
Subependymal Giant Cell Astrocytomas in Tuberous Sclerosis Complex—Current Views on Their Pathogenesis and Management
 ,
, 
Abstract
1. Introduction
2. Epidemiology of SEGA
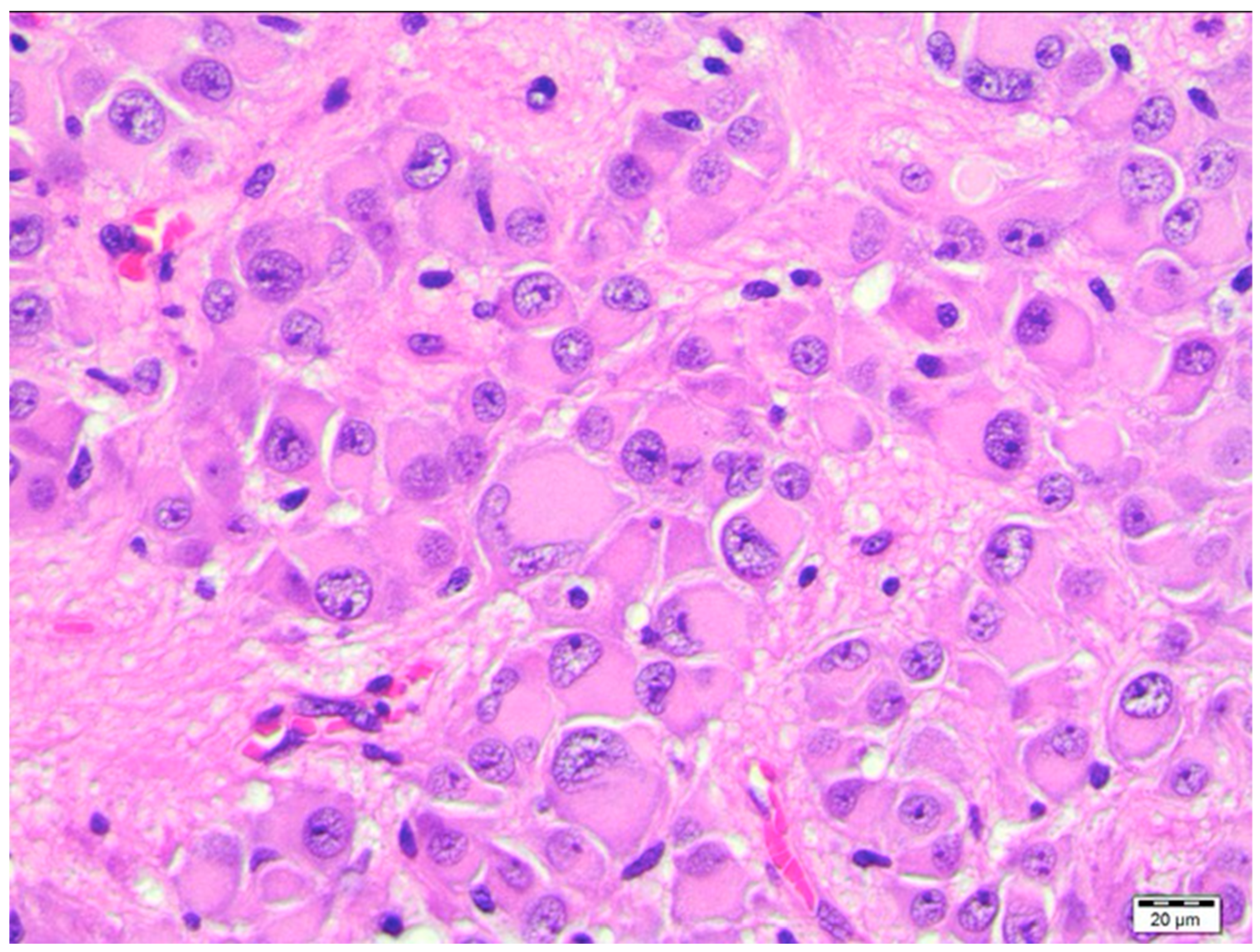
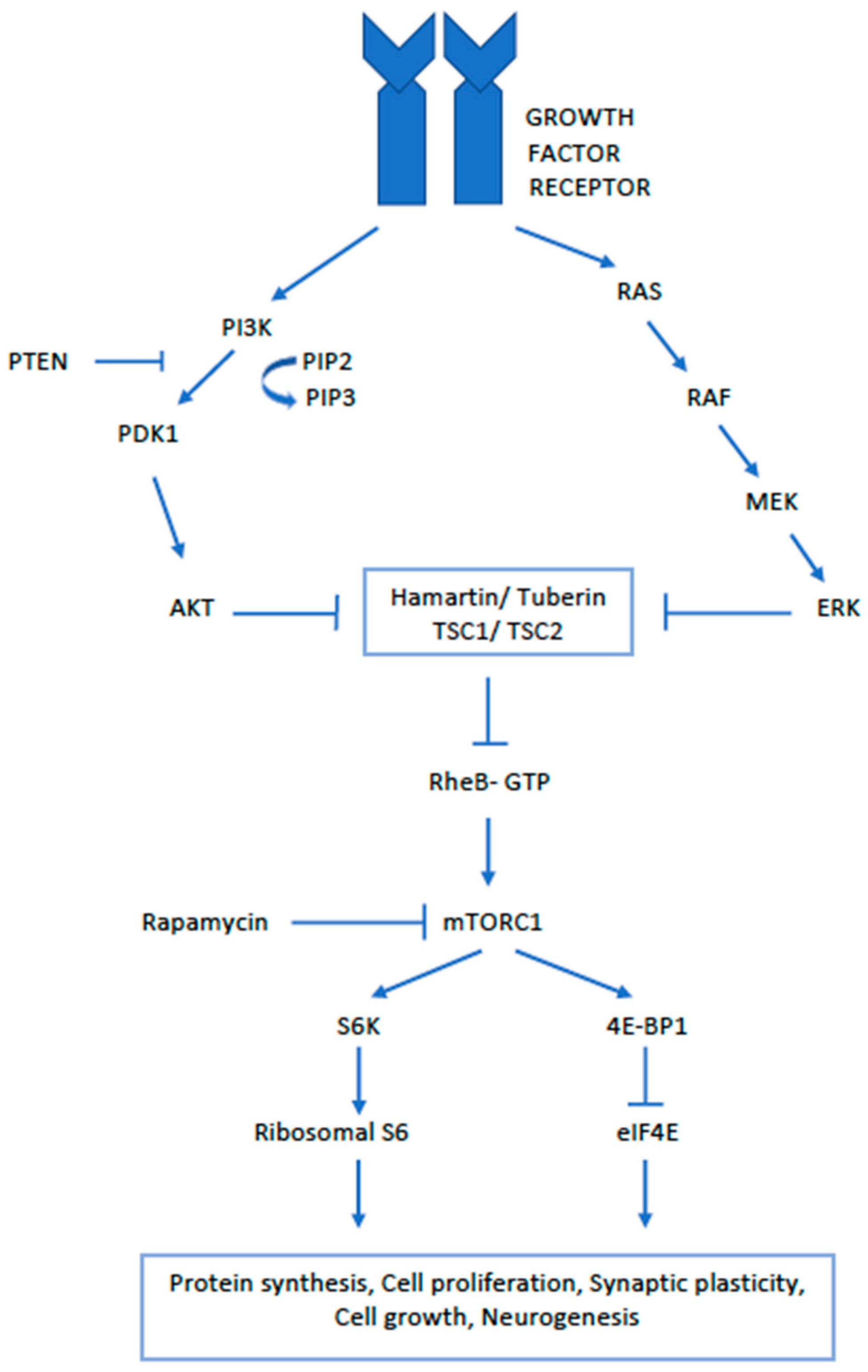
3. Pathology and Pathogenesis of SEGA
4. Non-TSC Associated SEGA
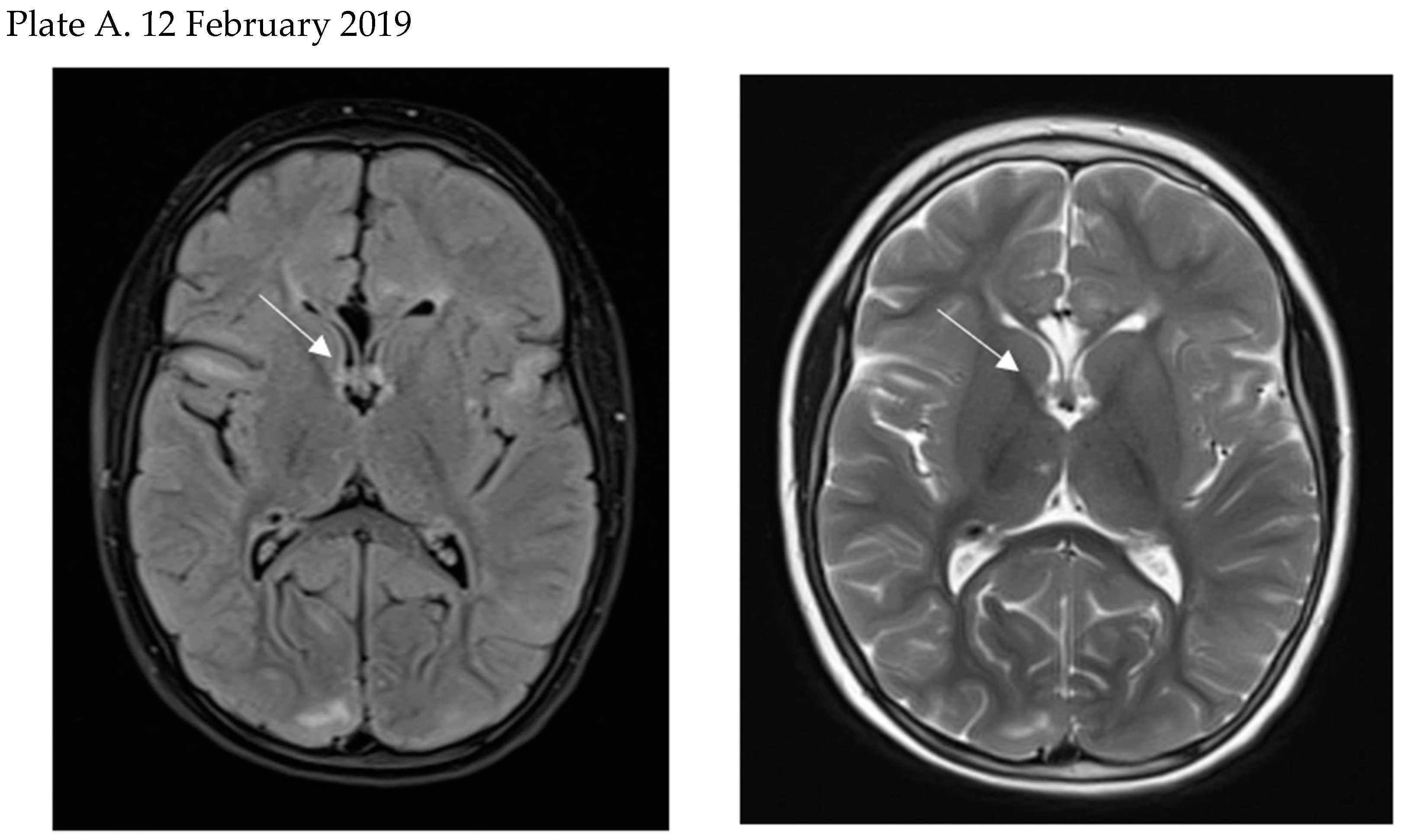
5. Clinical Presentation of SEGA and Patients’ Surveillance
6. Surgical and Pharmacological Treatment of SEGAs
7. Conclusions
- Today, thanks to recommended MRI follow-up, SEGA is diagnosed before the onset of neurological symptoms in the majority of TSC patients enabling early treatment. However, the optimal timing of SEGA treatment is not defined and requires further studies.
- Surgery and medical treatment with mTORi are used to treat SEGA and there is no consensus on the priority of one treatment over another. Currently, everolimus is approved for the treatment of unresectable tumors, but increasing evidence shows that it can be successfully used as a neoadjuvant therapy in combination with surgery.
- The treatment decision should be taken during the multidisciplinary discussion concerning the individual needs and challenges of the patient.
Author Contributions
Funding
Conflicts of Interest
References
- Curatolo, P.; Bombardieri, R.; Jozwiak, S. Tuberous sclerosis. Lancet 2008, 372, 657–668. [Google Scholar] [CrossRef]
- Osborne, J.P.; Fryer, A.; Webb, D. Epidemiology of tuberous sclerosis. Ann. N. Y. Acad. Sci. 1991, 615, 125–127. [Google Scholar] [CrossRef] [PubMed]
- Curatolo, P.; Moavero, R. mTOR inhibitors in tuberous sclerosis complex. Curr. Neuropharmacol. 2012, 10, 404–415. [Google Scholar] [CrossRef] [PubMed]
- Dabora, S.; Jozwiak, S.; Franz, D.; Roberts, P.S.; Nieto, A.; Chung, J.; Choy, Y.S.; Reeve, M.P.; Thiele, E.; Egelhoff, J.C.; et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am. J. Hum. Genet. 2001, 68, 64–80. [Google Scholar] [CrossRef]
- Specchio, N.; Di Micco, V.; Trivisano, M.; Ferretti, A.; Curatolo, P. The epilepsy-autism spectrum disorder phenotype in the era of molecular genetics and precision therapy. Epilepsia 2022, 63, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Bombardieri, R.; Pinci, M.; Moavero, R.; Cerminara, C.; Curatolo, P. Early control of seizures improves long-term outcome in children with tuberous sclerosis complex. Eur. J. Paediatr. Neurol. 2010, 14, 146–149. [Google Scholar] [CrossRef]
- Jóźwiak, S.; Mandera, M.; Młynarski, W. Natural History and Current Treatment Options for Subependymal Giant Cell Astrocytoma in Tuberous Sclerosis Complex. Semin. Pediatr. Neurol. 2015, 22, 274–281. [Google Scholar] [CrossRef]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Curatolo, P. Mechanistic target of rapamycin (mTOR) in tuberous sclerosis complex-associated epilepsy. Pediatr. Neurol. 2015, 52, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, S.; Mahalingam, R.; Melchiorre, J.; Harowitz, J.; Devinsky, O. Mortality in tuberous sclerosis complex. Epilepsy Behav. 2021, 121, 108032. [Google Scholar] [CrossRef]
- Shepherd, C.W.; Gomez, M.R.; Lie, J.T.; Crowson, C.S. Causes of death in patients with tuberous sclerosis. Mayo Clin. Proc. 1991, 66, 792–796. [Google Scholar] [CrossRef] [PubMed]
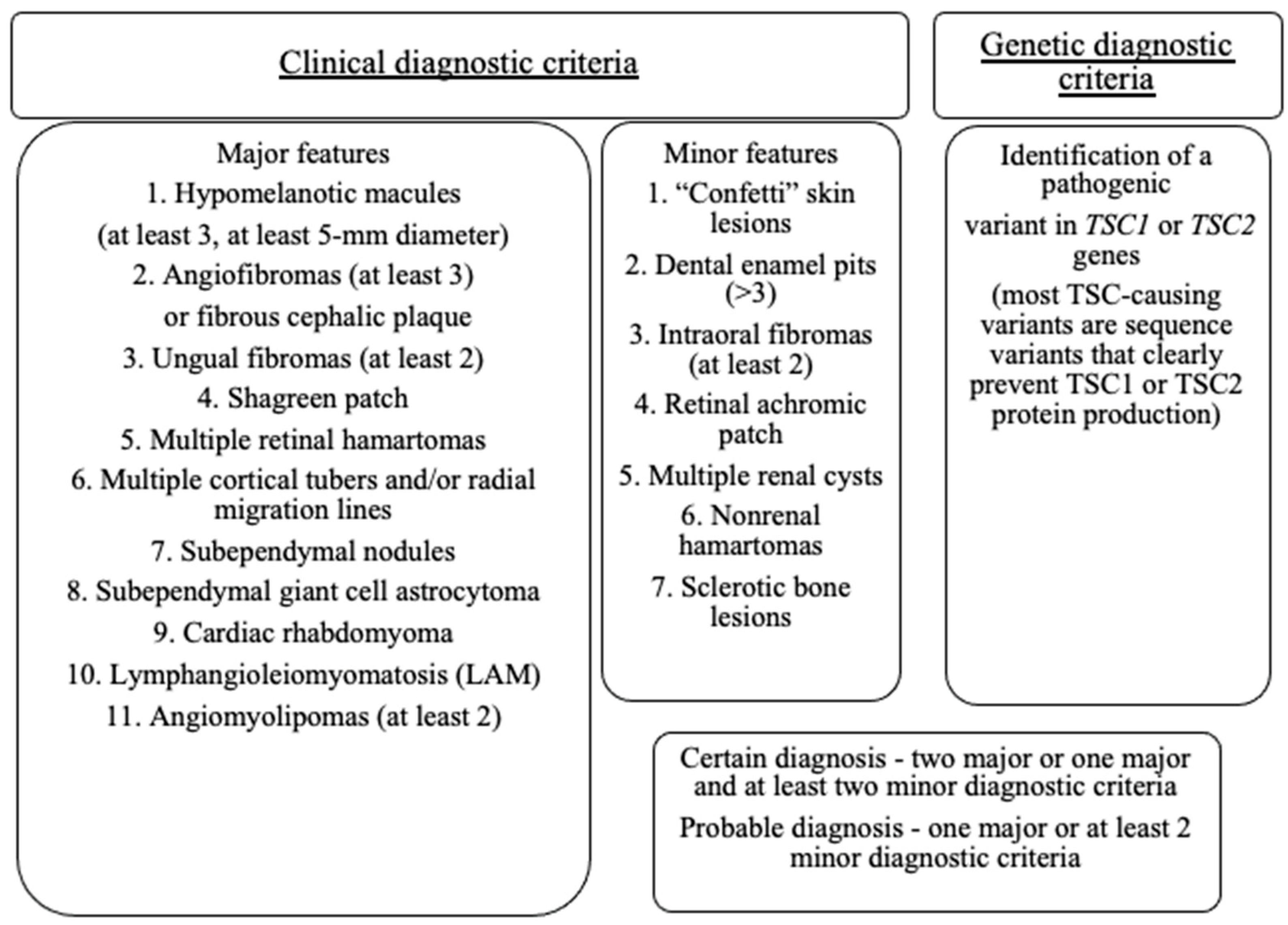
- Northrup, H.; Aronow, M.E.; Bebin, E.M.; Bissler, J.; Darling, T.N.; de Vries, P.J.; Frost, M.D.; Fuchs, Z.; Gosnell, E.S.; Gupta, N.; et al. Updated International Tuberous Sclerosis Complex Diagnostic Criteria and Surveillance and Management Recommendations. Pediatr. Neurol. 2021, 123, 50–66. [Google Scholar] [CrossRef]
- Moavero, R.; Coniglio, A.; Garaci, F.; Curatolo, P. Is mTOR inhibition a systemic treatment for tuberous sclerosis? Ital. J. Pediatr. 2013, 39, 57. [Google Scholar] [CrossRef]
- Giordano, F.; Moscheo, C.; Lenge, M.; Biagiotti, R.; Mari, R.; Sardi, I.; Buccoliero, L.; Aronica, E.; Guerrini, R.; Genitori, L. Neurosurgical treatment of subependymal giant cell astrocytomas in tuberous sclerosis complex: A series of 44 surgical procedures in 31 patients. Child’s Nerv. Syst. 2020, 36, 951–960. [Google Scholar] [CrossRef]
- Grajkowska, W.; Kotulska, K.; Jurkiewicz, E.; Matyja, E. Brain lesions in tuberous sclerosis complex. Folia Neuropathol. 2010, 48, 139–149. [Google Scholar] [PubMed]
- Jansen, A.C.; Belousova, E.; Benedik, M.P.; Carter, T.; Cottin, V.; Curatolo, P.; D’Amato, L.; d’Augères, G.B.; de Vries, P.J.; Ferreira, J.C.; et al. Clinical characteristics of subependymal giant-cell astrocytoma in tuberous sclerosis complex. Front. Neurol. 2019, 10, 705. [Google Scholar] [CrossRef] [PubMed]
- Kingswood, J.C.; D’Augères, G.B.; Belousova, E.; Ferreira, J.C.; Carter, T.; Castellana, R.; Cottin, V.; Curatolo, P.; Dahlin, M.; de Vries, P.J.; et al. TuberOus SClerosis registry to increase disease Awareness (TOSCA)—Baseline data on 2093 patients. Orphanet J. Rare Dis. 2017, 12, 2. [Google Scholar] [CrossRef] [PubMed]
- Adriaensen, M.E.A.P.M.; Schaefer-Prokop, C.M.; Stijnen, T.; Duyndam, D.A.C.; Zonnenberg, B.A.; Prokop, M. Prevalence of subependymal giant cell tumors in patients with tuberous sclerosis and a review of the literature. Eur. J. Neurol. 2009, 16, 691–696. [Google Scholar] [CrossRef]
- Rentz, A.M.; Skalicky, A.M.; Liu, Z.; Wheless, J.W.; Dunn, D.W.; Frost, M.D.; Nakagawa, J.; Magestro, M.; Prestifilippo, J. Tuberous sclerosis complex: A survey on health care resource use and health burden. Pediatr. Neurol. 2015, 52, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Kotulska, K.; Borkowska, J.; Roszkowski, M.; Mandera, M.; Daszkiewicz, P.; Drabik, K.; Jurkiewicz, E.; Larysz-Brysz, M.; Nowak, K.; Grajkowska, W.; et al. Surgical treatment of subependymal giant cell astrocytoma in tuberous sclerosis complex patients. Pediatr. Neurol. 2014, 50, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Karagianni, A.; Karydakis, P.; Giakoumettis, D.; Nikas, I.; Sfakianos, G.; Themistocleous, M. Fetal subependymal giant cell astrocytoma: A case report and review of the literature. Surg. Neurol. Int. 2020, 11, 1–5. [Google Scholar] [CrossRef]
- Gelot, A.B.; Represa, A. Progression of fetal brain lesions in tuberous sclerosis complex. Front. Neurosci. 2020, 14, 899. [Google Scholar] [CrossRef]
- Chan, D.L.; Kennedy, S.E.; Sarkozy, V.E.; Chung, C.W.T.; Flanagan, D.; Mowat, D.; Farrar, M.A.; Lawson, J.A. Congenital subpendymal giant cell astrocytoma in children with tuberous sclerosis complex: Growth patterns and neurological outcome. Pediatr. Res. 2021, 89, 1447–1451. [Google Scholar] [CrossRef]
- Kotulska, K.; Borkowska, J.; Mandera, M. Congenital subependymal giant cell astrocytomas in patients with tuberous sclerosis complex. Child’s Nerv. Syst. 2014, 30, 2037–2042. [Google Scholar] [CrossRef] [PubMed]
- Roszkowski, M.; Drabik, K.; Barszcz, S.; Jozwiak, S. Surgical treatment of intraventricular tumors associated with tuberous sclerosis. Child’s Nerv. Syst. 1995, 11, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, S.; Takaki, T.; Hikita, T.; Nishio, S. Subependymal giant-cell astrocytoma associated with tuberous sclerosis. Do subependymal nodules grow? Child’s Nerv. Syst. 1989, 5, 43–44. [Google Scholar] [CrossRef]
- Nabbout, R.; Santos, M.; Rolland, Y.; Delalande, O.; Dulac, O.; Chiron, C. Early diagnosis of subependymal giant cell astrocytoma in children with tuberous sclerosis. J. Neurol. Neurosurg. Psychiatry 1999, 66, 370–375. [Google Scholar] [CrossRef]
- Siedlecka, M.; Szlufik, S.; Grajkowska, W.; Roszkowski, M.; Jóźwiak, J. Erk activation as a possible mechanism of transformation of subependymal nodule into subependymal giant cell astrocytoma. Folia Neuropathol. 2015, 53, 8–14. [Google Scholar] [CrossRef]
- Bongaarts, A.; Giannikou, K.; Reinten, R.J.; Anink, J.J.; Mills, J.D.; Jansen, F.E.; Spliet, W.G.M.; den Dunnen, W.F.A.; Coras, R.; Blümcke, I.; et al. Subependymal giant cell astrocytomas in Tuberous sclerosis complex have consistent TSC1/TSC2 biallelic inactivation, and no BRAF mutations. Oncotarget 2017, 8, 95516–95529. [Google Scholar] [CrossRef] [PubMed]
- Grajkowska, W.; Kotulska, K.; Jurkiewicz, E.; Roszkowski, M.; Daszkiewicz, P.; Jóźwiak, S.; Matyja, E. Subependymal giant cell astrocytomas with atypical histological features mimicking malignant gliomas. Folia Neuropathol. 2011, 49, 39–46. [Google Scholar]
- Curatolo, P.; Specchio, N.; Aronica, E. Advances in the genetics and neuropathology of tuberous sclerosis complex: Edging closer to targeted therapy. Lancet Neurol 2022, 21, 843–856. [Google Scholar] [CrossRef]
- Jozwiak, S.; Nabbout, R.; Curatolo, P. Management of subependymal giant cell astrocytoma associated with tuberous sclerosis complex (TSC): Clinical recommendations. Eur. J. Paediatr. Neurol. 2013, 17, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Roth, J.; Roach, E.S.; Bartels, U.; Jóźwiak, S.; Koenig, M.K.; Weiner, H.L.; Franz, D.N.; Wang, H.Z. Subependymal giant cell astrocytoma: Diagnosis, screening, and treatment. Recommendations from the International tuberous sclerosis complex consensus conference. Pediatr. Neurol. 2013, 49, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Bongaarts, A.; van Scheppingen, J.; Korotkov, A.; Mijnsbergen, C.; Anink, J.J.; Jansen, F.E. The coding and non-coding transcriptional landscape of subependymal giant cell astrocytomas. Brain 2020, 143, 131–149. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowska, J.; Wigowska-Sowinska, J.; Napierala, D.; Slomski, R.; Kwiatkowski, D.J. Mosaicism in tuberous sclerosis as a potential cause of the failure of molecular diagnosis. N. Engl. J. Med. 1999, 340, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, T.; Wakisaka, A.; Daido, S.; Takao, S.; Tamiya, T.; Date, I.; Koizumi, S.; Niida, Y. A case of solitary subependymal giant cell astrocytoma: Two somatic hits of TSC2 in the tumor, without evidence of somatic mosaicism. J. Mol. Diagn. 2005, 7, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Giannikou, K.; Zhu, Z.; Kim, J.; Winden, K.D.; Tyburczy, M.E.; Marron, D.; Parker, J.S.; Hebert, Z.; Bongaarts, A.; Taing, L.; et al. Subependymal giant cell astrocytomas are characterized by mTORC1 hyperactivation, a very low somatic mutation rate, and a unique gene expression profile. Mod. Pathol. 2021, 34, 264–279. [Google Scholar] [CrossRef]
- Beaumont, T.L.; Godzik, J.; Dahiya, S.; Smyth, M.D. Subependymal giant cell astrocytoma in the absence of tuberous sclerosis complex: Case report. J. Neurosurg. Pediatr. 2015, 16, 134–137. [Google Scholar] [CrossRef] [PubMed]
- Jansen, A.C.; Belousova, E.; Benedik, M.P.; Carter, T.; Cottin, V.; Curatolo, P.; D’Amato, L.; d’Augères, G.B.; de Vries, P.J.; Ferreira, J.C.; et al. Newly Diagnosed and Growing Subependymal Giant Cell Astrocytoma in Adults With Tuberous Sclerosis Complex: Results From the International TOSCA Study. Front. Neurol. 2019, 10, 821. [Google Scholar] [CrossRef]
- Stawiski, K.; Trelińska, J.; Baranska, D.; Dachowska, I.; Kotulska, K.; Jóźwiak, S. Fendler, W.; Młynarski, W. What are the true volumes of SEGA tumors? Reliability of planimetric and popular semi-automated image segmentation methods. Magn. Reson. Mater. Phys. Biol. Med. 2017, 30, 397–405. [Google Scholar] [CrossRef] [PubMed]
- de Ribaupierre, S.; Dorfmuller, G.; Bulteau, C.; Fohlen, M.; Pinard, J.M.; Chiron, C.; Delalande, O. Subependymal giant-cell astrocytomas in pediatric tuberous sclerosis disease: When should we operate? Neurosurgery 2007, 60, 83–89. [Google Scholar] [CrossRef]
- Danassegarane, G.; Tinois, J.; Sahler, Y.; Aouaissia, S.; Riffaud, L. Subependymal giant-cell astrocytoma: A surgical review in the modern era of mTOR inhibitors. Neurochirurgie 2022, 68, 627–636. [Google Scholar] [CrossRef]
- Yamamoto, K.; Yamada, K.; Nakahara, T.; Ishihara, A.; Takaki, S.; Kochi, M.; Ushio, Y. Rapid regrowth of solitary subependymal giant cell astrocytoma—Case report. Neurol. Med. Chir. 2002, 42, 224–227. [Google Scholar] [CrossRef] [PubMed]
- Frassanito, P.; Noya, C.; Tamburrini, G. Current trends in the management of subependymal giant cell astrocytomas in tuberous sclerosis. Child’s Nerv. Syst. 2020, 36, 2527–2536. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, H.; Takimoto, H.; Shimada, N.; Hirata, M.; Ohnishi, T.; Hayakawa, T. Glioblastoma following radiotherapy in a patient with tuberous sclerosis. Neurol. Med. Chir. 1998, 38, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Beaumont, T.L.; Limbrick, D.D.; Smyth, M.D. Advances in the management of subependymal giant cell astrocytoma. Child’s Nerv. Syst. 2012, 28, 963–968. [Google Scholar] [CrossRef] [PubMed]
- Krueger, D.A.; Care, M.M.; Holland, K.; Agricola, K.; Tudor, C.; Mangeshkar, P.; Wilson, K.A.; Byars, A.; Sahmoud, T.; Franz, D.N. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N. Engl. J. Med. 2010, 363, 1801–1811. [Google Scholar] [CrossRef] [PubMed]
- Kotulska, K.; Chmielewski, D.; Borkowska, J.; Jurkiewicz, E.; Kuczyński, D.; Kmieć, T.; Łojszczyk, B.; Dunin-Wąsowicz, D.; Jozwiak, S. Long-term effect of everolimus on epilepsy and growth in children under 3 years of age treated for subependymal giant cell astrocytoma associated with tuberous sclerosis complex. Eur. J. Paediatr. Neurol. 2013, 17, 479–485. [Google Scholar] [CrossRef]
- Franz, D.N.; Belousova, E.; Sparagana, S. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): A multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2013, 381, 125–132. [Google Scholar] [CrossRef]
- Franz, D.N.; Belousova, E.; Sparagana, S.; Bebin, E.M.; Frost, M.D.; Kuperman, R.; Witt, O.; Kohrman, M.H.; Flamini, J.R.; Wu, J.Y.; et al. Long-term use of everolimus in patients with tuberous sclerosis complex: Final results from the EXIST-1 study. PLoS ONE 2016, 11, e0158476. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Fakhari, D.; Franz, D. Pharmacological treatment strategies for subependymal giant cell astrocytoma (SEGA). Expert Opin. Pharmacother. 2020, 21, 1329–1336. [Google Scholar] [CrossRef]
- Jiang, T.; Du, J.; Liu, R.; Wang, J.; Li, C. Presurgical administration of mTOR inhibitors in patients with large subependymal giant cell astrocytoma associated with tuberous sclerosis complex. World Neurosurg. 2017, 107, 1053.e1–1053.e6. [Google Scholar] [CrossRef]
- Weidman, D.R.; Palasamudram, S.; Zak, M.; Whitney, R.; McCoy, R.; Bouffet, E.; Taylor, M.; Schroff, M.; Bartels, U. The effect of mTOR inhibition on obstructive hydrocephalus in patients with tuberous sclerosis compolex related subependymal giant cell astrocytoma. J. Neurooncol. 2020, 147, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Moavero, R.; Carai, A.; Mastronuzzi, A.; Marciano, S.; Graziola, F.; Vigevano, F.; Curatolo, P. Everolimus alleviates obstructive hydrocephalus due to subependymal giant cell astrocytomas. Pediatr. Neurol. 2017, 68, 59–63. [Google Scholar] [CrossRef]
- Perek-Polnik, M.; Jozwiak, S.; Jurkiewicz, E.; Perek, D.; Kotulska, K. Effective everolimus treatment of inoperable, life-threatening subependymal giant cell astrocytoma and intractable epilepsy in a patient with tuberous sclerosis complex. Eur. J. Paediatr. Neurol. 2012, 16, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Franz, D.N.; Agricola, K.D.; Tudor, C.A.; Krueger, D.A. Everolimus for tumor recurrence after surgical resection for subependymal giant cell astrocytoma associated with tuberous sclerosis complex. J. Child Neurol. 2013, 28, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Sadowski, K.; Kotulska, K.; Schwartz, R.A.; Jozwiak, S. Systemic effects of treatment with mTOR inhibitors in tuberous sclerosis complex: A comprehensive review. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 586–594. [Google Scholar] [CrossRef]
- Yui, K.; Imataka, G.; Sasaki, H.; Kawasaki, Y.; Yoshihara, S. Contribution of Transferrin and Ceruloplasmin Neurotransmission and Oxidant/Antioxidant Status to the Effects of Everolimus: A Case Series. Cureus 2020, 12, e6920. [Google Scholar] [CrossRef]
- Trelinska, J.; Dachowska, I.; Kotulska, K.; Baranska, D.; Fendler, W.; Jozwiak, S.; Mlynarski, W. Factors affecting response to everolimus therapy for subependymal giant cell astrocytomas associated with tuberous sclerosis. Pediatr. Blood Cancer. 2015, 62, 616–621. [Google Scholar] [CrossRef]
- Sadowski, K.; Kotulska, K.; Jozwiak, S. Management of side effects of mTOR inhibitors in tuberous sclerosis patients. Pharmacol. Rep. 2016, 68, 536–542. [Google Scholar] [CrossRef]
- Vergès, B.; Cariou, B. mTOR inhibitors and diabetes. Diabetes Res. Clin. Pract. 2015, 110, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Bobeff, K.; Krajewska, K.; Baranska, D.; Kotulska, K.; Jozwiak, S.; Mlynarski, W.; Trelinska, J. Maintenance Therapy With Everolimus for Subependymal Giant Cell Astrocytoma in Patients With Tuberous Sclerosis—Final Results From the EMINENTS Study. Front. Neurol. 2021, 12, 581102. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Side Effects of Surgical Treatment | Side Effects of mTOR Inhibitors Treatment |
|---|---|
|
|
| Indications for Surgery | Indications for mTOR Inhibitors |
|---|---|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, C.; Zabielska, B.; Jiao, F.; Mei, D.; Wang, X.; Kotulska, K.; Jozwiak, S. Subependymal Giant Cell Astrocytomas in Tuberous Sclerosis Complex—Current Views on Their Pathogenesis and Management. J. Clin. Med. 2023, 12, 956. https://doi.org/10.3390/jcm12030956
Gao C, Zabielska B, Jiao F, Mei D, Wang X, Kotulska K, Jozwiak S. Subependymal Giant Cell Astrocytomas in Tuberous Sclerosis Complex—Current Views on Their Pathogenesis and Management. Journal of Clinical Medicine. 2023; 12(3):956. https://doi.org/10.3390/jcm12030956
Chicago/Turabian StyleGao, Chao, Bernadeta Zabielska, Fuyong Jiao, Daoqi Mei, Xiaona Wang, Katarzyna Kotulska, and Sergiusz Jozwiak. 2023. "Subependymal Giant Cell Astrocytomas in Tuberous Sclerosis Complex—Current Views on Their Pathogenesis and Management" Journal of Clinical Medicine 12, no. 3: 956. https://doi.org/10.3390/jcm12030956
APA StyleGao, C., Zabielska, B., Jiao, F., Mei, D., Wang, X., Kotulska, K., & Jozwiak, S. (2023). Subependymal Giant Cell Astrocytomas in Tuberous Sclerosis Complex—Current Views on Their Pathogenesis and Management. Journal of Clinical Medicine, 12(3), 956. https://doi.org/10.3390/jcm12030956