novel Alzheimer’s Disease Treatment to Traditional Approval. Available online: https://www.fda.gov/news-events/press-announcements/fda-converts-novel-Alzheimer’s-disease-treatment-traditional-approval (accessed on 8 August 2023).
- Ostrowitzki, S.; Lasser, R.A.; Dorflinger, E.; Scheltens, P.; Barkhof, F.; Nikolcheva, T.; Ashford, E.; Retout, S.; Hofmann, C.; Delmar, P.; et al. A Phase III Randomized Trial of Gantenerumab in Prodromal Alzheimer’s Disease. Alzheimer’s Res. Ther. 2017, 9, 95. [Google Scholar] [CrossRef]
- Crespi, G.A.N.; Hermans, S.J.; Parker, M.W.; Miles, L.A. Molecular Basis for Mid-Region Amyloid-β Capture by Leading Alzheimer’s Disease Immunotherapies. Sci. Rep. 2015, 5, 9649. [Google Scholar] [CrossRef]
- Antolini, L.; DiFrancesco, J.C.; Zedde, M.; Basso, G.; Arighi, A.; Shima, A.; Cagnin, A.; Caulo, M.; Carare, R.O.; Charidimou, A.; et al. Spontaneous ARIA-like Events in Cerebral Amyloid Angiopathy-Related Inflammation: A Multicenter Prospective Longitudinal Cohort Study. Neurology 2021, 97, e1809–e1822. [Google Scholar] [CrossRef]
- Ryan, N.S.; Lashley, T.; Revesz, T.; Dantu, K.; Fox, N.C.; Morris, H.R. Spontaneous ARIA (Amyloid-Related Imaging Abnormalities) and Cerebral Amyloid Angiopathy Related Inflammation in Presenilin 1-Associated Familial Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 44, 1069–1074. [Google Scholar] [CrossRef]
- Sperling, R.A.; Donohue, M.C.; Raman, R.; Rafii, M.S.; Johnson, K.; Masters, C.L.; van Dyck, C.H.; Iwatsubo, T.; Marshall, G.A.; Yaari, R.; et al. Trial of Solanezumab in Preclinical Alzheimer’s Disease. N. Engl. J. Med. 2023, 389, 1096–1107. [Google Scholar] [CrossRef]
- Ultsch, M.; Li, B.; Maurer, T.; Mathieu, M.; Adolfsson, O.; Muhs, A.; Pfeifer, A.; Pihlgren, M.; Bainbridge, T.W.; Reichelt, M.; et al. Structure of Crenezumab Complex with Aβ Shows Loss of β-Hairpin. Sci. Rep. 2016, 6, 39374. [Google Scholar] [CrossRef]
- Ostrowitzki, S.; Deptula, D.; Thurfjell, L.; Barkhof, F.; Bohrmann, B.; Brooks, D.J.; Klunk, W.E.; Ashford, E.; Yoo, K.; Xu, Z.-X.; et al. Mechanism of Amyloid Removal in Patients with Alzheimer Disease Treated with Gantenerumab. Arch. Neurol. 2012, 69, 198–207. [Google Scholar] [CrossRef]
- Lowe, S.L.; Duggan Evans, C.; Shcherbinin, S.; Cheng, Y.-J.; Willis, B.A.; Gueorguieva, I.; Lo, A.C.; Fleisher, A.S.; Dage, J.L.; Ardayfio, P.; et al. Donanemab (LY3002813) Phase 1b Study in Alzheimer’s Disease: Rapid and Sustained Reduction of Brain Amyloid Measured by Florbetapir F18 Imaging. J. Prev. Alzheimer’s Dis. 2021, 8, 414–424. [Google Scholar] [CrossRef]
- Sperling, R.; Salloway, S.; Brooks, D.J.; Tampieri, D.; Barakos, J.; Fox, N.C.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Porsteinsson, A.P.; et al. Amyloid-Related Imaging Abnormalities in Patients with Alzheimer’s Disease Treated with Bapineuzumab: A Retrospective Analysis. Lancet Neurol. 2012, 11, 241–249. [Google Scholar] [CrossRef]
- Salloway, S.; Chalkias, S.; Barkhof, F.; Burkett, P.; Barakos, J.; Purcell, D.; Suhy, J.; Forrestal, F.; Tian, Y.; Umans, K.; et al. Amyloid-Related Imaging Abnormalities in 2 Phase 3 Studies Evaluating Aducanumab in Patients With Early Alzheimer Disease. JAMA Neurol. 2022, 79, 13–21. [Google Scholar] [CrossRef]
- Honig, L.S.; Barakos, J.; Dhadda, S.; Kanekiyo, M.; Reyderman, L.; Irizarry, M.; Kramer, L.D.; Swanson, C.J.; Sabbagh, M. ARIA in Patients Treated with Lecanemab (BAN2401) in a Phase 2 Study in Early Alzheimer’s Disease. Alzheimer’s Dement. 2023, 9, e12377. [Google Scholar] [CrossRef]
- Mintun, M.A.; Lo, A.C.; Duggan Evans, C.; Wessels, A.M.; Ardayfio, P.A.; Andersen, S.W.; Shcherbinin, S.; Sparks, J.; Sims, J.R.; Brys, M.; et al. Donanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2021, 384, 1691–1704. [Google Scholar] [CrossRef]
- Barakos, J.; Purcell, D.; Suhy, J.; Chalkias, S.; Burkett, P.; Marsica Grassi, C.; Castrillo-Viguera, C.; Rubino, I.; Vijverberg, E. Detection and Management of Amyloid-Related Imaging Abnormalities in Patients with Alzheimer’s Disease Treated with Anti-Amyloid Beta Therapy. J. Prev. Alzheimer’s Dis. 2022, 9, 211–220. [Google Scholar] [CrossRef]
- Cummings, J.; Aisen, P.; Apostolova, L.G.; Atri, A.; Salloway, S.; Weiner, M. Aducanumab: Appropriate Use Recommendations. J. Prev. Alzheimer’s Dis. 2021, 8, 398–410. [Google Scholar] [CrossRef]
- Cummings, J.; Apostolova, L.; Rabinovici, G.D.; Atri, A.; Aisen, P.; Greenberg, S.; Hendrix, S.; Selkoe, D.; Weiner, M.; Petersen, R.C.; et al. Lecanemab: Appropriate Use Recommendations. J. Prev. Alzheimer’s Dis. 2023, 10, 362–377. [Google Scholar] [CrossRef] [PubMed]
- Leqembi [Package Insert]; Eisai Co., Ltd.: Tokyo, Japan, 2023.
- Serrano-Pozo, A.; Das, S.; Hyman, B.T. APOE and Alzheimer’s Disease: Advances in Genetics, Pathophysiology, and Therapeutic Approaches. Lancet Neurol. 2021, 20, 68–80. [Google Scholar] [CrossRef]
- Greenberg, S.M.; Rebeck, G.W.; Vonsattel, J.P.; Gomez-Isla, T.; Hyman, B.T. Apolipoprotein E Epsilon 4 and Cerebral Hemorrhage Associated with Amyloid Angiopathy. Ann. Neurol. 1995, 38, 254–259. [Google Scholar] [CrossRef]
- Kinnecom, C.; Lev, M.H.; Wendell, L.; Smith, E.E.; Rosand, J.; Frosch, M.P.; Greenberg, S.M. Course of Cerebral Amyloid Angiopathy-Related Inflammation. Neurology 2007, 68, 1411–1416. [Google Scholar] [CrossRef]
- Charidimou, A.; Boulouis, G.; Frosch, M.P.; Baron, J.-C.; Pasi, M.; Albucher, J.F.; Banerjee, G.; Barbato, C.; Bonneville, F.; Brandner, S.; et al. The Boston Criteria Version 2.0 for Cerebral Amyloid Angiopathy: A Multicentre, Retrospective, MRI-Neuropathology Diagnostic Accuracy Study. Lancet Neurol. 2022, 21, 714–725. [Google Scholar] [CrossRef]
- Chung, K.K.; Anderson, N.E.; Hutchinson, D.; Synek, B.; Barber, P.A. Cerebral Amyloid Angiopathy Related Inflammation: Three Case Reports and a Review. J. Neurol. Neurosurg. Psychiatry 2011, 82, 20–26. [Google Scholar] [CrossRef]
- Greenberg, S.M.; Ardayfio, P.; Battioui, C.; Zimmer, J.A.; Evans, C.D.; Wang, H.; Serap, E.; Lu, M.; Sparks, J.D.; Andersen, S.; et al. ARIA Insights from the Donanemab Trials. 16th Clinical Trials on Alzheimer’s Disease (CTAD) Boston, MA (USA) October 24–27, 2023: Symposia. J. Prev. Alzheimer’s Dis. 2023, 10 (Suppl. 1), 4–55. [Google Scholar] [CrossRef]
- Kirshner, H.; Bradshaw, M. The Inflammatory Form of Cerebral Amyloid Angiopathy or “Cerebral Amyloid Angiopathy-Related Inflammation” (CAARI). Curr. Neurol. Neurosci. Rep. 2015, 15, 572. [Google Scholar] [CrossRef]
- Eng, J.A.; Frosch, M.P.; Choi, K.; Rebeck, G.W.; Greenberg, S.M. Clinical Manifestations of Cerebral Amyloid Angiopathy–Related Inflammation. Ann. Neurol. 2004, 55, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Auriel, E.; Charidimou, A.; Gurol, M.E.; Ni, J.; Van Etten, E.S.; Martinez-Ramirez, S.; Boulouis, G.; Piazza, F.; DiFrancesco, J.C.; Frosch, M.P.; et al. Validation of Clinicoradiological Criteria for the Diagnosis of Cerebral Amyloid Angiopathy-Related Inflammation. JAMA Neurol. 2016, 73, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Plotzker, A.S.; Henson, R.L.; Fagan, A.M.; Morris, J.C.; Day, G.S. Clinical and Paraclinical Measures Associated with Outcome in Cerebral Amyloid Angiopathy with Related Inflammation. J. Alzheimer’s Dis. 2021, 80, 133–142. [Google Scholar] [CrossRef]
- Yamada, M.; Itoh, Y.; Shintaku, M.; Kawamura, J.; Jensson, O.; Thorsteinsson, L.; Suematsu, N.; Matsushita, M.; Otomo, E. Immune Reactions Associated with Cerebral Amyloid Angiopathy. Stroke 1996, 27, 1155–1162. [Google Scholar] [CrossRef]
- Piazza, F.; Caminiti, S.P.; Zedde, M.; Presotto, L.; DiFrancesco, J.C.; Pascarella, R.; Giossi, A.; Sessa, M.; Poli, L.; Basso, G.; et al. Association of Microglial Activation With Spontaneous ARIA-E and CSF Levels of Anti-Aβ Autoantibodies. Neurology 2022, 99, e1265–e1277. [Google Scholar] [CrossRef]
- Moussaddy, A.; Levy, A.; Strbian, D.; Sundararajan, S.; Berthelet, F.; Lanthier, S. Inflammatory Cerebral Amyloid Angiopathy, Amyloid-β–Related Angiitis, and Primary Angiitis of the Central Nervous System. Stroke 2015, 46, e210–e213. [Google Scholar] [CrossRef] [PubMed]
- Scolding, N.J.; Joseph, F.; Kirby, P.A.; Mazanti, I.; Gray, F.; Mikol, J.; Ellison, D.; Hilton, D.A.; Williams, T.L.; MacKenzie, J.M.; et al. Aβ-Related Angiitis: Primary Angiitis of the Central Nervous System Associated with Cerebral Amyloid Angiopathy. Brain 2005, 128, 500–515. [Google Scholar] [CrossRef] [PubMed]
- Salvarani, C.; Hunder, G.G.; Morris, J.M.; Brown, R.D.; Christianson, T.; Giannini, C. Aβ-Related Angiitis: Comparison with CAA without Inflammation and Primary CNS Vasculitis. Neurology 2013, 81, 1596–1603. [Google Scholar] [CrossRef]
- Kozberg, M.G.; Yi, I.; Freeze, W.M.; Auger, C.A.; Scherlek, A.A.; Greenberg, S.M.; van Veluw, S.J. Blood-Brain Barrier Leakage and Perivascular Inflammation in Cerebral Amyloid Angiopathy. Brain Commun. 2022, 4, fcac245. [Google Scholar] [CrossRef] [PubMed]
- DiFrancesco, J.C.; Brioschi, M.; Brighina, L.; Ruffmann, C.; Saracchi, E.; Costantino, G.; Galimberti, G.; Conti, E.; Curtò, N.A.; Marzorati, L.; et al. Anti-Aβ Autoantibodies in the CSF of a Patient with CAA-Related Inflammation: A Case Report. Neurology 2011, 76, 842–844. [Google Scholar] [CrossRef] [PubMed]
- DiFrancesco, J.C.; Touat, M.; Caulo, M.; Gallucci, M.; Garcin, B.; Levy, R.; Uncini, A.; Piazza, F. Recurrence of Cerebral Amyloid Angiopathy-Related Inflammation: A Report of Two Cases from the iCAβ International Network. J. Alzheimer’s Dis. 2015, 46, 1071–1077. [Google Scholar] [CrossRef]
- Nicoll, J.A.R.; Wilkinson, D.; Holmes, C.; Steart, P.; Markham, H.; Weller, R.O. Neuropathology of Human Alzheimer Disease after Immunization with Amyloid-Beta Peptide: A Case Report. Nat. Med. 2003, 9, 448–452. [Google Scholar] [CrossRef]
- Castellani, R.J.; Shanes, E.D.; McCord, M.; Reish, N.J.; Flanagan, M.E.; Mesulam, M.-M.; Jamshidi, P. Neuropathology of Anti-Amyloid-β Immunotherapy: A Case Report. J. Alzheimer’s Dis. 2023, 93, 803–813. [Google Scholar] [CrossRef]
- Solopova, E.; Romero-Fernandez, W.; Harmsen, H.; Ventura-Antunes, L.; Wang, E.; Shostak, A.; Maldonado, J.; Donahue, M.J.; Schultz, D.; Coyne, T.M.; et al. Fatal Iatrogenic Cerebral β-Amyloid-Related Arteritis in a Woman Treated with Lecanemab for Alzheimer’s Disease. Nat. Commun. 2023, 14, 8220. [Google Scholar] [CrossRef]
- Scherlek, A.A.; Kozberg, M.G.; Nicoll, J.A.R.; Perosa, V.; Freeze, W.M.; van der Weerd, L.; Bacskai, B.J.; Greenberg, S.M.; Frosch, M.P.; Boche, D.; et al. Histopathological Correlates of Haemorrhagic Lesions on Ex Vivo Magnetic Resonance Imaging in Immunized Alzheimer’s Disease Cases. Brain Commun. 2022, 4, fcac021. [Google Scholar] [CrossRef]
- Taylor, X.; Clark, I.M.; Fitzgerald, G.J.; Oluoch, H.; Hole, J.T.; Demattos, R.B. Amyloid-β (Aβ) Immunotherapy Induced Microhemorrhages Are Associated with Activated Perivascular Macrophages and Peripheral Monocyte Recruitment in Alzheimer’s Disease Mice. Mol. Neurodegener. 2023, 18, 59. [Google Scholar] [CrossRef]
- Loeffler, D.A. Antibody-Mediated Clearance of Brain Amyloid-β: Mechanisms of Action, Effects of Natural and Monoclonal Anti-Aβ Antibodies, and Downstream Effects. J. Alzheimer’s Dis. Rep. 2023, 7, 873–899. [Google Scholar] [CrossRef]
- Batista, A.F.; Khan, K.A.; Papavergi, M.-T.; Lemere, C.A. The Importance of Complement-Mediated Immune Signaling in Alzheimer’s Disease Pathogenesis. Int. J. Mol. Sci. 2024, 25, 817. [Google Scholar] [CrossRef]
- Cenina, A.R.F.; De Leon, J.; Tay, K.Y.; Wong, C.F.; Kandiah, N. Cerebral Amyloid Angiopathy–Related Inflammation Presenting With Rapidly Progressive Dementia, Responsive to IVIg. Alzheimer Dis. Assoc. Disord. 2015, 29, 347. [Google Scholar] [CrossRef]
- Regenhardt, R.W.; Thon, J.M.; Das, A.S.; Thon, O.R.; Charidimou, A.; Viswanathan, A.; Gurol, M.E.; Chwalisz, B.K.; Frosch, M.P.; Cho, T.A.; et al. Association between Immunosuppressive Treatment and Outcomes of Cerebral Amyloid Angiopathy-Related Inflammation. JAMA Neurol. 2020, 77, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Charidimou, A.; Boulouis, G.; Greenberg, S.M.; Viswanathan, A. Cortical Superficial Siderosis and Bleeding Risk in Cerebral Amyloid Angiopathy: A Meta-Analysis. Neurology 2019, 93, E2192–E2202. [Google Scholar] [CrossRef] [PubMed]
- Charidimou, A.; Boulouis, G.; Roongpiboonsopit, D.; Xiong, L.; Pasi, M.; Schwab, K.M.; Rosand, J.; Gurol, M.E.; Greenberg, S.M.; Viswanathan, A. Cortical Superficial Siderosis and Recurrent Intracerebral Hemorrhage Risk in Cerebral Amyloid Angiopathy: Large Prospective Cohort and Preliminary Meta-Analysis. Int. J. Stroke 2019, 14, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Charidimou, A.; Boulouis, G.; Xiong, L.; Jessel, M.J.; Roongpiboonsopit, D.; Ayres, A.; Schwab, K.M.; Rosand, J.; Gurol, M.E.; Greenberg, S.M.; et al. Cortical Superficial Siderosis and First-Ever Cerebral Hemorrhage in Cerebral Amyloid Angiopathy. Neurology 2017, 88, 1607–1614. [Google Scholar] [CrossRef] [PubMed]
- Charidimou, A.; Boulouis, G.; Roongpiboonsopit, D.; Auriel, E.; Pasi, M.; Haley, K.; Van Etten, E.S.; Martinez-Ramirez, S.; Ayres, A.; Vashkevich, A.; et al. Cortical Superficial Siderosis Multifocality in Cerebral Amyloid Angiopathy: A Prospective Study. Neurology 2017, 89, 2128–2135. [Google Scholar] [CrossRef] [PubMed]
- Kozberg, M.G.; Perosa, V.; Gurol, M.E.; van Veluw, S.J. A Practical Approach to the Management of Cerebral Amyloid Angiopathy. Int. J. Stroke 2021, 16, 356–369. [Google Scholar] [CrossRef]
- Al-Shahi Salman, R.; Dennis, M.S.; Sandercock, P.A.G.; Sudlow, C.L.M.; Wardlaw, J.M.; Whiteley, W.N.; Murray, G.D.; Stephen, J.; Newby, D.E.; Sprigg, N.; et al. Effects of Antiplatelet Therapy after Stroke Due to Intracerebral Haemorrhage (RESTART): A Randomised, Open-Label Trial. Lancet 2019, 393, 2613–2623. [Google Scholar] [CrossRef]
- Greenberg, S.M.; Ziai, W.C.; Cordonnier, C.; Dowlatshahi, D.; Francis, B.; Goldstein, J.N.; Hemphill, J.C.; Johnson, R.; Keigher, K.M.; Mack, W.J.; et al. 2022 Guideline for the Management of Patients with Spontaneous Intracerebral Hemorrhage: A Guideline from the American Heart Association/American Stroke Association. Stroke 2022, 53, e282–e361. [Google Scholar] [CrossRef]
- Sabbagh, M.; van Dyck, C.H. Response to: Multiple Cerebral Hemorrhages in a Patient Receiving Lecanemab and Treated with t-PA for Stroke. N. Engl. J. Med. 2023, 388, 480. [Google Scholar] [CrossRef] [PubMed]
- Schreuder, F.H.B.M.; van Nieuwenhuizen, K.M.; Hofmeijer, J.; Vermeer, S.E.; Kerkhoff, H.; Zock, E.; Luijckx, G.J.; Messchendorp, G.P.; van Tuijl, J.; Bienfait, H.P.; et al. Apixaban versus No Anticoagulation after Anticoagulation-Associated Intracerebral Haemorrhage in Patients with Atrial Fibrillation in the Netherlands (APACHE-AF): A Randomised, Open-Label, Phase 2 Trial. Lancet Neurol. 2021, 20, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, P.B.; Skjøth, F.; Søgaard, M.; Kjældgaard, J.N.; Lip, G.Y.H.; Larsen, T.B. Non–Vitamin K Antagonist Oral Anticoagulants Versus Warfarin in Atrial Fibrillation Patients with Intracerebral Hemorrhage. Stroke 2019, 50, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Salman, R.A.-S.; Stephen, J.; Tierney, J.F.; Lewis, S.C.; Newby, D.E.; Parry-Jones, A.R.; White, P.M.; Connolly, S.J.; Benavente, O.R.; Dowlatshahi, D.; et al. Effects of Oral Anticoagulation in People with Atrial Fibrillation after Spontaneous Intracranial Haemorrhage (COCROACH): Prospective, Individual Participant Data Meta-Analysis of Randomised Trials. Lancet Neurol. 2023, 22, 1140–1149. [Google Scholar] [CrossRef] [PubMed]
- Shoamanesh, A. Anticoagulation in Patients with Cerebral Amyloid Angiopathy. Lancet 2023, 402, 1418–1419. [Google Scholar] [CrossRef]
- Holmes, D.R.; Kar, S.; Price, M.J.; Whisenant, B.; Sievert, H.; Doshi, S.K.; Huber, K.; Reddy, V.Y. Prospective Randomized Evaluation of the Watchman Left Atrial Appendage Closure Device in Patients with Atrial Fibrillation versus Long-Term Warfarin Therapy: The PREVAIL Trial. J. Am. Coll. Cardiol. 2014, 64, 1–12. [Google Scholar] [CrossRef]
- Reddy, V.Y.; Doshi, S.K.; Sievert, H.; Buchbinder, M.; Neuzil, P.; Huber, K.; Halperin, J.L.; Holmes, D. Percutaneous Left Atrial Appendage Closure for Stroke Prophylaxis in Patients with Atrial Fibrillation 2.3-Year Follow-up of the PROTECT AF (Watchman Left Atrial Appendage System for Embolic Protection in Patients with Atrial Fibrillation) Trial. Circulation 2013, 127, 720–729. [Google Scholar] [CrossRef]
- Osmancik, P.; Herman, D.; Neuzil, P.; Hala, P.; Taborsky, M.; Kala, P.; Poloczek, M.; Stasek, J.; Haman, L.; Branny, M.; et al. Left Atrial Appendage Closure Versus Direct Oral Anticoagulants in High-Risk Patients with Atrial Fibrillation. J. Am. Coll. Cardiol. 2020, 75, 3122–3135. [Google Scholar] [CrossRef]
- Schrag, M.; Grory, B.M.; Nackenoff, A.; Eaton, J.; Mistry, E.; Kirshner, H.; Yaghi, S.; Ellis, C.R. Left Atrial Appendage Closure for Patients with Cerebral Amyloid Angiopathy and Atrial Fibrillation: The LAA-CAA Cohort. Transl. Stroke Res. 2021, 12, 259–265. [Google Scholar] [CrossRef]
- Amarenco, P.; Bogoussla, J.; Rudolph, A.E.; Sillesen, H.; Simunovic, L.; Szarek, M.; Welch, K.M.A.; Zivsky, J.A.; Callahan, A., 3rd; Goldstein, L.B.; et al. High-Dose Atorvastatin after Stroke or Transient Ischemic Attack. N. Engl. J. Med. 2006, 355, 549–559. [Google Scholar] [CrossRef]
- Ma, C.; Gurol, M.E.; Huang, Z.; Lichtenstein, A.H.; Wang, X.; Wang, Y.; Neumann, S.; Wu, S.; Gao, X. Low-Density Lipoprotein Cholesterol and Risk of Intracerebral Hemorrhage: A Prospective Study. Neurology 2019, 93, e445–e457. [Google Scholar] [CrossRef]
- Falcone, G.J.; Kirsch, E.; Acosta, J.N.; Noche, R.B.; Leasure, A.; Marini, S.; Chung, J.; Selim, M.; Meschia, J.F.; Brown, D.L.; et al. Genetically Elevated LDL Associates with Lower Risk of Intracerebral Hemorrhage. Ann. Neurol. 2020, 88, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Ribe, A.R.; Vestergaard, C.H.; Vestergaard, M.; Pedersen, H.S.; Prior, A.; Lietzen, L.W.; Brynningsen, P.K.; Fenger-Grøn, M. Statins and Risk of Intracerebral Hemorrhage in Individuals with a History of Stroke. Stroke 2020, 51, 1111–1119. [Google Scholar] [CrossRef] [PubMed]
- Charidimou, A.; Turc, G.; Oppenheim, C.; Yan, S.; Scheitz, J.F.; Erdur, H.; Klinger-Gratz, P.P.; El-Koussy, M.; Takahashi, W.; Moriya, Y.; et al. Microbleeds, Cerebral Hemorrhage, and Functional Outcome After Stroke Thrombolysis. Stroke 2017, 48, 2084–2090. [Google Scholar] [CrossRef]
- Zand, R.; Tsivgoulis, G.; Singh, M.; McCormack, M.; Goyal, N.; Ishfaq, M.F.; Shahripour, R.B.; Nearing, K.; Elijovich, L.; Alexandrov, A.W.; et al. Cerebral Microbleeds and Risk of Intracerebral Hemorrhage Post Intravenous Thrombolysis. J. Stroke Cerebrovasc. Dis. 2017, 26, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Powers, W.J.; Rabinstein, A.A.; Ackerson, T.; Adeoye, O.M.; Bambakidis, N.C.; Becker, K.; Biller, J.; Brown, M.; Demaerschalk, B.M.; Hoh, B.; et al. Guidelines for the Early Management of Patients with Acute Ischemic Stroke: 2019 Update to the 2018 Guidelines for the Early Management of Acute Ischemic Stroke a Guideline for Healthcare Professionals from the American Heart Association/American Stroke A. Stroke 2019, 50, e344–e418. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Mild | Moderate | Severe | Location of Increased Vascular Permeability | ||
|---|---|---|---|---|---|
| Parenchyma | Leptomeninges | ||||
| ARIA-E | FLAIR hyperintensity confined to sulcus and or cortex/subcortical white matter in one location < 5 cm | FLAIR hyperintensity 5 to 10 cm, or more than 1 site of involvement, each measuring <10 cm | FLAIR hyperintensity measuring >10 cm, often with significant subcortical white matter/sulcal involvement. May involve one or more separate sites | “Vasogenic edema” | Sulcal effusion/exudate |
| ARIA-H | ≤4 new microhemorrhages on T2*-GRE OR 1 focal area of superficial siderosis on T2*-GRE | 5 to 9 new microhemorrhages OR 2 focal areas of superficial siderosis | 10 or more new microhemorrhages OR >2 focal areas of superficial siderosis | Microhemorrhages | Superficial hemosiderosis |
| Bapineuzumab [6] | Solanezumab [7,8] | Gantenerumab [11,26] | Crenezumab [10] | Aducanumab [3] | Lecanemab [5] | Donanemab [4] | |
|---|---|---|---|---|---|---|---|
| ARIA-E rate | |||||||
| Treatment arm | 15.3% (APOE ε4 carrier study); 4.2–14.2% (APOE ε4 noncarrier study) | 0.9% [7]; 1% [8] | 6.6% (105 mg dose) [26]; 13.5% (225 mg dose) [26]; 24.9% (pooled) [11] | 0.3% (CREAD and CREAD2) | 26–35% (EMERGE); 26–36% (ENGAGE) | 12.6% | 24.0% |
| Placebo arm | 0.2% (APOE ε4 carrier study); 0.2% (APOE ε4 noncarrier study) | 0.4% [7]; 2% [8] | 0.8% [26]; 2.7% (pooled) [11] | 0.3% (CREAD); 0% (CREAD2) | 2% (EMERGE); 3% (ENGAGE) | 1.7% | 1.9% |
| ARIA-H rate | |||||||
| Treatment arm | Not reported | 4.9% [7]; 3.5% [8] | 22.9% (105 mg dose) [26]; 16.2% (225 mg dose) [26]; 22.9% (pooled) [11] | 9.8% (CREAD); 5.0% (CREAD2) | 10–20% (EMERGE); 9–19% (ENGAGE) | 17.3% | 19.7% |
| Placebo arm | Not reported | 5.6% [7]; 2.8% [8] | 13.2% [26]; 12.3% (pooled) [11] | 7.8% (CREAD); 5.9% (CREAD2) | 7% (EMERGE); 6% (ENGAGE) | 9.0% | 7.4% |
| Anti-amyloid antibody treatment (except solanezumab and crenezumab) |
| Higher anti-amyloid antibody dose |
| Early timepoint in treatment course (especially first 6 months) |
| Presence of APOE ε4 allele |
| Underlying cerebral amyloid angiopathy |
| Bapineuzumab [6] | Solanezumab [7,8] | Gantenerumab [11,26] | Crenezumab [10] | Aducanumab [3] | Lecanemab [5] | Donanemab [4] | |
|---|---|---|---|---|---|---|---|
| Exclusion based on small vessel disease markers | |||||||
| Microhemorrhages | >1 | Not specified [7]); >4 [8] | >2 [26]; >5 micro-hemorrhages + superficial siderosis [11] | >4 | >4 | >4 | >4 |
| Cortical superficial siderosis | Not specified | Not specified [7,8] | Not specified [26]; >5 micro-hemorrhages + superficial siderosis [11] | Yes | Yes | Yes | >1 |
| White matter changes | Not specified | Not specified [7,8] | Extensive/ Confluent [26]; Fazekas score 3 [11] | Not specified | Diffuse involvement | Severe | Severe |
| Exclusion based on ischemic or hemorrhagic stroke | |||||||
| History of clinical stroke | Yes | Not specified [7,8] | Yes [26]; Yes, within 1 year [11] | Yes | Yes, within 1 year | Yes, within 1 year | Not specified |
| Cortical infarcts on imaging | >1 cm3 | Not specified [7,8] | Not specified [26]; territorial infarct > 1 cm3 [11] | Yes | >1.5 cm | Yes | Not specified |
| Lacunar infarcts on imaging | >1 | Not specified [7,8] | >1 [26]; >2 [11] | Not specified | >1 | Multiple | Not specified |
| ICH on imaging | >1 cm3 | Not specified [7,8] | Not specified [11,26] | Yes | Yes | Yes | >1 cm |
| Exclusion based on antithrombotic use | |||||||
| Aspirin use | Allowed | Not specified [7,8] | Not specified [26]; Allowed [11] | Not specified | Allowed | Allowed | Allowed |
| Other antiplatelet use | Clopidogrel and dipyridamole allowed | Not specified [7,8] | Not specified [26]; Allowed [11] | Not specified | Excluded | Allowed | Allowed |
| Anticoagulant use | Excluded | Not specified [7,8] | Not specified [26]; Excluded [11] | Not specified | Excluded | Allowed | Allowed |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bilodeau, P.A.; Dickson, J.R.; Kozberg, M.G. The Impact of Anti-Amyloid Immunotherapies on Stroke Care. J. Clin. Med. 2024, 13, 1245. https://doi.org/10.3390/jcm13051245
Bilodeau PA, Dickson JR, Kozberg MG. The Impact of Anti-Amyloid Immunotherapies on Stroke Care. Journal of Clinical Medicine. 2024; 13(5):1245. https://doi.org/10.3390/jcm13051245
Chicago/Turabian StyleBilodeau, Philippe A., John R. Dickson, and Mariel G. Kozberg. 2024. "The Impact of Anti-Amyloid Immunotherapies on Stroke Care" Journal of Clinical Medicine 13, no. 5: 1245. https://doi.org/10.3390/jcm13051245
APA StyleBilodeau, P. A., Dickson, J. R., & Kozberg, M. G. (2024). The Impact of Anti-Amyloid Immunotherapies on Stroke Care. Journal of Clinical Medicine, 13(5), 1245. https://doi.org/10.3390/jcm13051245