
Pulmonary Hypertension in Underrepresented Minorities: A Narrative Review

 , and
, and
Abstract
1. Introduction
2. Disparities in Pulmonary Hypertension
2.1. Prevalence and Presentation
2.2. Health Outcomes
2.3. Sex Differences
3. Pulmonary Hypertension Groups and Associated Conditions
3.1. Group 1 Pulmonary Hypertension
3.2. Group 2 Pulmonary Hypertension
3.3. Group 3 Pulmonary Hypertension
3.4. Group 4 Pulmonary Hypertension
4. Vasodilation Physiology and Treatment Considerations
4.1. cGMP and Nitric Oxide
4.2. cAMP and Prostacyclins
4.3. ERA Pathway
4.4. Treatment Approach
5. Representation in Pulmonary Hypertension Trials
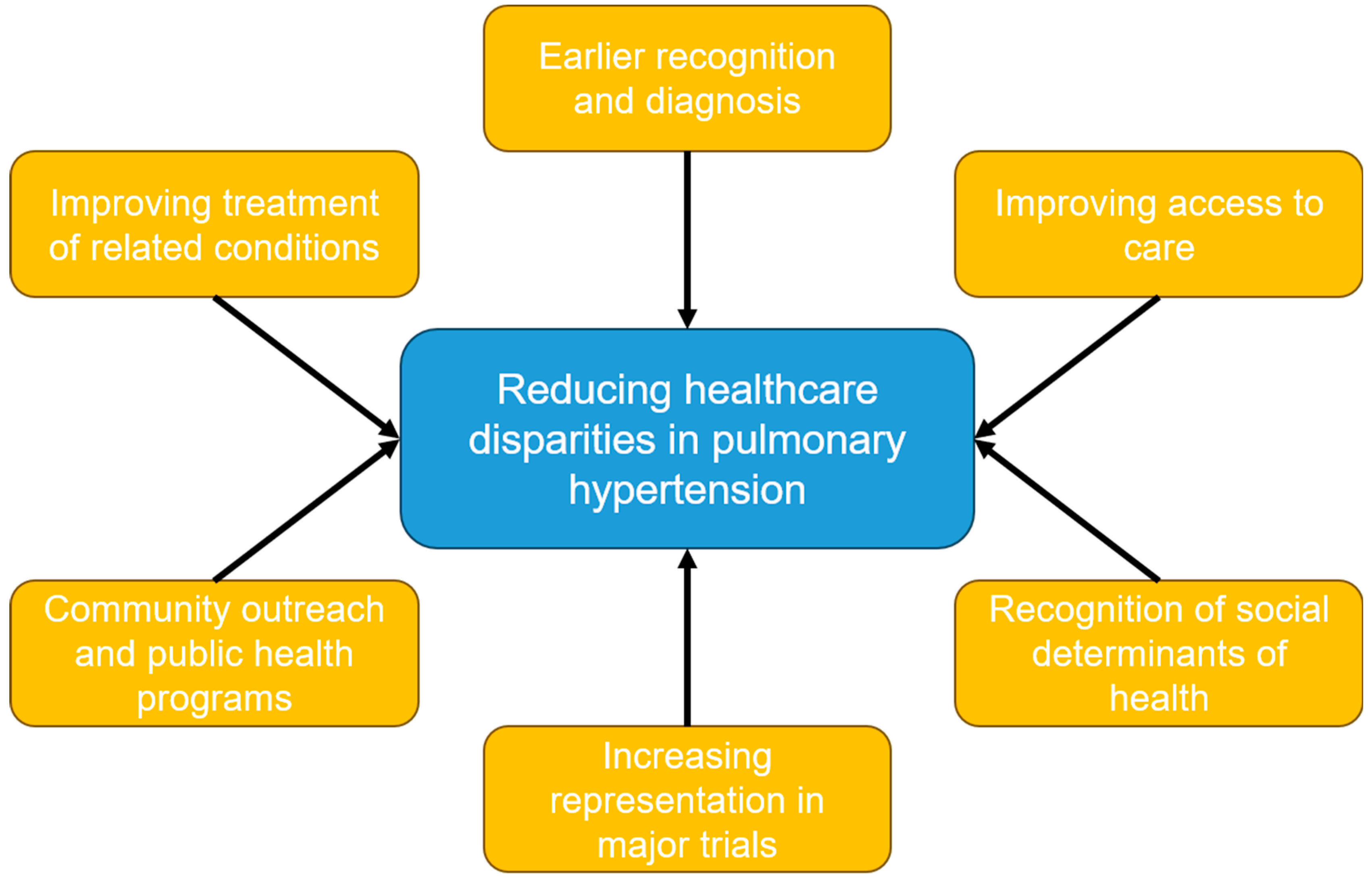
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Poch, D.; Mandel, J. Pulmonary Hypertension. Ann. Intern. Med. 2021, 174, Itc49–Itc64. [Google Scholar] [CrossRef] [PubMed]
- Flanagin, A.; Frey, T.; Christiansen, S.L. Updated Guidance on the Reporting of Race and Ethnicity in Medical and Science Journals. JAMA 2021, 326, 621. [Google Scholar] [CrossRef] [PubMed]
- Houlihan, J.; Leffler, S. Assessing and Addressing Social Determinants of Health: A Key Competency for Succeeding in Value-Based Care. Prim. Care Clin. Off. Pract. 2019, 46, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Fiscella, K.; Sanders, M.R. Racial and Ethnic Disparities in the Quality of Health Care. Annu. Rev. Public Health 2016, 37, 375–394. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Kovacs, G.; Hoeper, M.M.; Badagliacca, R.; Berger, R.; Brida, M.; Carlsen, J.; Coats, A.; Escribano-Subias, P.; Ferrari, P.; et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Heart J. 2022, 43, 3618–3731. [Google Scholar] [CrossRef]
- Hoeper, M.M.; Humbert, M.; Souza, R.; Idrees, M.; Kawut, S.M.; Sliwa-Hahnle, K.; Jing, Z.; Gibbs, J.S. A global view of pulmonary hypertension. Lancet Respir. Med. 2016, 4, 306–322. [Google Scholar] [CrossRef]
- Hassoun, P.M. Pulmonary Arterial Hypertension. N. Engl. J. Med. 2021, 385, 2361–2376. [Google Scholar] [CrossRef]
- Bernardo, R.J.; de Jesus Perez, V.A. Health Care Disparities in Pulmonary Arterial Hypertension. Clin. Chest Med. 2023, 44, 543–554. [Google Scholar] [CrossRef]
- Brown, L.M.; Chen, H.; Halpern, S.; Taichman, D.; McGoon, M.; Farber, H.; Frost, A.; Liou, T.; Turner, M.; Feldkircher, K.; et al. Delay in recognition of pulmonary arterial hypertension: Factors identified from the REVEAL Registry. Chest 2011, 140, 19–26. [Google Scholar] [CrossRef]
- Rich, S.; Dantzker, D.R.; Ayres, S.M.; Bergofsy, E.; Brundage, B.; Detre, K.; Fishman, A.; Goldring, R.; Groves, B.; Koerner, S.; et al. Primary pulmonary hypertension. A national prospective study. Ann. Intern. Med. 1987, 107, 216–223. [Google Scholar] [CrossRef]
- Blanco, I.; Mathai, S.; Shafiq, M.; Boyce, D.; Kolb, T.; Chami, H.; Hummers, L.; Housten, T.; Chaisson, N.; Zaiman, A.; et al. Severity of systemic sclerosis-associated pulmonary arterial hypertension in African Americans. Medicine 2014, 93, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Baughman, R.P.; Shlobin, O.A.; Wells, A.U.; Alhamad, E.H.; Culver, D.A.; Barney, J.; Cordova, F.; Carmona, E.; Scholand, M.B.; Wijsenbeek, M.; et al. Clinical features of sarcoidosis associated pulmonary hypertension: Results of a multi-national registry. Respir. Med. 2018, 139, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Klings, E.S.; Machado, R.F.; Barst, R.J.; Morris, C.R.; Mubarak, K.; Gordeuk, V.; Kato, G.; Ataga, K.; Gibbs, J.S.; Castro, O.; et al. An official American Thoracic Society clinical practice guideline: Diagnosis, risk stratification, and management of pulmonary hypertension of sickle cell disease. Am. J. Respir. Crit. Care Med. 2014, 189, 727–740. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, V.V.; Langer, A.; Tan, M.; Clements, P.J.; Oudiz, R.; Tapson, V.F.; Channick, R.; Rubin, L.J. Contemporary trends in the diagnosis and management of pulmonary arterial hypertension: An initiative to close the care gap. Chest 2013, 143, 324–332. [Google Scholar] [CrossRef]
- Talwar, A.; Sahni, S.; Talwar, A.; Kohn, N.; Klinger, J.R. Socioeconomic status affects pulmonary hypertension disease severity at time of first evaluation. Pulm. Circ. 2016, 6, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.H.; Yang, L.; Peng, F.H.; Yao, J.; Zou, L.; Liu, D.; Jiang, X.; Li, J.; Gao, L.; Qu, J.; et al. Lower socioeconomic status is associated with worse outcomes in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2013, 187, 303–310. [Google Scholar] [CrossRef]
- Medrek, S.; Sahay, S.; Zhao, C.; Selej, M.; Frost, A. Impact of race on survival in pulmonary arterial hypertension: Results from the REVEAL registry. J. Heart Lung Transplant. 2020, 39, 321–330. [Google Scholar] [CrossRef]
- Parikh, K.S.; Stackhouse, K.A.; Hart, S.A.; Bashore, T.M.; Krasuski, R.A. Health insurance and racial disparities in pulmonary hypertension outcomes. Am. J. Manag. Care 2017, 23, 474–480. [Google Scholar]
- Hjalmarsson, C.; Rådegran, G.; Kylhammar, D.; Rundqvist, B.; Multing, J.; Nisell, M.D.; Kjellström, B. Impact of age and comorbidity on risk stratification in idiopathic pulmonary arterial hypertension. Eur. Respir. J. 2018, 51, 1702310. [Google Scholar] [CrossRef]
- Morris, H.; Denver, N.; Gaw, R.; Labazi, H.; Mair, K.; MacLean, M.R. Sex Differences in Pulmonary Hypertension. Clin. Chest Med. 2021, 42, 217–228. [Google Scholar] [CrossRef]
- Rodriguez-Arias, J.J.; García-Álvarez, A. Sex Differences in Pulmonary Hypertension. Front. Aging 2021, 2, 727558. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; Huscher, D.; Ghofrani, H.A.; Delcroix, M.; Distler, O.; Schweiger, C.; Grunig, E.; Staehler, G.; Rosenkranz, S.; Halank, M.; et al. Elderly patients diagnosed with idiopathic pulmonary arterial hypertension: Results from the COMPERA registry. Int. J. Cardiol. 2013, 168, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Olsson, K.M.; Delcroix, M.; Ghofrani, H.A.; Tiede, H.; Huscher, D.; Speich, R.; Grunig, E.; Staehler, G.; Rosenkranz, S.; Halank, M.; et al. Anticoagulation and survival in pulmonary arterial hypertension: Results from the Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA). Circulation 2014, 129, 57–65. [Google Scholar] [CrossRef]
- Benza, R.L.; Miller, D.P.; Gomberg-Maitland, M.; Frantz, R.P.; Foreman, A.J.; Coffey, C.S.; Frost, A.; Barst, R.J.; Badesch, D.B.; Elliott, C.G.; et al. Predicting survival in pulmonary arterial hypertension: Insights from the Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management (REVEAL). Circulation 2010, 122, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Kawut, S.M.; Barr, R.G.; Lima, J.A.; Praestgaard, A.; Johnson, W.C.; Chahal, H.; Ogunyankin, K.O.; Bristow, M.R.; Kizer, J.R.; Tandri, H.; et al. Right ventricular structure is associated with the risk of heart failure and cardiovascular death: The Multi-Ethnic Study of Atherosclerosis (MESA)—Right ventricle study. Circulation 2012, 126, 1681–1688. [Google Scholar] [CrossRef]
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Frost, A.E.; Badesch, D.B.; Barst, R.J.; Benza, R.L.; Elliott, G.C.; Farber, H.W.; Krichman, A.; Liou, T.G.; Raskob, G.E.; Wason, P.; et al. The changing picture of patients with pulmonary arterial hypertension in the United States: How REVEAL differs from historic and non-US Contemporary Registries. Chest 2011, 139, 128–137. [Google Scholar] [CrossRef]
- Al-Naamani, N.; Paulus, J.K.; Roberts, K.E.; Pauciulo, M.W.; Lutz, K.; Nichols, W.C.; Kawut, S.M. Racial and ethnic differences in pulmonary arterial hypertension. Pulm. Circ. 2017, 7, 793–796. [Google Scholar] [CrossRef]
- Zanatta, E.; Polito, P.; Famoso, G.; Larosa, M.; De Zorzi, E.; Scarpieri, E.; Cozzi, F.; Doria, A. Pulmonary arterial hypertension in connective tissue disorders: Pathophysiology and treatment. Exp. Biol. Med. 2019, 244, 120–131. [Google Scholar] [CrossRef]
- Mayes, M.D.; Lacey, J.V.; Beebe-Dimmer, J., Jr.; Gillespie, B.W.; Cooper, B.; Laing, T.J.; Schottenfield, D. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum. 2003, 48, 2246–2255. [Google Scholar] [CrossRef]
- Reveille, J.D.; Fischbach, M.; McNearney, T.; Friedman, A.W.; Aguilar, M.B.; Lisse, J.; Fritzler, M.J.; Ahn, C.; Arnett, F.C. Systemic sclerosis in 3 US ethnic groups: A comparison of clinical, sociodemographic, serologic, and immunogenetic determinants. Semin. Arthritis Rheum. 2001, 30, 332–346. [Google Scholar] [CrossRef]
- Izmirly, P.M.; Ferucci, E.D.; Somers, E.C.; Wang, L.; Lim, S.S.; Drenkard, C.; Dall’Era, M.; McCune, W.J.; Gordon, C.; Helmick, C.; et al. Incidence rates of systemic lupus erythematosus in the USA: Estimates from a meta-analysis of the Centers for Disease Control and Prevention national lupus registries. Lupus Sci. Med. 2021, 8, e000614. [Google Scholar] [CrossRef] [PubMed]
- Mizus, M.; Li, J.; Goldman, D.; Petri, M.A. Autoantibody clustering of lupus-associated pulmonary hypertension. Lupus Sci. Med. 2019, 6, e000356. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, S.; Gibbs, J.S.; Wachter, R.; De Marco, T.; Vonk-Noordegraaf, A.; Vachiéry, J.L. Left ventricular heart failure and pulmonary hypertension. Eur. Heart J. 2016, 37, 942–954. [Google Scholar] [CrossRef] [PubMed]
- Bahrami, H.; Kronmal, R.; Bluemke, D.A.; Olson, J.; Shea, S.; Liu, K.; Burke, G.L.; Lima, J.A. Differences in the incidence of congestive heart failure by ethnicity: The multi-ethnic study of atherosclerosis. Arch. Intern. Med. 2008, 168, 2138–2145. [Google Scholar] [CrossRef] [PubMed]
- Flack, J.M.; Ferdinand, K.C.; Nasser, S.A. Epidemiology of hypertension and cardiovascular disease in African Americans. J. Clin. Hypertens. 2003, 5 (Suppl. 1), 5–11. [Google Scholar] [CrossRef]
- Yang, B.Q.; Assad, T.R.; O’Leary, J.M.; Xu, M.; Halliday, S.J.; D’Amico, R.W.; Farber-Eger, E.H.; Wells, Q.S.; Hemmes, A.R.; Brittan, E.L. Racial differences in patients referred for right heart catheterization and risk of pulmonary hypertension. Pulm. Circ. 2018, 8, 2045894018764273. [Google Scholar] [CrossRef]
- Chaouat, A.; Bugnet, A.S.; Kadaoui, N.; Schott, R.; Enache, I.; Ducoloné, A.; Ehrhart, M.; Kessler, R.; Weitzenblum, E. Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2005, 172, 189–194. [Google Scholar] [CrossRef]
- Nathan, S.D.; Shlobin, O.A.; Ahmad, S.; Koch, J.; Barnett, S.D.; Ad, N.; Burton, N.; Leslie, K. Serial development of pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Respiration 2008, 76, 288–294. [Google Scholar] [CrossRef]
- Pleasants, R.A.; Riley, I.L.; Mannino, D.M. Defining and targeting health disparities in chronic obstructive pulmonary disease. Int. J. Chronic Obstr. Pulm. Dis. 2016, 11, 2475–2496. [Google Scholar] [CrossRef]
- Prescott, E.; Godtfredsen, N.; Vestbo, J.; Osler, M. Social position and mortality from respiratory diseases in males and females. Eur. Respir. J. 2003, 21, 821–826. [Google Scholar] [CrossRef] [PubMed]
- Waxman, A.; Restrepo-Jaramillo, R.; Thenappan, T.; Ravichandran, A.; Engel, P.; Bajwa, A.; Allen, R.; Feldman, J.; Argula, R.; Smith, P.; et al. Inhaled Treprostinil in Pulmonary Hypertension Due to Interstitial Lung Disease. N. Engl. J. Med. 2021, 384, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Collard, H.R.; Anstrom, K.J.; Schwarz, M.I.; Zisman, D.A. Sildenafil improves walk distance in idiopathic pulmonary fibrosis. Chest 2007, 131, 897–899. [Google Scholar] [CrossRef] [PubMed]
- Corte, T.J.; Gatzoulis, M.A.; Parfitt, L.; Harries, C.; Wells, A.U.; Wort, S.J. The use of sildenafil to treat pulmonary hypertension associated with interstitial lung disease. Respirology 2010, 15, 1226–1232. [Google Scholar] [CrossRef] [PubMed]
- Corte, T.J.; Keir, G.J.; Dimopoulos, K.; Howard, L.; Corris, P.A.; Parfitt, L.; Foley, C.; Yanez-Lopez, M.; Babalis, D.; Marino, P.; et al. Bosentan in pulmonary hypertension associated with fibrotic idiopathic interstitial pneumonia. Am. J. Respir. Crit. Care Med. 2014, 190, 208–217. [Google Scholar] [CrossRef]
- Goudie, A.R.; Lipworth, B.J.; Hopkinson, P.J.; Wei, L.; Struthers, A.D. Tadalafil in patients with chronic obstructive pulmonary disease: A randomised, double-blind, parallel-group, placebo-controlled trial. Lancet Respir. Med. 2014, 2, 293–300. [Google Scholar] [CrossRef]
- Hoeper, M.M.; Halank, M.; Wilkens, H.; Gunther, A.; Weimann, G.; Gebert, I.; Leuchte, H.H.; Behr, J. Riociguat for interstitial lung disease and pulmonary hypertension: A pilot trial. Eur. Respir. J. 2013, 41, 853–860. [Google Scholar] [CrossRef]
- Rao, R.S.; Singh, S.; Sharma, B.B.; Agarwal, V.V.; Singh, V. Sildenafil improves six-minute walk distance in chronic obstructive pulmonary disease: A randomised, double-blind, placebo-controlled trial. Indian J. Chest Dis. Allied Sci. 2011, 53, 81–85. [Google Scholar]
- Valerio, G.; Bracciale, P.; Grazia D’Agostino, A. Effect of bosentan upon pulmonary hypertension in chronic obstructive pulmonary disease. Ther. Adv. Respir. Dis. 2009, 3, 15–21. [Google Scholar] [CrossRef]
- Vitulo, P.; Stanziola, A.; Confalonieri, M.; Libertucci, D.; Oggionni, T.; Rottoli, P.; Paciocco, G.; Tuzzolino, F.; Martino, L.; Beretta, M.; et al. Sildenafil in severe pulmonary hypertension associated with chronic obstructive pulmonary disease: A randomized controlled multicenter clinical trial. J. Heart Lung Transplant. 2017, 36, 166–174. [Google Scholar] [CrossRef]
- Brakefield, W.S.; Olusanya, O.A.; White, B.; Shaban-Nejad, A. Social Determinants and Indicators of COVID-19 among Marginalized Communities: A Scientific Review and Call to Action for Pandemic Response and Recovery. Disaster Med. Public Health Prep. 2022, 17, e193. [Google Scholar] [CrossRef] [PubMed]
- Egom, E.À.; Shiwani, H.A.; Nouthe, B. From acute SARS-CoV-2 infection to pulmonary hypertension. Front. Physiol. 2022, 13, 1023758. [Google Scholar] [CrossRef] [PubMed]
- Rossi, R.; Coppi, F.; Monopoli, D.E.; Sgura, F.A.; Arrotti, S.; Boriani, G. Pulmonary arterial hypertension and right ventricular systolic dysfunction in COVID-19 survivors. Cardiol. J. 2022, 29, 163–165. [Google Scholar] [CrossRef] [PubMed]
- Su, A.Y.; Vinogradsky, A.; Wang, A.S.; Ning, Y.; Abrahams, E.; Bacchetta, M.; Kurlansky, P.; Rosenzweig, E.; Takeda, K. Impact of sex, race and socioeconomic status on survival after pulmonary thromboendarterectomy for chronic thromboembolic pulmonary hypertension. Eur. J. Cardiothorac. Surg. 2022, 62, ezac364. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.C.Y.; Man, H.S.J.; Asghar, U.M.; McRae, K.; Zhao, Y.; Donahoe, L.L.; Wu, L.; Granton, J.; de Perrot, M. Impact of sex on outcome after pulmonary endarterectomy for chronic thromboembolic pulmonary hypertension. J. Heart Lung Transplant. 2023, 42, 1578–1586. [Google Scholar] [CrossRef] [PubMed]
- Abramov, A.Y.; Fraley, C.; Diao, C.T.; Winkfein, R.; Colicos, M.A.; Duchen, M.R.; French, R.J.; Pavlov, E. Targeted polyphosphatase expression alters mitochondrial metabolism and inhibits calcium-dependent cell death. Proc. Natl. Acad. Sci. USA 2007, 104, 18091–18096. [Google Scholar] [CrossRef]
- Lo, C.C.W.; Moosavi, S.M.; Bubb, K.J. The Regulation of Pulmonary Vascular Tone by Neuropeptides and the Implications for Pulmonary Hypertension. Front. Physiol. 2018, 9, 1167. [Google Scholar] [CrossRef]
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357 Pt 3, 593–615. [Google Scholar] [CrossRef]
- Klinger, J.R. The nitric oxide/cGMP signaling pathway in pulmonary hypertension. Clin. Chest Med. 2007, 28, 143–167. [Google Scholar] [CrossRef]
- Ruopp, N.F.; Cockrill, B.A. Diagnosis and Treatment of Pulmonary Arterial Hypertension: A Review. JAMA 2022, 327, 1379–1391. [Google Scholar] [CrossRef]
- Galiè, N.; Ghofrani, H.A.; Torbicki, A.; Barst, R.J.; Rubin, L.J.; Badesch, D.; Fleming, T.; Parpia, T.; Burgess, G.; Branzi, A.; et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N. Engl. J. Med. 2005, 353, 2148–2157. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Brundage, B.H.; Ghofrani, H.A.; Oudiz, R.J.; Simonneau, G.; Safdar, Z.; Shapiro, S.; White, R.J.; Chan, M.; Beardsworth, A.; et al. Tadalafil therapy for pulmonary arterial hypertension. Circulation 2009, 119, 2894–2903. [Google Scholar] [CrossRef]
- Stasch, J.-P.; Pacher, P.; Evgenov, O.V. Soluble Guanylate Cyclase as an Emerging Therapeutic Target in Cardiopulmonary Disease. Circulation 2011, 123, 2263–2273. [Google Scholar] [CrossRef] [PubMed]
- Ghofrani, H.A.; Galiè, N.; Grimminger, F.; Grunig, E.; Humbert, M.; Jing, Z.; Keogh, A.M.; Langelben, D.; Kilama, M.O.; Fritsch, A.; et al. Riociguat for the treatment of pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Ghofrani, H.A.; D’Armini, A.M.; Grimminger, F.; Hoeper, M.M.; Jansa, P.; Kim, N.H.; Mayer, E.; Simonneau, G.; Wilkins, M.R.; Fritsch, A.; et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N. Engl. J. Med. 2013, 369, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Ruan, C.H.; Dixon, R.A.; Willerson, J.T.; Ruan, K.H. Prostacyclin therapy for pulmonary arterial hypertension. Tex. Heart Inst. J. 2010, 37, 391–399. [Google Scholar] [PubMed]
- Barst, R.J.; Rubin, L.J.; Long, W.A.; McGoon, M.D.; Rich, S.; Badesch, D.B.; Groves, B.M.; Tapson, V.F.; Bourge, R.C.; Brundage, B.H.; et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. N. Engl. J. Med. 1996, 334, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Simonneau, G.; Barst, R.J.; Galie, N.; Naeije, R.; Rich, S.; Bourge, R.C.; Keogh, A.; Oudiz, R.; Frost, A.; Blackburn, S.D.; et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: A double-blind, randomized, placebo-controlled trial. Am. J. Respir. Crit. Care Med. 2002, 165, 800–804. [Google Scholar] [CrossRef]
- Jing, Z.C.; Parikh, K.; Pulido, T.; Jerjes-Sanchez, C.; White, R.J.; Allen, R.; Torbicki, A.; Xu, K.; Yehle, D.; Laliberte, K.; et al. Efficacy and safety of oral treprostinil monotherapy for the treatment of pulmonary arterial hypertension: A randomized, controlled trial. Circulation 2013, 127, 624–633. [Google Scholar] [CrossRef]
- Channick, R.N.; Olschewski, H.; Seeger, W.; Staub, T.; Voswinckel, R.; Rubin, L.J. Safety and efficacy of inhaled treprostinil as add-on therapy to bosentan in pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2006, 48, 1433–1437. [Google Scholar] [CrossRef]
- Olschewski, H.; Simonneau, G.; Galiè, N.; Higenbottam, T.; Naeije, R.; Rubin, L.J.; Nikkho, S.; Speich, R.; Hoeper, M.M.; Behr, J.; et al. Inhaled iloprost for severe pulmonary hypertension. N. Engl. J. Med. 2002, 347, 322–329. [Google Scholar] [CrossRef] [PubMed]
- Sitbon, O.; Channick, R.; Chin, K.M.; Frey, A.; Gaine, S.; Galié, A.; Ghofrani, H.; Hoeper, M.M.; Lang, I.M.; Preiss, R.; et al. Selexipag for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2015, 373, 2522–2533. [Google Scholar] [CrossRef] [PubMed]
- Channick, R.N.; Simonneau, G.; Sitbon, O.; Robbins, I.M.; Frost, A.; Tapson, V.F.; Badesch, D.B.; Roux, S.; Rainisio, M.; Bodin, F.; et al. Effects of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: A randomised placebo-controlled study. Lancet 2001, 358, 1119–1123. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.J.; Badesch, D.B.; Barst, R.J.; Galie, N.; Black, C.M.; Keogh, A.; Pulido, T.; Frost, A.; Roux, S.; Leconte, I.; et al. Bosentan therapy for pulmonary arterial hypertension. N. Engl. J. Med. 2002, 346, 896–903. [Google Scholar] [CrossRef]
- Pulido, T.; Adzerikho, I.; Channick, R.N.; Delcroix, M.; Galié, N.; Ghofrani, H.; Jansa, P.; Jing, Z.; Le Brun, F.; Mehta, S.; et al. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 809–818. [Google Scholar] [CrossRef]
- Galiè, N.; Olschewski, H.; Oudiz, R.J.; Torres, F.; Frost, A.; Ghofrani, H.; Badesch, D.B.; McGoon, M.D.; McLaughlin, V.V.; Roecker, E.B.; et al. Ambrisentan for the treatment of pulmonary arterial hypertension: Results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 2008, 117, 3010–3019. [Google Scholar] [CrossRef]
- Lajoie, A.C.; Lauzière, G.; Lega, J.C.; Lacasse, Y.; Martin, S.; Simard, S.; Bonnet, S.; Provencher, S. Combination therapy versus monotherapy for pulmonary arterial hypertension: A meta-analysis. Lancet Respir. Med. 2016, 4, 291–305. [Google Scholar] [CrossRef]
- Galiè, N.; Barberà, J.A.; Frost, A.E.; Frost, A.E.; Ghofrani, H.; Hoeper, M.M.; McLaughlin, V.V.; Peacock, A.J.; Simonneau, G.; Blair, C.; et al. Initial Use of Ambrisentan plus Tadalafil in Pulmonary Arterial Hypertension. N. Engl. J. Med. 2015, 373, 834–844. [Google Scholar] [CrossRef]
- Weill, D.; Benden, C.; Corris, P.A.; Dark, J.H.; Davis, R.D.; Keshavjee, S.; Lederer, D.J.; Mulligan, M.J.; Patterson, G.A.; Singer, L.G.; et al. A consensus document for the selection of lung transplant candidates: 2014—An update from the Pulmonary Transplantation Council of the International Society for Heart and Lung Transplantation. J. Heart Lung Transplant. 2015, 34, 1–15. [Google Scholar] [CrossRef]
- Benza, R.L.; Gomberg-Maitland, M.; Elliott, C.G.; Farber, H.W.; Foreman, A.J.; Frost, A.E.; McGoon, M.D.; Pasta, D.J.; Selej, M.; Burger, C.D.; et al. Predicting Survival in Patients With Pulmonary Arterial Hypertension: The REVEAL Risk Score Calculator 2.0 and Comparison With ESC/ERS-Based Risk Assessment Strategies. Chest 2019, 156, 323–337. [Google Scholar] [CrossRef]
- George, M.P.; Champion, H.C.; Pilewski, J.M. Lung transplantation for pulmonary hypertension. Pulm. Circ. 2011, 1, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Christie, J.D.; Edwards, L.B.; Kucheryavaya, A.Y.; Aurora, P.; Christie, J.D.; Kirk, R.; Dobbels, F.; Rahmel, A.O.; Hertz, M.I. The Registry of the International Society for Heart and Lung Transplantation: Twenty-seventh official adult lung and heart-lung transplant report—2010. J. Heart Lung Transplant. 2010, 29, 1104–1118. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Hu, H.; Dong, H. Systematic Review of the Economic Burden of Pulmonary Arterial Hypertension. Pharmacoeconomics 2016, 34, 533–550. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.A.; Kalbaugh, C.A. Challenging assumptions about minority participation in US clinical research. Am. J. Public Health 2011, 101, 2217–2222. [Google Scholar] [CrossRef] [PubMed]
- Corbie-Smith, G.; Thomas, S.B.; Williams, M.V.; Moody-Ayers, S. Attitudes and beliefs of African Americans toward participation in medical research. J. Gen. Intern. Med. 1999, 14, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Shavers-Hornaday, V.L.; Lynch, C.F.; Burmeister, L.F.; Torner, J.C. Why are African Americans under-represented in medical research studies? Impediments to participation. Ethn. Health 1997, 2, 31–45. [Google Scholar] [CrossRef]
- Nelson, A. Unequal treatment: Confronting racial and ethnic disparities in health care. J. Natl. Med. Assoc. 2002, 94, 666–668. [Google Scholar]
- Wendler, D.; Kington, R.; Madans, J.; Van Wye, G.; Christ-Schmidt, H.; Pratt, L.A.; Brawley, O.W.; Gross, C.P.; Emanuel, E. Are racial and ethnic minorities less willing to participate in health research? PLoS Med. 2006, 3, e19. [Google Scholar] [CrossRef]
- Humbert, M.; Barst, R.J.; Robbins, I.M.; Channick, R.M.; Galie, N.; Boonstra, A.; Rubin, L.J.; Horn, E.M.; Manes, A.; Simonneau, G. Combination of bosentan with epoprostenol in pulmonary arterial hypertension: BREATHE-2. Eur. Respir. J. 2004, 24, 353–359. [Google Scholar] [CrossRef]
- Said, K. Macitentan in pulmonary arterial hypertension: The SERAPHIN trial. Glob. Cardiol. Sci. Pract. 2014, 2014, 26–30. [Google Scholar] [CrossRef]
- Simonneau, G.; D’Armini, A.M.; Ghofrani, H.A.; Grimminger, F.; Hoeper, M.M.; Jansa, P.; Kim, N.H.; Wang, C.; Wilkins, M.R.; Fritsch, A.; et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension: A long-term extension study (CHEST-2). Eur. Respir. J. 2015, 45, 1293–1302. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, P.W.; Pieske, B.; Anstrom, K.J.; Ezekowitz, J.; Hernandez, A.F.; Butler, J.; Lam, C.S.P.; Ponikowski, P.; Voors, A.A.; Jia, G.; et al. Vericiguat in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2020, 382, 1883–1893. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; McLaughlin, V.; Gibbs, J.S.R.; Gomberg-Maitland, M.; Hoeper, M.M.; Preston, I.R.; Souza, R.; Waxman, A.; Escribano Subias, P.; Feldman, J.; et al. Sotatercept for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2021, 384, 1204–1215. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Trial Name | Major Conclusions | Minority Recruitment |
|---|---|---|
| Channick et al. [73] | Bosentan improves exercise capacity and hemodynamics in patients with pulmonary hypertension. | <18% Black patients |
| Rubin et al. [74] | Bosentan is an effective medication in patients with pulmonary arterial hypertension. | >77% White patients |
| BREATHE-2 [89] | Trial suggested a trend towards hemodynamic or clinical improvement in patients with pulmonary arterial hypertension who were treated with a combination of epoprostenol and bosentan. | <10% Black patients |
| SUPER-1 [61] | In patients with pulmonary arterial hypertension, sildenafil improves exercise capacity, WHO functional class, and hemodynamics. | <10% non-White patients >80% White patients |
| ARIES 1 and 2 [76] | Ambrisentan improves exercise capacity in pulmonary arterial hypertension. | 0–6% Black patients 66–92% White patients |
| SERAPHIN [90] | Macitentan reduced morbidity and mortality amongst patients with pulmonary arterial hypertension. | 2.6% Black patients 54.5% White patients |
| CHEST1 [65] and CHEST2 [91] | Riociguat improved exercise capacity and pulmonary vascular resistance in patients with chronic thromboembolic pulmonary hypertension. | 3% Black patients 71% White patients |
| GRIPHON [72] | In patients with pulmonary arterial hypertension, treatment with selexipag led to a lower risk of death and pulmonary hypertension complication. | <10% enrollment in Latin America Predominantly enrolled in Europe |
| AMBITION [78] | Amongst those with pulmonary arterial hypertension, combination therapy with ambrisentan and tadalafil led to less risk of composite death, hospitalization for pulmonary arterial hypertension, disease progression, and unsatisfactory long-term clinical response than monotherapy with either agent. | <15% non-White patients |
| VICTORIA [92] | Amongst patients with a high risk of heart failure, those who received vericiguat had less risk of death due to cardiovascular causes or heart failure hospitalizations than those who received a placebo. | 4.9% Black patients 64% White patients |
| PULSAR [93] | Amongst patients receiving background therapy for pulmonary arterial hypertension, treatment with sotatercept resulted in a reduction in pulmonary vascular resistance. | 4% Black patients 92% White patients |
| INCREASE [42] | In patients with pulmonary hypertension secondary to interstitial lung disease, inhaled treprostinil improved exercise capacity. | 71 Black patients 238 White patients |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Contreras, J.; Nussbaum, J.; Cangialosi, P.; Thapi, S.; Radakrishnan, A.; Hall, J.; Ramesh, P.; Trivieri, M.G.; Sandoval, A.F. Pulmonary Hypertension in Underrepresented Minorities: A Narrative Review. J. Clin. Med. 2024, 13, 285. https://doi.org/10.3390/jcm13010285
Contreras J, Nussbaum J, Cangialosi P, Thapi S, Radakrishnan A, Hall J, Ramesh P, Trivieri MG, Sandoval AF. Pulmonary Hypertension in Underrepresented Minorities: A Narrative Review. Journal of Clinical Medicine. 2024; 13(1):285. https://doi.org/10.3390/jcm13010285
Chicago/Turabian StyleContreras, Johanna, Jeremy Nussbaum, Peter Cangialosi, Sahityasri Thapi, Ankitha Radakrishnan, Jillian Hall, Prashasthi Ramesh, Maria Giovanna Trivieri, and Alejandro Folch Sandoval. 2024. "Pulmonary Hypertension in Underrepresented Minorities: A Narrative Review" Journal of Clinical Medicine 13, no. 1: 285. https://doi.org/10.3390/jcm13010285
APA StyleContreras, J., Nussbaum, J., Cangialosi, P., Thapi, S., Radakrishnan, A., Hall, J., Ramesh, P., Trivieri, M. G., & Sandoval, A. F. (2024). Pulmonary Hypertension in Underrepresented Minorities: A Narrative Review. Journal of Clinical Medicine, 13(1), 285. https://doi.org/10.3390/jcm13010285