
CNS Ageing in Health and Neurodegenerative Disorders


 , ,
, , 
 and
and 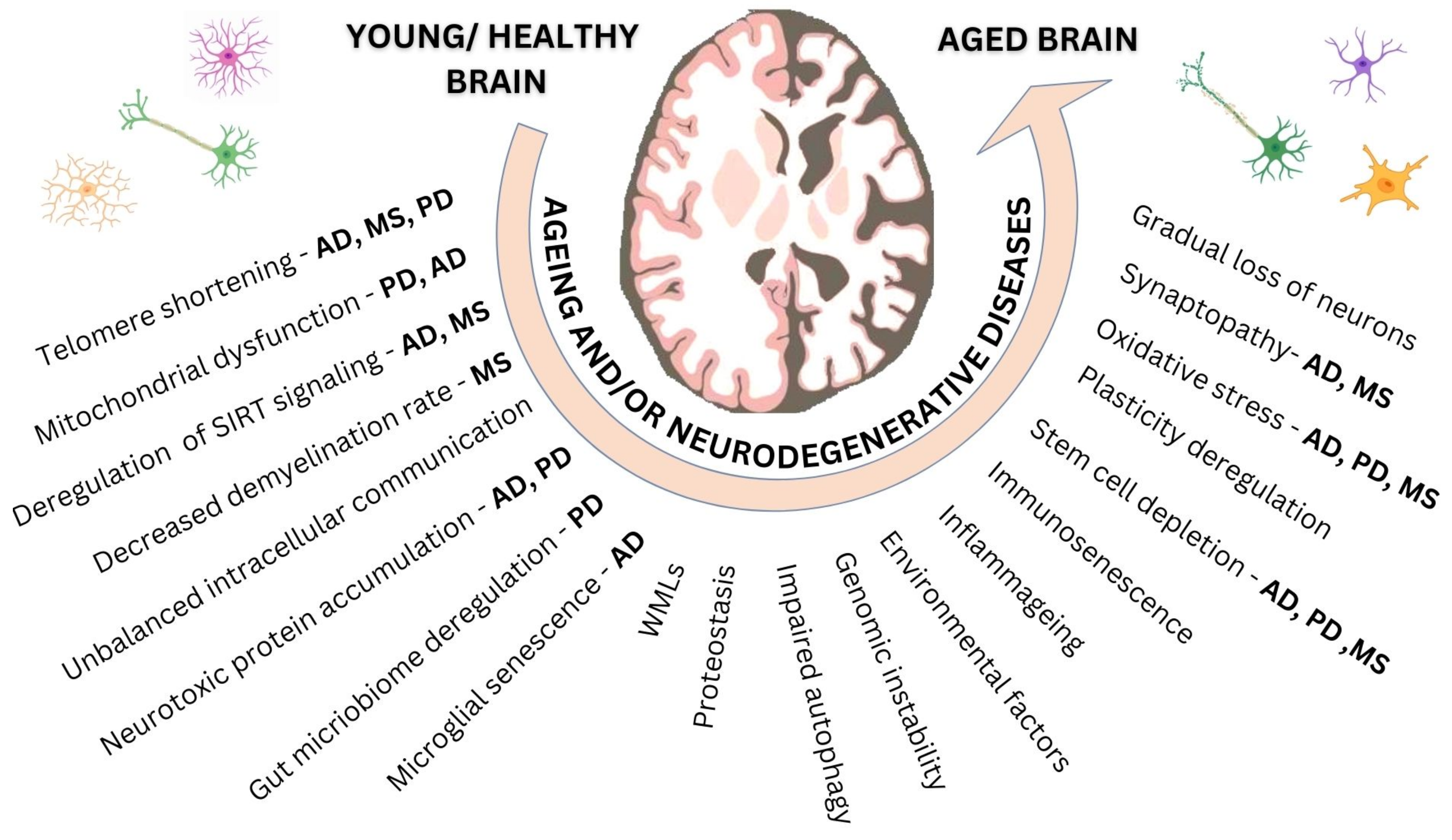{kind=link}
Abstract
1. Introduction
2. Ageing at the Molecular and the Cellular Level
2.1. Ageing Damaging Mechanisms
2.2. Responses to Damage
2.3. Alterations That Affect Phenotypic Manifestations
3. Ageing, CNS Architecture and Function
4. Ageing and CNS Neurodegenerative Disorders
5. Early and Late CNS Ageing Processes: The Case of MS
6. Ageing, Comorbidities and DMTs
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Melzer, D.; Pilling, L.C.; Ferrucci, L. The genetics of human ageing. Nat. Rev. Genet. 2020, 21, 88–101. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Salvioli, S.; Garagnani, P.; de Eguileor, M.; Monti, D.; Capri, M. Immunobiography and the Heterogeneity of Immune Responses in the Elderly: A Focus on Inflammaging and Trained Immunity. Front. Immunol. 2017, 8, 982. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Morsiani, C.; Conte, M.; Santoro, A.; Grignolio, A.; Monti, D.; Capri, M.; Salvioli, S. The Continuum of Aging and Age-Related Diseases: Common Mechanisms but Different Rates. Front. Med. 2018, 5, 61. [Google Scholar] [CrossRef] [PubMed]
- Satoh, A.; Imai, S.I.; Guarente, L. The brain, sirtuins, and ageing. Nat. Rev. Neurosci. 2017, 18, 362–374. [Google Scholar] [CrossRef]
- Partridge, L.; Deelen, J.; Slagboom, P.E. Facing up to the global challenges of ageing. Nature 2018, 561, 45–56. [Google Scholar] [CrossRef]
- Hung, C.W.; Chen, Y.C.; Hsieh, W.L.; Chiou, S.H.; Kao, C.L. Ageing and neurodegenerative diseases. Ageing Res. Rev. 2010, 9 (Suppl. 1), S36–S46. [Google Scholar] [CrossRef]
- Mattson, M.P.; Magnus, T. Ageing and neuronal vulnerability. Nat. Rev. Neurosci. 2006, 7, 278–294. [Google Scholar] [CrossRef]
- Ahadi, S.; Zhou, W.; Rose, S.M.S.-F.; Sailani, M.R.; Contrepois, K.; Avina, M.; Ashland, M.; Brunet, A.; Snyder, M. Personal aging markers and ageotypes revealed by deep longitudinal profiling. Nat. Med. 2020, 26, 83–90. [Google Scholar] [CrossRef]
- Piening, B.D.; Lovejoy, J.; Earls, J.C. Ageotypes: Distinct Biomolecular Trajectories in Human Aging. Trends Pharmacol. Sci. 2020, 41, 299–301. [Google Scholar] [CrossRef]
- Jin, K. Modern Biological Theories of Aging. Aging Dis. 2010, 1, 72–74. [Google Scholar]
- Kenyon, C.J. The genetics of ageing. Nature 2010, 464, 504–512. [Google Scholar] [CrossRef]
- Liochev, S.I. Which is the most significant cause of aging? Antioxidants 2015, 4, 793–810. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Proteostasis and aging. Nat. Med. 2015, 21, 1406–1415. [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the Autophagy-Inflammation-Cell Death Axis in Organismal Aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef]
- Heydari, A.R.; Unnikrishnan, A.; Lucente, L.V.; Richardson, A. Caloric restriction and genomic stability. Nucleic Acids Res. 2007, 35, 7485–7496. [Google Scholar] [CrossRef]
- Iourov, I.; Yurov, Y.; Vorsanova, S.; Kutsev, S. Chromosome Instability, Aging and Brain Diseases. Cells 2021, 10, 1256. [Google Scholar] [CrossRef]
- Coleman, C.; Martin, I. Unraveling Parkinson’s Disease Neurodegeneration: Does Aging Hold the Clues? J. Park. Dis. 2022, 12, 2321–2338. [Google Scholar] [CrossRef]
- Aunan, J.R.; Watson, M.M.; Hagland, H.R.; Søreide, K. Molecular and biological hallmarks of ageing. Br. J. Surg. 2016, 103, e29–e46. [Google Scholar] [CrossRef] [PubMed]
- Marioni, R.E.; Shah, S.; McRae, A.F.; Chen, B.H.; Colicino, E.; Harris, S.E.; Gibson, J.; Henders, A.K.; Redmond, P.; Cox, S.R.; et al. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 2015, 16, 25. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Chan, A.T.; Fuchs, C.S.; Giovannucci, E. Molecular pathological epidemiology of colorectal neoplasia: An emerging transdisciplinary and interdisciplinary field. Gut 2011, 60, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liu, H.; Hu, Q.; Wang, L.; Liu, J.; Zheng, Z.; Zhang, W.; Ren, J.; Zhu, F.; Liu, G.-H. Epigenetic regulation of aging: Implications for interventions of aging and diseases. Signal Transduct. Target. Ther. 2022, 7, 374. [Google Scholar] [CrossRef]
- Cawthon, R.M.; Smith, K.R.; O’Brien, E.; Sivatchenko, A.; Kerber, R.A. Association between telomere length in blood and mortality in people aged 60 years or older. Lancet 2003, 361, 393–395. [Google Scholar] [CrossRef]
- Zecca, L.; Youdim, M.B.H.; Riederer, P.; Connor, J.R.; Crichton, R.R. Iron, brain ageing and neurodegenerative disorders. Nat. Rev. Neurosci. 2004, 5, 863–873. [Google Scholar] [CrossRef]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef]
- Lu, T.; Pan, Y.; Kao, S.-Y.; Li, C.; Kohane, I.; Chan, J.; Yankner, B.A. Gene regulation and DNA damage in the ageing human brain. Nat. Cell Biol. 2004, 429, 883–891. [Google Scholar] [CrossRef]
- Lehtonen, Š.; Sonninen, T.-M.; Wojciechowski, S.; Goldsteins, G.; Koistinaho, J. Dysfunction of Cellular Proteostasis in Parkinson’s Disease. Front. Neurosci. 2019, 13, 457. [Google Scholar] [CrossRef]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef]
- Kesidou, E.; Lagoudaki, R.; Touloumi, O.; Poulatsidou, K.-N.; Simeonidou, C. Autophagy and neurodegenerative disorders. Neural Regen. Res. 2013, 8, 2275–2283. [Google Scholar] [CrossRef]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef]
- Pera, A.; Campos, C.; López, N.; Hassouneh, F.; Alonso, C.; Tarazona, R.; Solana, R. Immunosenescence: Implications for response to infection and vaccination in older people. Maturitas 2015, 82, 50–55. [Google Scholar] [CrossRef]
- Harman, D. Origin and evolution of the free radical theory of aging: A brief personal history, 1954–2009. Biogerontology 2009, 10, 773. [Google Scholar] [CrossRef]
- Bishop, N.A.; Lu, T.; Yankner, B.A. Neural mechanisms of ageing and cognitive decline. Nature 2010, 464, 529–535. [Google Scholar] [CrossRef]
- Palmeira, C.M.; Teodoro, J.S.; Amorim, J.A.; Steegborn, C.; Sinclair, D.A.; Rolo, A.P. Mitohormesis and metabolic health: The interplay between ROS, cAMP and sirtuins. Free. Radic. Biol. Med. 2019, 141, 483–491. [Google Scholar] [CrossRef]
- Lautrup, S.; Sinclair, D.A.; Mattson, M.P.; Fang, E.F. NAD+ in Brain Aging and Neurodegenerative Disorders. Cell Metab. 2019, 30, 630–655. [Google Scholar] [CrossRef]
- Zong, H.; Ren, J.M.; Young, L.H.; Pypaert, M.; Mu, J.; Birnbaum, M.J.; Shulman, G.I. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc. Natl. Acad. Sci. USA 2002, 99, 15983–15987. [Google Scholar] [CrossRef]
- Anton, S.; Leeuwenburgh, C. Fasting or caloric restriction for Healthy Aging. Exp. Gerontol. 2013, 48, 1003–1005. [Google Scholar] [CrossRef]
- Di Giosia, P.; Stamerra, C.A.; Giorgini, P.; Jamialahamdi, T.; Butler, A.E.; Sahebkar, A. The role of nutrition in inflammaging. Ageing Res. Rev. 2022, 77, 101596. [Google Scholar] [CrossRef]
- Satoh, A.; Brace, C.S.; Rensing, N.; Cliften, P.; Wozniak, D.F.; Herzog, E.D.; Yamada, K.A.; Imai, S.-I. Sirt1 Extends Life Span and Delays Aging in Mice through the Regulation of Nk2 Homeobox 1 in the DMH and LH. Cell Metab. 2013, 18, 416–430. [Google Scholar] [CrossRef] [PubMed]
- Segel, M.; Neumann, B.; Hill, M.F.; Weber, I.P.; Viscomi, C.; Zhao, C.; Chalut, K.J. Niche stiffness underlies the ageing of central nervous system progenitor cells. Nature 2019, 573, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Neumann, B.; Baror, R.; Zhao, C.; Segel, M.; Dietmann, S.; Rawji, K.S.; Franklin, R.J. Metformin restores CNS remyelination capacity by rejuvenating aged stem cells. Cell Stem Cell 2019, 25, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Sandoval, J.; A Swiss, V.; Li, J.; Dupree, J.; Franklin, R.J.M.; Casaccia-Bonnefil, P. Age-dependent epigenetic control of differentiation inhibitors is critical for remyelination efficiency. Nat. Neurosci. 2008, 11, 1024–1034. [Google Scholar] [CrossRef] [PubMed]
- Cantuti-Castelvetri, L.; Fitzner, D.; Bosch-Queralt, M.; Weil, M.-T.; Su, M.; Sen, P.; Ruhwedel, T.; Mitkovski, M.; Trendelenburg, G.; Lütjohann, D.; et al. Defective cholesterol clearance limits remyelination in the aged central nervous system. Science 2018, 359, 684–688. [Google Scholar] [CrossRef]
- Berghoff, S.A.; Spieth, L.; Saher, G. Local cholesterol metabolism orchestrates remyelination. Trends Neurosci. 2022, 45, 272–283. [Google Scholar] [CrossRef]
- Duggal, N.A.; Niemiro, G.; Harridge, S.D.; Simpson, R.J.; Lord, J.M. Can physical activity ameliorate immunosenescence and thereby reduce age-related multi-morbidity? Nat. Rev. Immunol. 2019, 19, 563–572. [Google Scholar] [CrossRef]
- Vaughn, C.B.; Jakimovski, D.; Kavak, K.S.; Ramanathan, M.; Benedict, R.H.B.; Zivadinov, R.; Weinstock-Guttman, B. Epidemiology and treatment of multiple sclerosis in elderly populations. Nat. Rev. Neurol. 2019, 15, 329–342. [Google Scholar] [CrossRef]
- Feehan, J.; Tripodi, N.; Apostolopoulos, V. The twilight of the immune system: The impact of immunosenescence in aging. Maturitas 2021, 147, 7–13. [Google Scholar] [CrossRef]
- Ponnappan, S.; Ponnappan, U.; Zheng, Y.; Joyce, B.T.; Colicino, E.; Liu, L.; Zhang, W.; Dai, Q.; Shrubsole, M.J.; Kibbe, W.A.; et al. Aging and Immune Function: Molecular Mechanisms to Interventions. Antioxidants Redox Signal. 2011, 14, 1551–1585. [Google Scholar] [CrossRef]
- Wagner, K.-D.; Wagner, N. The Senescence Markers p16INK4A, p14ARF/p19ARF, and p21 in Organ Development and Homeostasis. Cells 2022, 11, 1966. [Google Scholar] [CrossRef]
- Liu, X.; Liu, Z.; Wu, Z.; Ren, J.; Fan, Y.; Sun, L.; Cao, G.; Niu, Y.; Zhang, B.; Ji, Q.; et al. Resurrection of endogenous retroviruses during aging reinforces senescence. Cell 2023, 186, 287–304.e26. [Google Scholar] [CrossRef]
- Bürkle, A.; Caselli, G.; Franceschi, C.; Mariani, E.; Sansoni, P.; Santoni, A.; Vecchio, G.; Witkowski, J.M.; Caruso, C. Pathophysiology of ageing, longevity and age related diseases. Immun. Ageing 2007, 4, 4. [Google Scholar] [CrossRef]
- Nikolich-Žugich, J. The twilight of immunity: Emerging concepts in aging of the immune system. Nat. Immunol. 2018, 19, 10–19. [Google Scholar] [CrossRef]
- Aw, D.; Silva, A.B.; Palmer, D.B. Immunosenescence: Emerging challenges for an ageing population. Immunology 2007, 120, 435–446. [Google Scholar] [CrossRef]
- Del Giudice, G.; Goronzy, J.J.; Grubeck-Loebenstein, B.; Lambert, P.-H.; Mrkvan, T.; Stoddard, J.J.; Doherty, T.M. Fighting against a protean enemy: Immunosenescence, vaccines, and healthy aging. NPJ Aging Mech. Dis. 2018, 4, 1. [Google Scholar] [CrossRef]
- Musella, A.; Gentile, A.; Rizzo, F.R.; De Vito, F.; Fresegna, D.; Bullitta, S.; Mandolesi, G. Interplay between age and neuroinflammation in multiple sclerosis: Effects on motor and cognitive functions. Front. Aging Neurosci. 2018, 10, 238. [Google Scholar] [CrossRef]
- Schwartz, M.; Baruch, K. The resolution of neuroinflammation in neurodegeneration: Leukocyte recruitment via the choroid plexus. EMBO J. 2014, 33, 7–22. [Google Scholar] [CrossRef]
- Andrews-Hanna, J.R.; Snyder, A.Z.; Vincent, J.L.; Lustig, C.; Head, D.; Raichle, M.E.; Buckner, R.L. Disruption of Large-Scale Brain Systems in Advanced Aging. Neuron 2007, 56, 924–935. [Google Scholar] [CrossRef]
- Petralia, R.S.; Mattson, M.P.; Yao, P.J. Communication breakdown: The impact of ageing on synapse structure. Ageing Res. Rev. 2014, 14, 31–42. [Google Scholar] [CrossRef]
- Grajauskas, L.A.; Siu, W.; Medvedev, G.; Guo, H.; D’Arcy, R.C.; Song, X. MRI-based evaluation of structural degeneration in the ageing brain: Pathophysiology and assessment. Ageing Res. Rev. 2019, 49, 67–82. [Google Scholar] [CrossRef] [PubMed]
- Hedman, A.M.; Van Haren, N.E.; Schnack, H.G.; Kahn, R.S.; Pol, H.H. Human brain changes across the life span: A review of 56 longitudinal magnetic resonance imaging studies. Hum. Brain Mapp. 2011, 33, 1987–2002. [Google Scholar] [CrossRef] [PubMed]
- Wegner, C.; Esiri, M.M.; Chance, S.A.; Palace, J.; Matthews, P.M. Neocortical neuronal, synaptic, and glial loss in multiple sclerosis. Neurology 2006, 67, 960–967. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Sachdev, P.; Lipnicki, D.M.; Zhang, H.; Liu, T.; Zhu, W.; Suo, C.; Zhuang, L.; Crawford, J.; Reppermund, S.; et al. A longitudinal study of brain atrophy over two years in community-dwelling older individuals. Neuroimage 2014, 86, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.N.; Barnes, C.A. Neural plasticity in the ageing brain. Nat. Rev. Neurosci. 2006, 7, 30–40. [Google Scholar] [CrossRef]
- E Grueter, B.; Schulz, U.G. Age-related cerebral white matter disease (leukoaraiosis): A review. Postgrad. Med. J. 2011, 88, 79–87. [Google Scholar] [CrossRef]
- Vernooij, M.W.; Ikram, M.A.; Tanghe, H.L.; Vincent, A.J.; Hofman, A.; Krestin, G.P.; Niessen, W.J.; Breteler, M.M.; van der Lugt, A. Incidental Findings on Brain MRI in the General Population. N. Engl. J. Med. 2007, 357, 1821–1828. [Google Scholar] [CrossRef]
- Lampe, L.; Kharabian-Masouleh, S.; Kynast, J.; Arelin, K.; Steele, C.J.; Löffler, M.; Witte, A.V.; Schroeter, M.L.; Villringer, A.; Bazin, P.-L. Lesion location matters: The relationships between white matter hyperintensities on cognition in the healthy elderly. J. Cereb. Blood Flow Metab. 2017, 39, 36–43. [Google Scholar] [CrossRef]
- Satizabal, C.L.; Zhu, Y.-C.; Dufouil, C.; Tzourio, C. Inflammatory Proteins and the Severity of Dilated Virchow-Robin Spaces in the Elderly. J. Alzheimer’s Dis. 2012, 33, 323–328. [Google Scholar] [CrossRef]
- Favaretto, A.; Lazzarotto, A.; Riccardi, A.; Pravato, S.; Margoni, M.; Causin, F.; Anglani, M.G.; Seppi, D.; Poggiali, D.; Gallo, P. Enlarged Virchow Robin spaces associate with cognitive decline in multiple sclerosis. PLoS ONE 2017, 12, e0185626. [Google Scholar] [CrossRef]
- Greenberg, S.M.; Vernooij, M.W.; Cordonnier, C.; Viswanathan, A.; Salman, R.A.S.; Warach, S.; Breteler, M.M. Cerebral microbleeds: A guide to detection and interpretation. Lancet Neurol. 2009, 8, 165–174. [Google Scholar] [CrossRef]
- Ding, J.; Sigurðsson, S.; Jónsson, P.V.; Eiriksdottir, G.; Charidimou, A.; Lopez, O.L.; A Van Buchem, M.; Guðnason, V.; Launer, L.J. Large Perivascular Spaces Visible on Magnetic Resonance Imaging, Cerebral Small Vessel Disease Progression, and Risk of Dementia: The Age, Gene/Environment Susceptibility-Reykjavik Study. JAMA Neurol. 2017, 74, 1105–1112. [Google Scholar] [CrossRef]
- Ding, Z.; Huang, Y.; Bailey, S.K.; Gao, Y.; Cutting, L.E.; Rogers, B.P.; Newton, A.T.; Gore, J.C. Detection of synchronous brain activity in white matter tracts at rest and under functional loading. Proc. Natl. Acad. Sci. USA 2018, 115, 595–600. [Google Scholar] [CrossRef]
- Adalbert, R.; Coleman, M.P. Review: Axon pathology in age-related neurodegenerative disorders. Neuropathol. Appl. Neurobiol. 2013, 39, 90–108. [Google Scholar] [CrossRef]
- Grillo, F.W. Long live the axon. Parallels between ageing and pathology from a presynaptic point of view. J. Chem. Neuroanat. 2016, 76, 28–34. [Google Scholar] [CrossRef]
- Bowley, M.P.; Cabral, H.; Rosene, D.; Peters, A. Age changes in myelinated nerve fibers of the cingulate bundle and corpus callosum in the rhesus monkey. J. Comp. Neurol. 2010, 518, 3046–3064. [Google Scholar] [CrossRef]
- Samuel, M.A.; Zhang, Y.; Meister, M.; Sanes, J.R. Age-Related Alterations in Neurons of the Mouse Retina. J. Neurosci. 2011, 31, 16033–16044. [Google Scholar] [CrossRef]
- Yang, Y.; Coleman, M.; Zhang, L.; Zheng, X.; Yue, Z. Autophagy in axonal and dendritic degeneration. Trends Neurosci. 2013, 36, 418–428. [Google Scholar] [CrossRef]
- Morrison, J.H.; Baxter, M.G. The ageing cortical synapse: Hallmarks and implications for cognitive decline. Nat. Rev. Neurosci. 2012, 13, 240–250. [Google Scholar] [CrossRef]
- Smith, T.D.; Adams, M.M.; Gallagher, M.; Morrison, J.H.; Rapp, P.R. Circuit-specific alterations in hippocampal synaptophysin immunoreactivity predict spatial learning impairment in aged rats. J. Neurosci. 2000, 20, 6587–6593. [Google Scholar] [CrossRef]
- Jang, Y.C.; Van Remmen, H. Age-associated alterations of the neuromuscular junction. Exp. Gerontol. 2011, 46, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Higgins-Chen, A.T.; Thrush, K.L.; Levine, M.E. Aging biomarkers and the brain. Semin. Cell Dev. Biol. 2021, 116, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Theodoropoulou, E.; Alfredsson, L.; Piehl, F.; Marabita, F.; Jagodic, M. Different epigenetic clocks reflect distinct pathophysiological features of multiple sclerosis. Epigenomics 2019, 11, 1429–1439. [Google Scholar] [CrossRef]
- Xiang, Y.; Xin, J.; Le, W.; Yang, Y. Neurogranin: A Potential Biomarker of Neurological and Mental Diseases. Front. Aging Neurosci. 2020, 12, 584743. [Google Scholar] [CrossRef] [PubMed]
- Chee, S.E.J.; Solito, E. The Impact of Ageing on the CNS Immune Response in Alzheimer’s Disease. Front. Immunol. 2021, 12, 738511. [Google Scholar] [CrossRef]
- Braidy, N.; Jayasena, T.; Poljak, A.; Sachdev, P.S. Sirtuins in cognitive ageing and Alzheimer’s disease. Curr. Opin. Psychiatry 2012, 25, 226–230. [Google Scholar] [CrossRef]
- Bonda, D.J.; Lee, H.-G.; Camins, A.; Pallàs, M.; Casadesus, G.; Smith, M.A.; Zhu, X. The sirtuin pathway in ageing and Alzheimer disease: Mechanistic and therapeutic considerations. Lancet Neurol. 2011, 10, 275–279. [Google Scholar] [CrossRef]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Neill, D. Should Alzheimer’s disease be equated with human brain ageing?: A maladaptive interaction between brain evolution and senescence. Ageing Res. Rev. 2012, 11, 104–122. [Google Scholar] [CrossRef]
- Spittau, B. Aging microglia—Phenotypes, functions and implications for age-related neurodegenerative diseases. Front. Aging Neurosci. 2017, 9, 194. [Google Scholar] [CrossRef]
- Habib, N.; McCabe, C.; Medina, S.; Varshavsky, M.; Kitsberg, D.; Dvir-Szternfeld, R.; Green, G.; Dionne, D.; Nguyen, L.; Marshall, J.L.; et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat. Neurosci. 2020, 23, 701–706. [Google Scholar] [CrossRef]
- Reeve, A.; Simcox, E.; Turnbull, D. Ageing and Parkinson’s disease: Why is advancing age the biggest risk factor? Ageing Res. Rev. 2014, 14, 19–30. [Google Scholar] [CrossRef]
- Levy, G. The Relationship of Parkinson Disease With Aging. Arch. Neurol. 2007, 64, 1242–1246. [Google Scholar] [CrossRef]
- Pirazzini, C.; Azevedo, T.; Baldelli, L.; Bartoletti-Stella, A.; Calandra-Buonaura, G.; Molin, A.D.; Dimitri, G.M.; Doykov, I.; Gómez-Garre, P.; Hägg, S.; et al. A geroscience approach for Parkinson’s disease: Conceptual framework and design of PROPAG-AGEING project. Mech. Ageing Dev. 2020, 194, 111426. [Google Scholar] [CrossRef]
- Pang, S.Y.Y.; Ho, P.W.L.; Liu, H.F.; Leung, C.T.; Li, L.; Chang, E.E.S.; Ho, S.L. The interplay of aging, genetics and environmental factors in the pathogenesis of Parkinson’s disease. Transl. Neurodegener. 2019, 8, 23. [Google Scholar] [CrossRef]
- Surmeier, D.J. Calcium, ageing, and neuronal vulnerability in Parkinson’s disease. Lancet Neurol. 2007, 6, 933–938. [Google Scholar] [CrossRef]
- Sepe, S.; Milanese, C.; Gabriels, S.; Derks, K.W.; Payan-Gomez, C.; van Ijcken, W.F.; Rijksen, Y.M.; Nigg, A.L.; Moreno, S.; Cerri, S.; et al. Inefficient DNA Repair Is an Aging-Related Modifier of Parkinson’s Disease. Cell Rep. 2016, 15, 1866–1875. [Google Scholar] [CrossRef]
- Regensburger, M.; Prots, I.; Winner, B. Adult Hippocampal Neurogenesis in Parkinson’s Disease: Impact on Neuronal Survival and Plasticity. Neural Plast. 2014, 2014, 454696. [Google Scholar] [CrossRef]
- Marchetti, B.; Tirolo, C.; L’Episcopo, F.; Caniglia, S.; Testa, N.; Smith, J.A.; Serapide, M.F. Parkinson’s disease, aging and adult neurogenesis: Wnt/β-catenin signalling as the key to unlock the mystery of endogenous brain repair. Aging Cell 2020, 19, e13101. [Google Scholar] [CrossRef]
- Bassi, M.S.; Gilio, L.; Iezzi, E.; Moscatelli, A.; Pekmezovic, T.; Drulovic, J.; Furlan, R.; Finardi, A.; Mandolesi, G.; Musella, A.; et al. Age at Disease Onset Associates With Oxidative Stress, Neuroinflammation, and Impaired Synaptic Plasticity in Relapsing-Remitting Multiple Sclerosis. Front. Aging Neurosci. 2021, 13, 694651. [Google Scholar] [CrossRef]
- Hecker, M.; Bühring, J.; Fitzner, B.; Rommer, P.S.; Zettl, U.K. Genetic, Environmental and Lifestyle Determinants of Accelerated Telomere Attrition as Contributors to Risk and Severity of Multiple Sclerosis. Biomolecules 2021, 11, 1510. [Google Scholar] [CrossRef] [PubMed]
- Hecker, L.J.; Edwards, M.A.; Nielsen, S.E. Assessing the nutritional consequences of switching foraging behavior in wood bison. Ecol. Evol. 2021, 11, 16165–16176. [Google Scholar] [CrossRef] [PubMed]
- Kritsilis, M.; Rizou, S.V.; Koutsoudaki, P.N.; Evangelou, K.; Gorgoulis, V.G.; Papadopoulos, D. Ageing, Cellular Senescence and Neurodegenerative Disease. Int. J. Mol. Sci. 2018, 19, 2937. [Google Scholar] [CrossRef] [PubMed]
- Azam, S.; Haque, E.; Balakrishnan, R.; Kim, I.-S.; Choi, D.-K. The Ageing Brain: Molecular and Cellular Basis of Neurodegeneration. Front. Cell Dev. Biol. 2021, 9, 683459. [Google Scholar] [CrossRef] [PubMed]
- Cespón, J.; Miniussi, C.; Pellicciari, M.C. Interventional programmes to improve cognition during healthy and pathological ageing: Cortical modulations and evidence for brain plasticity. Ageing Res. Rev. 2018, 43, 81–98. [Google Scholar] [CrossRef]
- Mandolesi, G.; Gentile, A.; Musella, A.; Fresegna, D.; De Vito, F.; Bullitta, S.; Sepman, H.; Marfia, G.A.; Centonze, D. Synaptopathy connects inflammation and neurodegeneration in multiple sclerosis. Nat. Rev. Neurol. 2015, 11, 711–724. [Google Scholar] [CrossRef]
- Rossi, S.; Motta, C.; Studer, V.; Barbieri, F.; Buttari, F.; Bergami, A.; Sancesario, G.; Bernardini, S.; De Angelis, G.; Martino, G.; et al. Tumor necrosis factor is elevated in progressive multiple sclerosis and causes excitotoxic neurodegeneration. Mult. Scler. J. 2014, 20, 304–312. [Google Scholar] [CrossRef]
- Di Benedetto, S.; Müller, L.; Wenger, E.; Düzel, S.; Pawelec, G. Contribution of neuroinflammation and immunity to brain aging and the mitigating effects of physical and cognitive interventions. Neurosci. Biobehav. Rev. 2017, 75, 114–128. [Google Scholar] [CrossRef]
- Landi, D.; Rossini, P.M. Cerebral restorative plasticity from normal ageing to brain diseases: A “never ending story”. Restor. Neurol. Neurosci. 2010, 28, 349–366. [Google Scholar] [CrossRef]
- Gutchess, A. Plasticity of the aging brain: New directions in cognitive neuroscience. Science 2014, 346, 579–582. [Google Scholar] [CrossRef]
- Zhao, C.; Deng, W.; Gage, F.H. Mechanisms and Functional Implications of Adult Neurogenesis. Cell 2008, 132, 645–660. [Google Scholar] [CrossRef]
- Martino, G.; Pluchino, S. The therapeutic potential of neural stem cells. Nat. Rev. Neurosci. 2006, 7, 395–406. [Google Scholar] [CrossRef]
- Ernst, A.; Frisén, J. Adult Neurogenesis in Humans- Common and Unique Traits in Mammals. PLOS Biol. 2015, 13, e1002045. [Google Scholar] [CrossRef]
- Gao, J.; Wang, W.-Y.; Mao, Y.W.; Gräff, J.; Guan, J.-S.; Pan, L.; Mak, G.; Kim, D.; Su, S.C.; Tsai, L.-H. A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature 2010, 466, 1105–1109. [Google Scholar] [CrossRef]
- Popov, A.; Brazhe, A.; Denisov, P.; Sutyagina, O.; Li, L.; Lazareva, N.; Verkhratsky, A.; Semyanov, A. Astrocyte dystrophy in ageing brain parallels impaired synaptic plasticity. Aging Cell 2021, 20, e13334. [Google Scholar] [CrossRef]
- Duda, P.; Wójcicka, O.; Wiśniewski, J.R.; Rakus, D. Global quantitative TPA-based proteomics of mouse brain structures reveals significant alterations in expression of proteins involved in neuronal plasticity during aging. Aging 2018, 10, 1682–1697. [Google Scholar] [CrossRef]
- Fulop, T.; Witkowski, J.M.; Olivieri, F.; Larbi, A. The integration of inflammaging in age-related diseases. Semin. Immunol. 2018, 40, 17–35. [Google Scholar] [CrossRef]
- Fulop, T.; Larbi, A.; Dupuis, G.; Le Page, A.; Frost, E.H.; Cohen, A.A.; Franceschi, C. Immunosenescence and inflamm-aging as two sides of the same coin: Friends or foes? Front. Immunol. 2018, 8, 1960. [Google Scholar] [CrossRef]
- Fülöp, T.; Dupuis, G.; Witkowski, J.M.; Larbi, A. The Role of Immunosenescence in the Development of Age-Related Diseases. Rev. Investig. Clin. 2016, 68, 84–91. [Google Scholar]
- Confavreux, C.; Vukusic, S. Age at disability milestones in multiple sclerosis. Brain 2006, 129, 595–605. [Google Scholar] [CrossRef]
- Guillemin, F.; Baumann, C.; Epstein, J.; Kerschen, P.; Garot, T.; Mathey, G.; Debouverie, M.; on behalf of the LORSEP Group. Older Age at Multiple Sclerosis Onset Is an Independent Factor of Poor Prognosis: A Population-Based Cohort Study. Neuroepidemiology 2017, 48, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Daltrozzo, T.; Hapfelmeier, A.; Donnachie, E.; Schneider, A.; Hemmer, B. A Systematic Assessment of Prevalence, Incidence and Regional Distribution of Multiple Sclerosis in Bavaria From 2006 to 2015. Front. Neurol. 2018, 9, 871. [Google Scholar] [CrossRef]
- Marrie, R.A.; Yu, N.; Blanchard, J.; Leung, S.; Elliott, L. The rising prevalence and changing age distribution of multiple sclerosis in Manitoba. Neurology 2010, 74, 465–471. [Google Scholar] [CrossRef]
- Marrie, R.A.; Miller, A.; Sormani, M.P.; Thompson, A.; Waubant, E.; Trojano, M.; O’Connor, P.; Reingold, S.; Cohen, J.A.; For the attendees of the International Workshop on Comorbidity in Multiple Sclerosis. The challenge of comorbidity in clinical trials for multiple sclerosis. Neurology 2016, 86, 1437–1445. [Google Scholar] [CrossRef] [PubMed]
- Marrie, R.A. Comorbidity in multiple sclerosis: Implications for patient care. Nat. Rev. Neurol. 2017, 13, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Marrie, R.A.; Rudick, R.; Horwitz, R.; Cutter, G.; Tyry, T.; Campagnolo, D.; Vollmer, T. Vascular comorbidity is associated with more rapid disability progression in multiple sclerosis. Neurology 2010, 74, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.C.; Damian, A.; Conway, D.; Mowry, E.M. Vascular comorbidity is associated with lower brain volumes and lower neuroperformance in a large multiple sclerosis cohort. Mult. Scler. J. 2021, 27, 1914–1923. [Google Scholar] [CrossRef]
- Walton, C.; King, R.; Rechtman, L.; Kaye, W.; Leray, E.; Marrie, R.A.; Robertson, N.; La Rocca, N.; Uitdehaag, B.; Van Der Mei, I.; et al. Rising prevalence of multiple sclerosis worldwide: Insights from the Atlas of MS, third edition. Mult. Scler. J. 2020, 26, 1816–1821. [Google Scholar] [CrossRef]
- Sumelahti, M.-L.; Holmberg, M.H.A.; Murtonen, A.; Huhtala, H.; Elovaara, I. Increasing Incidence in Relapsing-Remitting MS and High Rates among Young Women in Finland: A Thirty-Year Follow-Up. Mult. Scler. Int. 2014, 2014, 186950. [Google Scholar] [CrossRef]
- Kappos, L.; Bar-Or, A.; Cree, B.A.C.; Fox, R.J.; Giovannoni, G.; Gold, R.; Vermersch, P.; Arnold, D.L.; Arnould, S.; Scherz, T.; et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): A double-blind, randomised, phase 3 study. Lancet 2018, 391, 1263–1273. [Google Scholar] [CrossRef]
- Medicine, N.L.O. Nonrelapsing Secondary Progressive Multiple Sclerosis (NRSPMS) Study of Bruton’s Tyrosine Kinase (BTK) Inhibitor Tolebrutinib (SAR442168) (HERCULES). 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04411641 (accessed on 1 September 2022).
- Lynch, S.; Baker, S.; Nashatizadeh, M.; Thuringer, A.; Thelen, J.; Bruce, J. Disability measurement in Multiple Sclerosis patients 55 years and older: What is the Expanded Disability Status Scale really telling clinicians? Mult. Scler. Relat. Disord. 2021, 49, 102724. [Google Scholar] [CrossRef]
- Manouchehrinia, A.; Westerlind, H.; Kingwell, E.; Zhu, F.; Carruthers, R.; Ramanujam, R.; Ban, M.; Glaser, A.; Sawcer, S.; Tremlett, H.; et al. Age Related Multiple Sclerosis Severity Score: Disability ranked by age. Mult. Scler. J. 2017, 23, 1938–1946. [Google Scholar] [CrossRef]
- Weideman, A.M.; Barbour, C.; Tapia-Maltos, M.A.; Tran, T.; Jackson, K.; Kosa, P.; Komori, M.; Wichman, A.; Johnson, K.; Greenwood, M.; et al. New Multiple Sclerosis Disease Severity Scale Predicts Future Accumulation of Disability. Front. Neurol. 2017, 8, 598. [Google Scholar] [CrossRef]
- Weideman, A.M.; Maltos, M.A.T.; Johnson, K.; Greenwood, M.; Bielekova, B. Meta-analysis of the Age-Dependent Efficacy of Multiple Sclerosis Treatments. Front. Neurol. 2017, 8, 577. [Google Scholar] [CrossRef]
- Schweitzer, F.; Laurent, S.; Fink, G.R.; Barnett, M.H.; Reddel, S.; Hartung, H.-P.; Warnke, C. Age and the risks of high-efficacy disease modifying drugs in multiple sclerosis. Curr. Opin. Neurol. 2019, 32, 305–312. [Google Scholar] [CrossRef]
- Knox, K.B.; Saini, A.; Levin, M.C. The dilemma of when to stop disease-modifying therapy in multiple sclerosis: A narrative review and Canadian regional reimbursement policies. Int. J. MS Care 2020, 22, 75–84. [Google Scholar] [CrossRef]
- Hua, L.H.; Harris, H.; Conway, D.; Thompson, N.R. Changes in patient-reported outcomes between continuers and discontinuers of disease modifying therapy in patients with multiple sclerosis over age 60. Mult. Scler. Relat. Disord. 2019, 30, 252–256. [Google Scholar] [CrossRef]
- Zhang, Y.; Caldito, N.G.; Shirani, A.; Salter, A.; Cutter, G.; Culpepper, I.W.; Wallin, M.; Kosa, P.; Bielekova, B.; Lublin, F.; et al. Aging and efficacy of disease-modifying therapies in multiple sclerosis: A meta-analysis of clinical trials. Ther. Adv. Neurol. Disord. 2020, 13, 1756286420969016. [Google Scholar] [CrossRef]
- Schwehr, N.A.; The BeAMS Study group; Kuntz, K.M.; Enns, E.A.; Shippee, N.D.; Kingwell, E.; Tremlett, H.; Carpenter, A.F.; Butler, M. Informing Medication Discontinuation Decisions among Older Adults with Relapsing-Onset Multiple Sclerosis. Drugs Aging 2020, 37, 225–235. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kesidou, E.; Theotokis, P.; Damianidou, O.; Boziki, M.; Konstantinidou, N.; Taloumtzis, C.; Sintila, S.-A.; Grigoriadis, P.; Evangelopoulos, M.E.; Bakirtzis, C.; et al. CNS Ageing in Health and Neurodegenerative Disorders. J. Clin. Med. 2023, 12, 2255. https://doi.org/10.3390/jcm12062255
Kesidou E, Theotokis P, Damianidou O, Boziki M, Konstantinidou N, Taloumtzis C, Sintila S-A, Grigoriadis P, Evangelopoulos ME, Bakirtzis C, et al. CNS Ageing in Health and Neurodegenerative Disorders. Journal of Clinical Medicine. 2023; 12(6):2255. https://doi.org/10.3390/jcm12062255
Chicago/Turabian StyleKesidou, Evangelia, Paschalis Theotokis, Olympia Damianidou, Marina Boziki, Natalia Konstantinidou, Charilaos Taloumtzis, Styliani-Aggeliki Sintila, Panagiotis Grigoriadis, Maria Eleptheria Evangelopoulos, Christos Bakirtzis, and et al. 2023. "CNS Ageing in Health and Neurodegenerative Disorders" Journal of Clinical Medicine 12, no. 6: 2255. https://doi.org/10.3390/jcm12062255
APA StyleKesidou, E., Theotokis, P., Damianidou, O., Boziki, M., Konstantinidou, N., Taloumtzis, C., Sintila, S.-A., Grigoriadis, P., Evangelopoulos, M. E., Bakirtzis, C., & Simeonidou, C. (2023). CNS Ageing in Health and Neurodegenerative Disorders. Journal of Clinical Medicine, 12(6), 2255. https://doi.org/10.3390/jcm12062255