
Pharmacogenomic Considerations for Anticoagulant Prescription in Patients with Hereditary Haemorrhagic Telangiectasia

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Gene Identification
2.2. Gene Location and Variant Identification
2.3. Data Analysis
3. Results
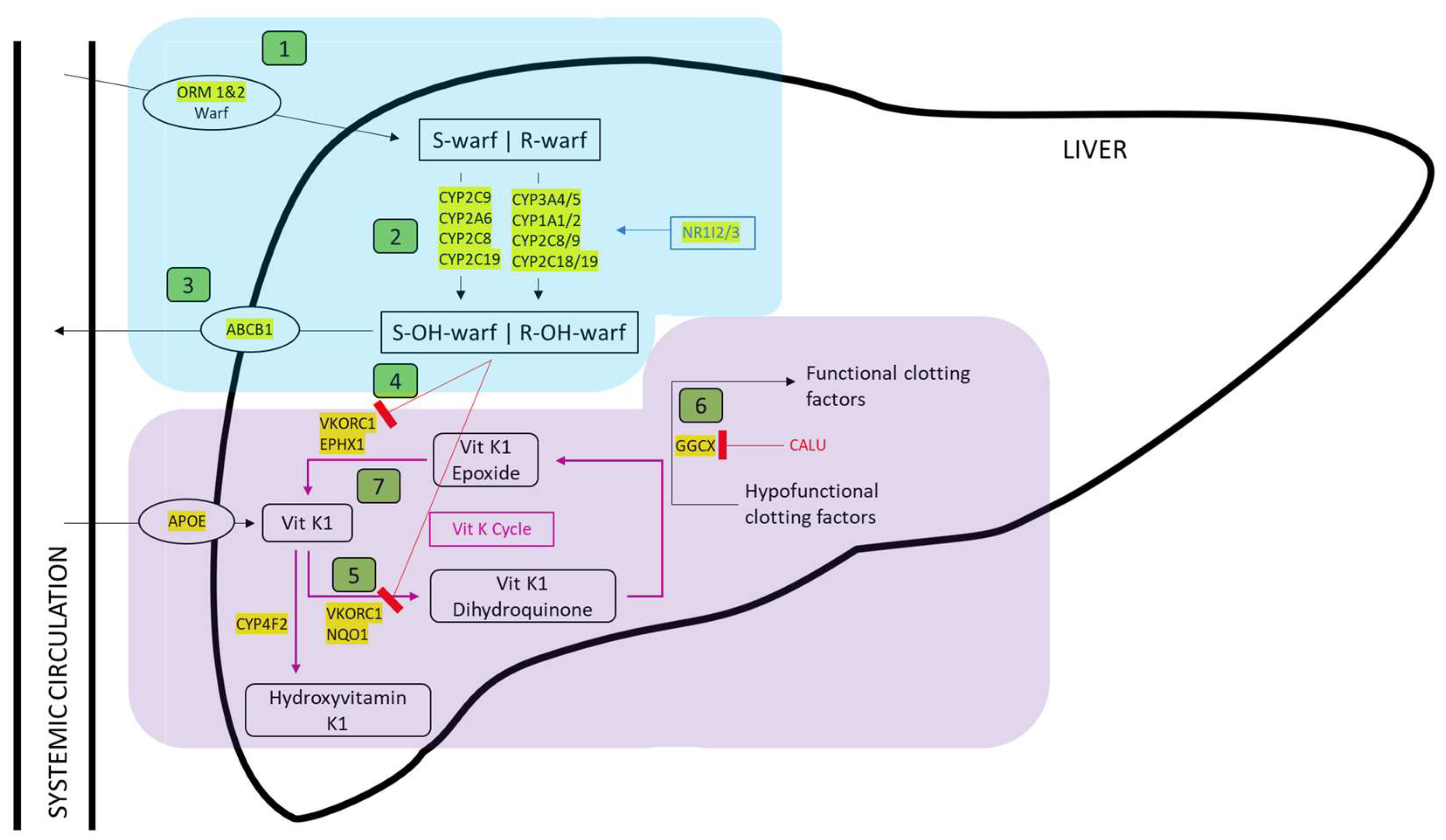
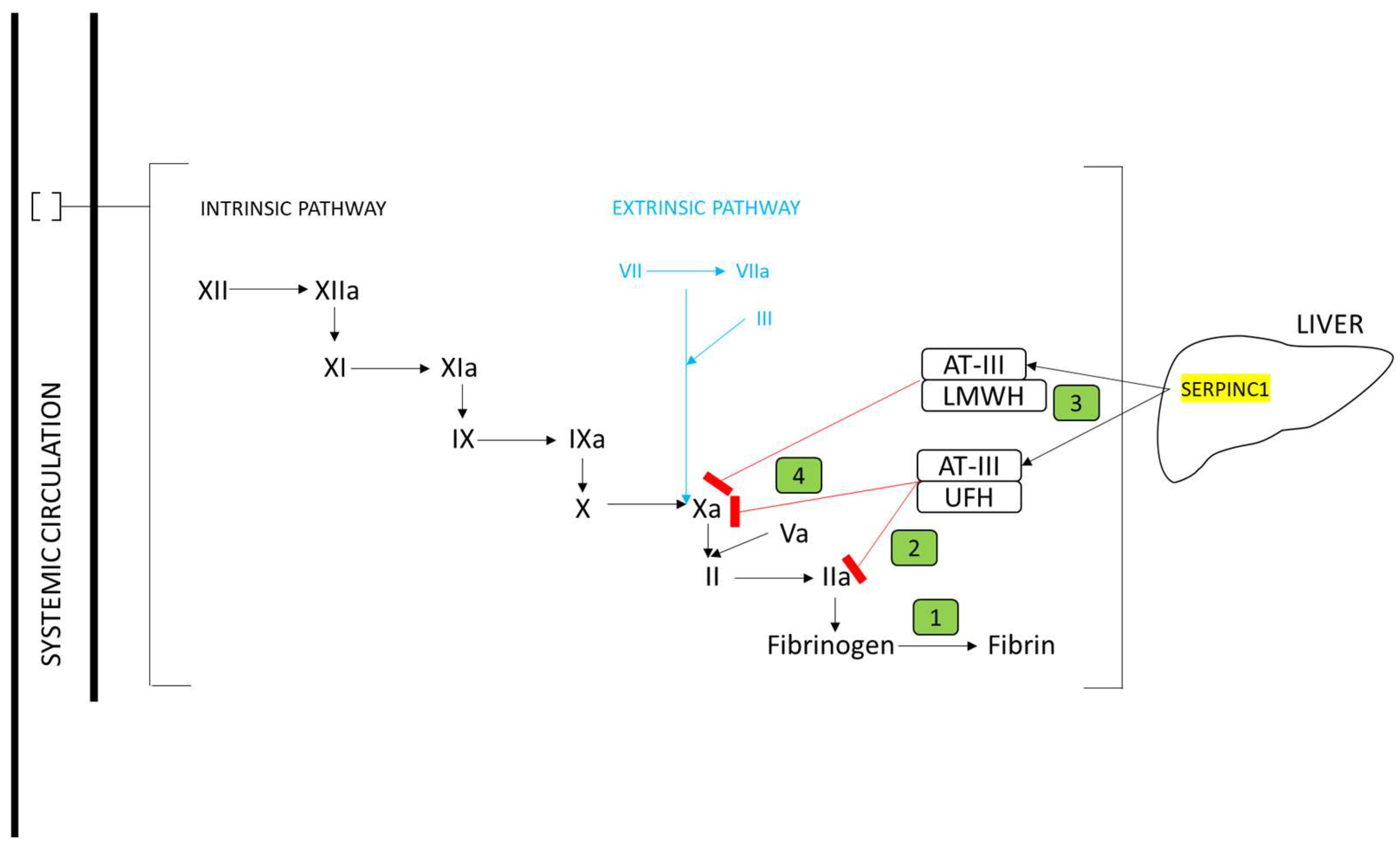
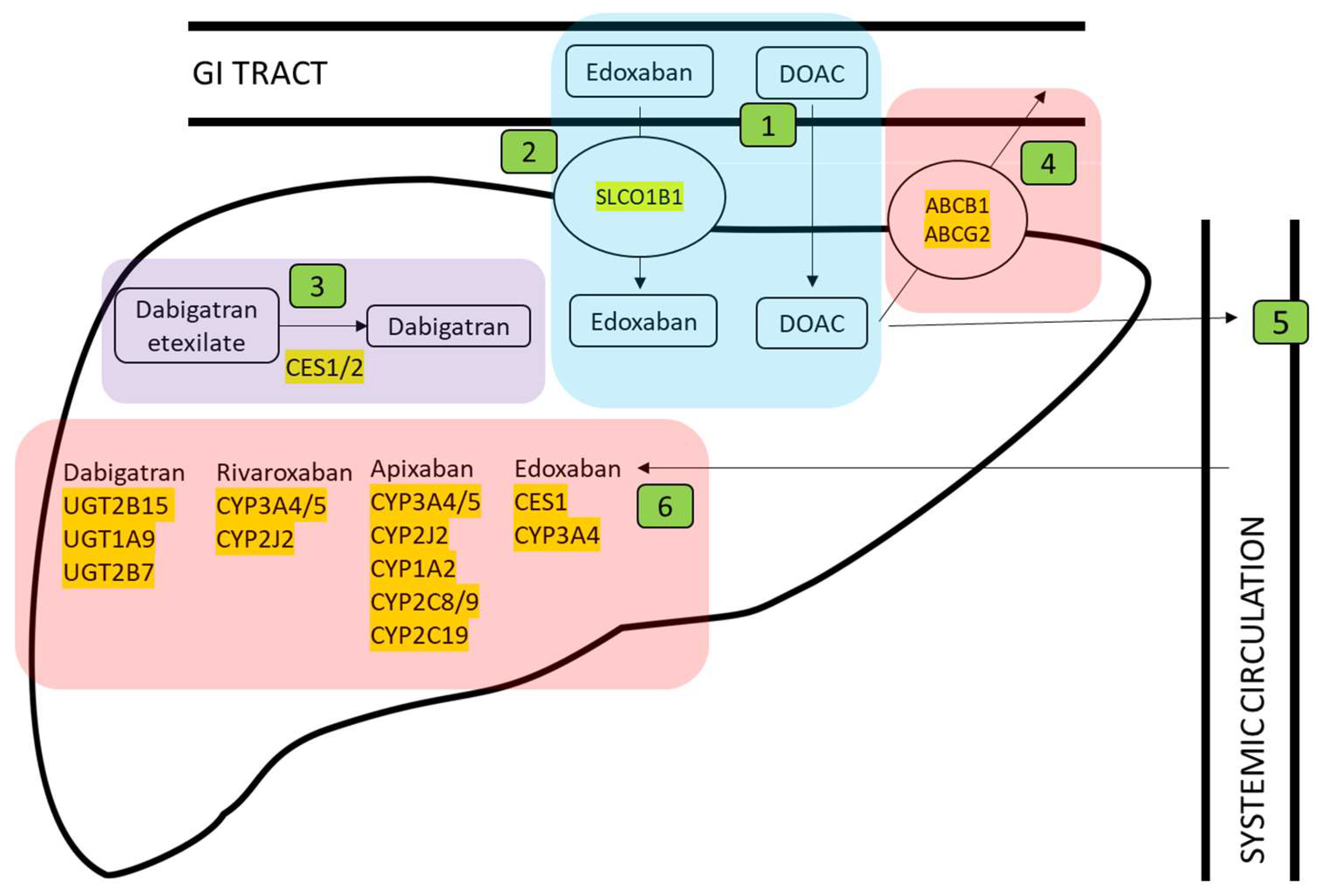
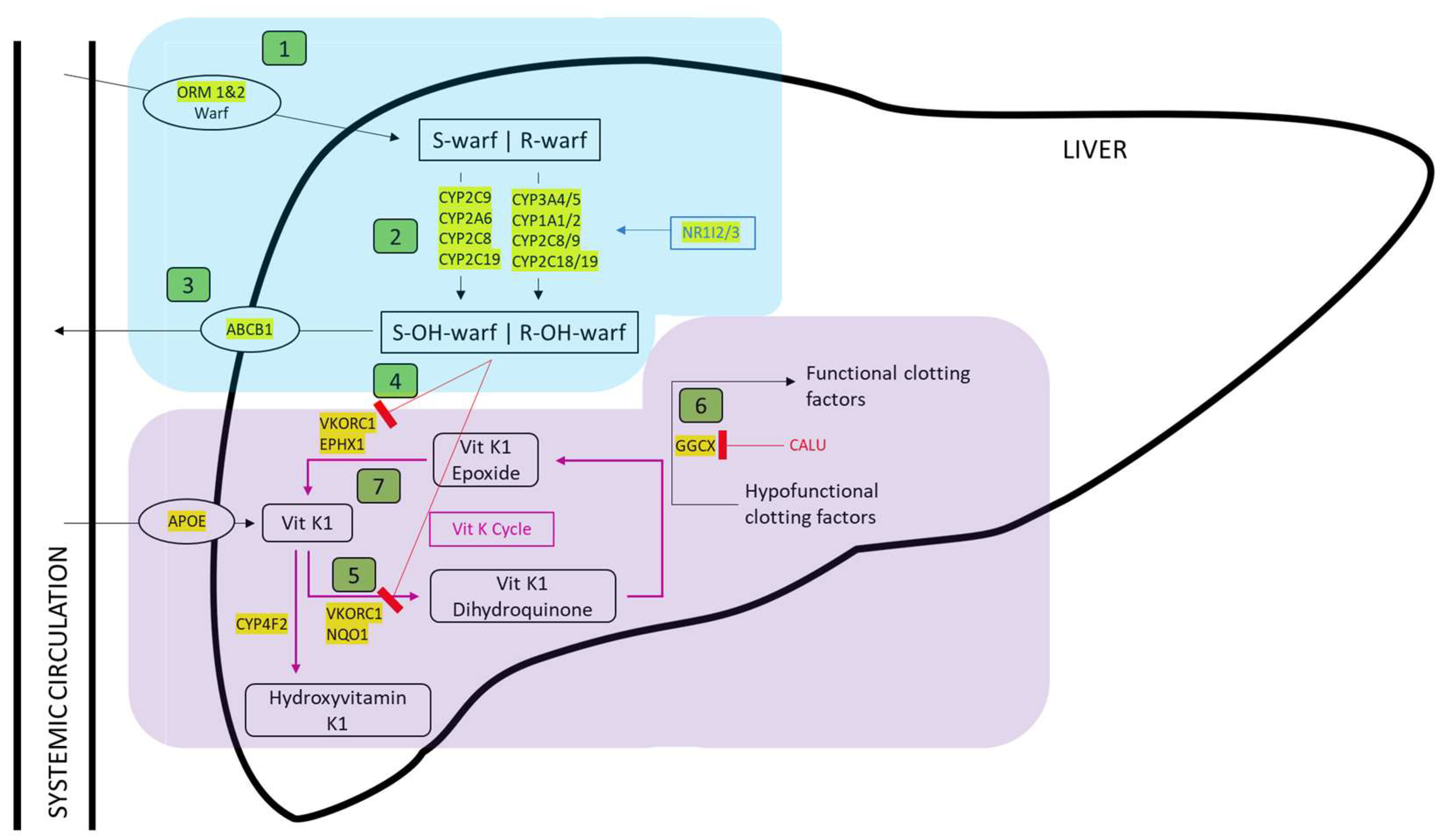
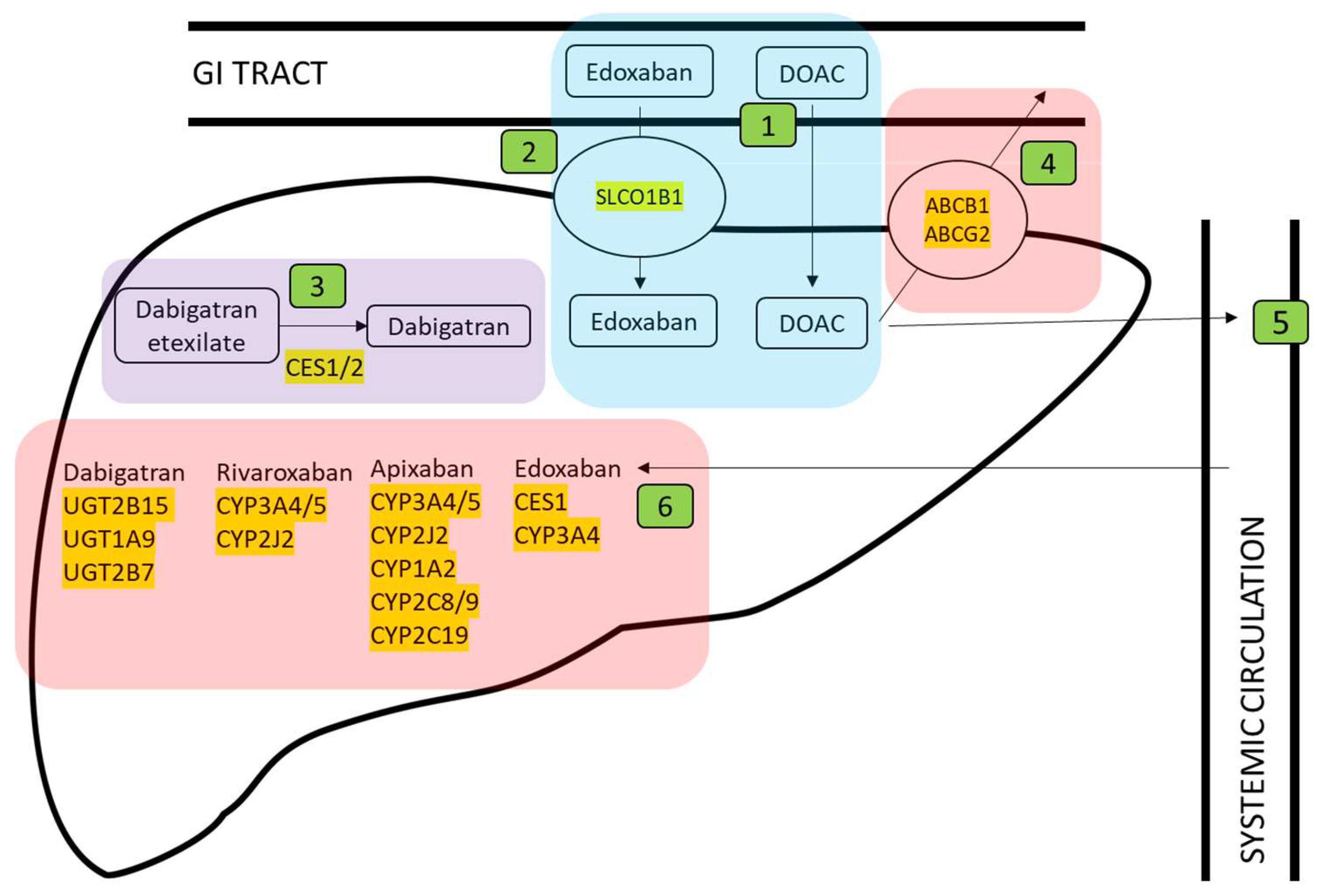
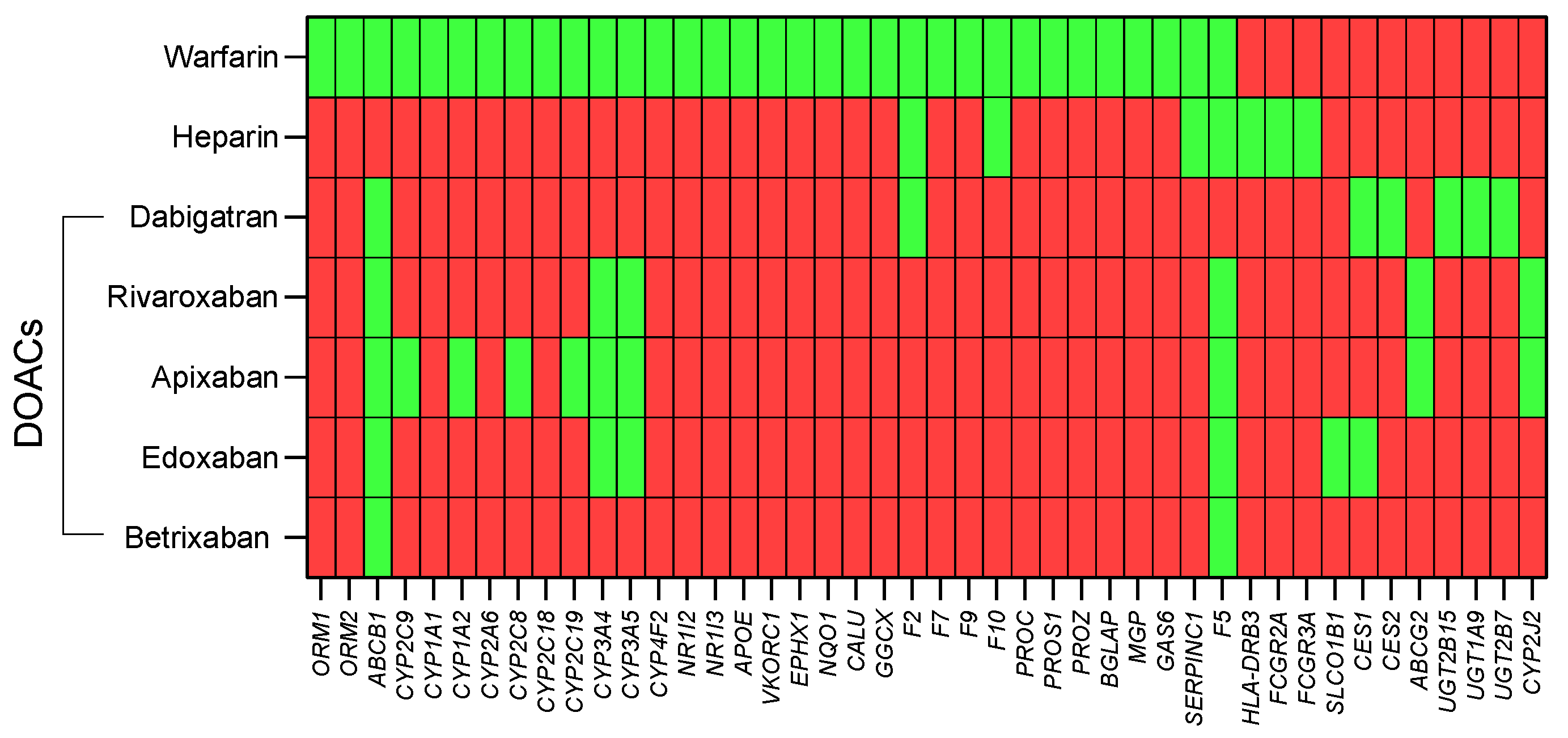
3.1. Identification of Genes Involved in Anticoagulant Metabolism
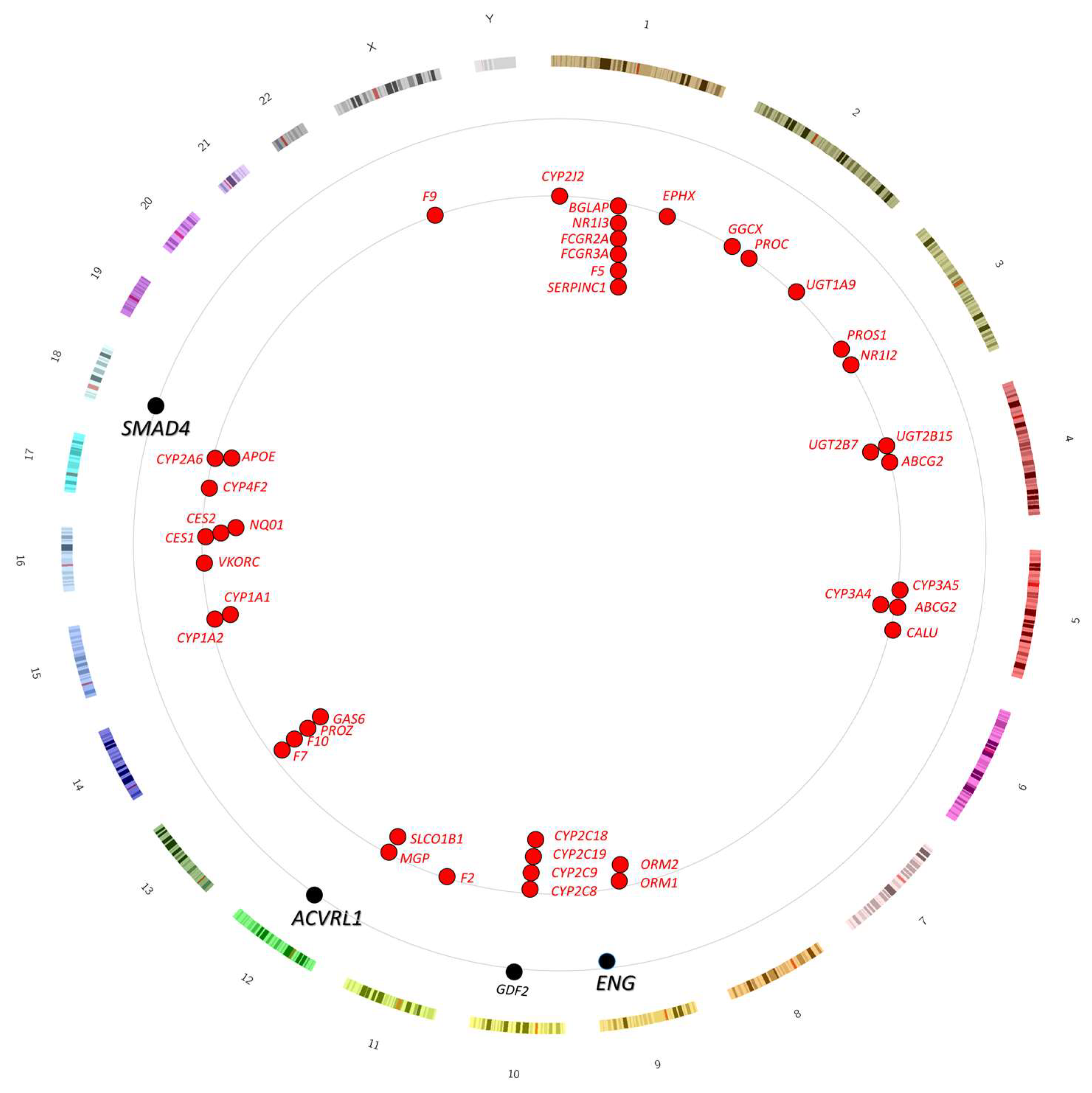
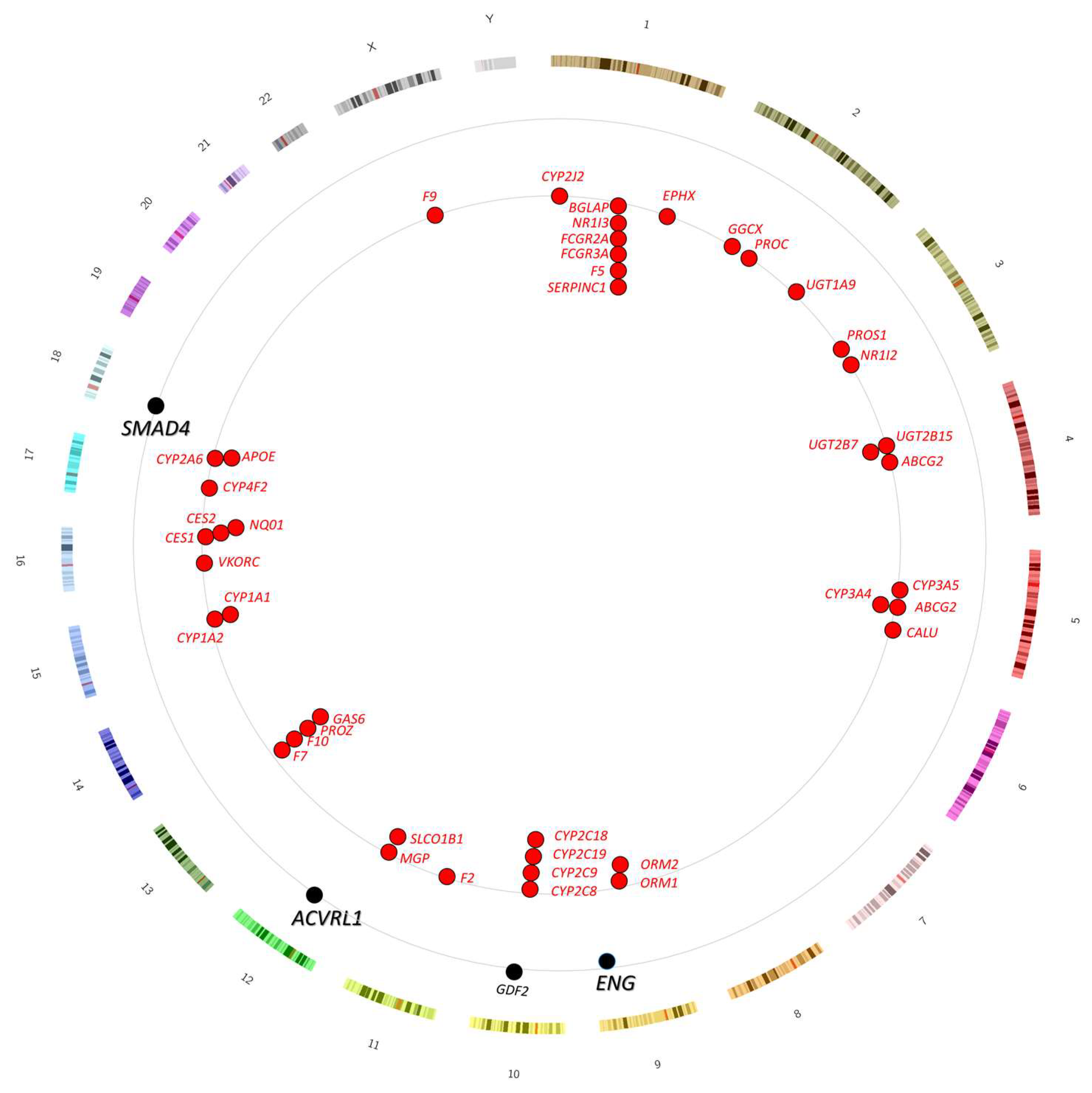
3.2. Genomic Location of Genes Involved in Anticoagulant Metabolism
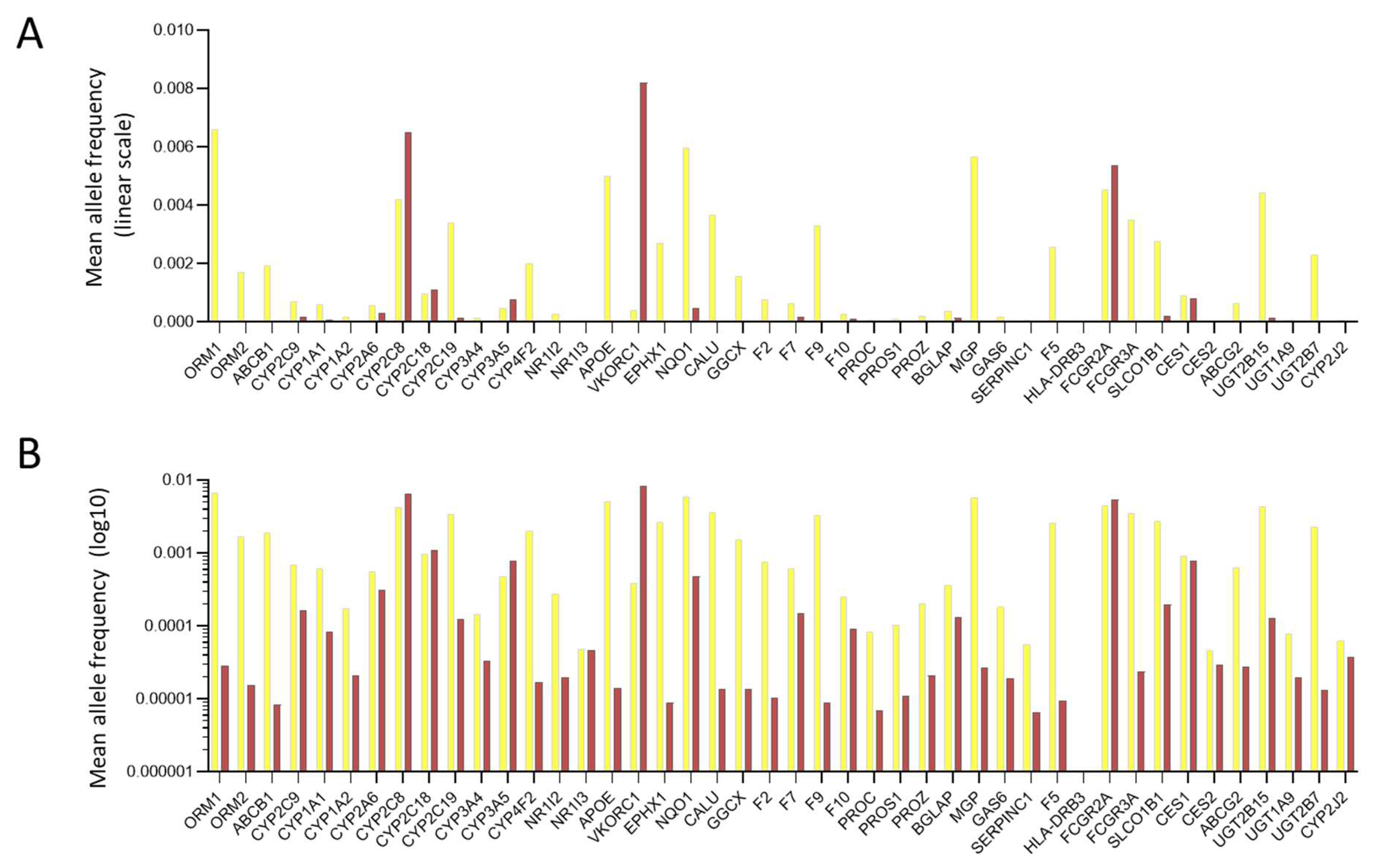
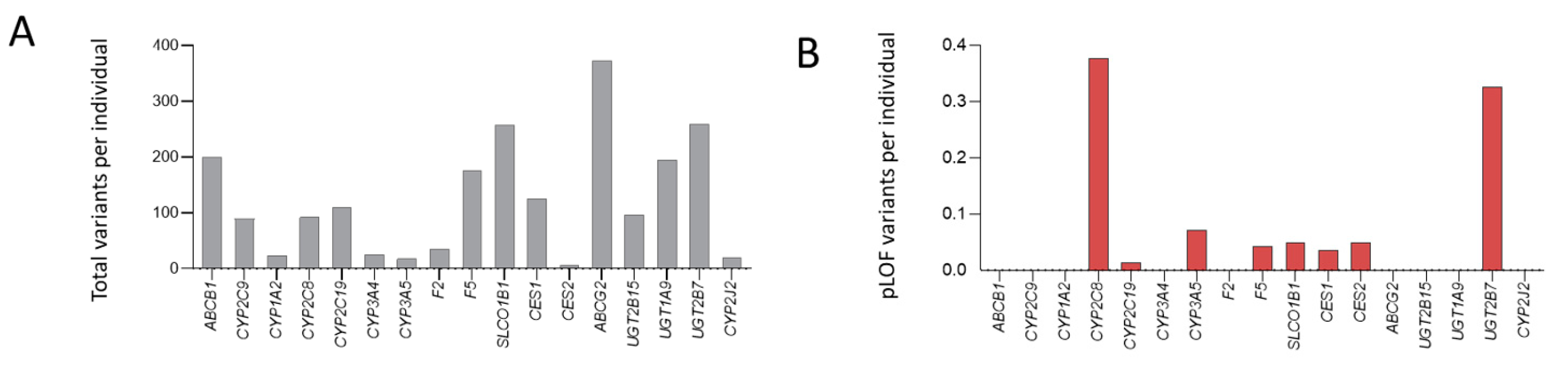
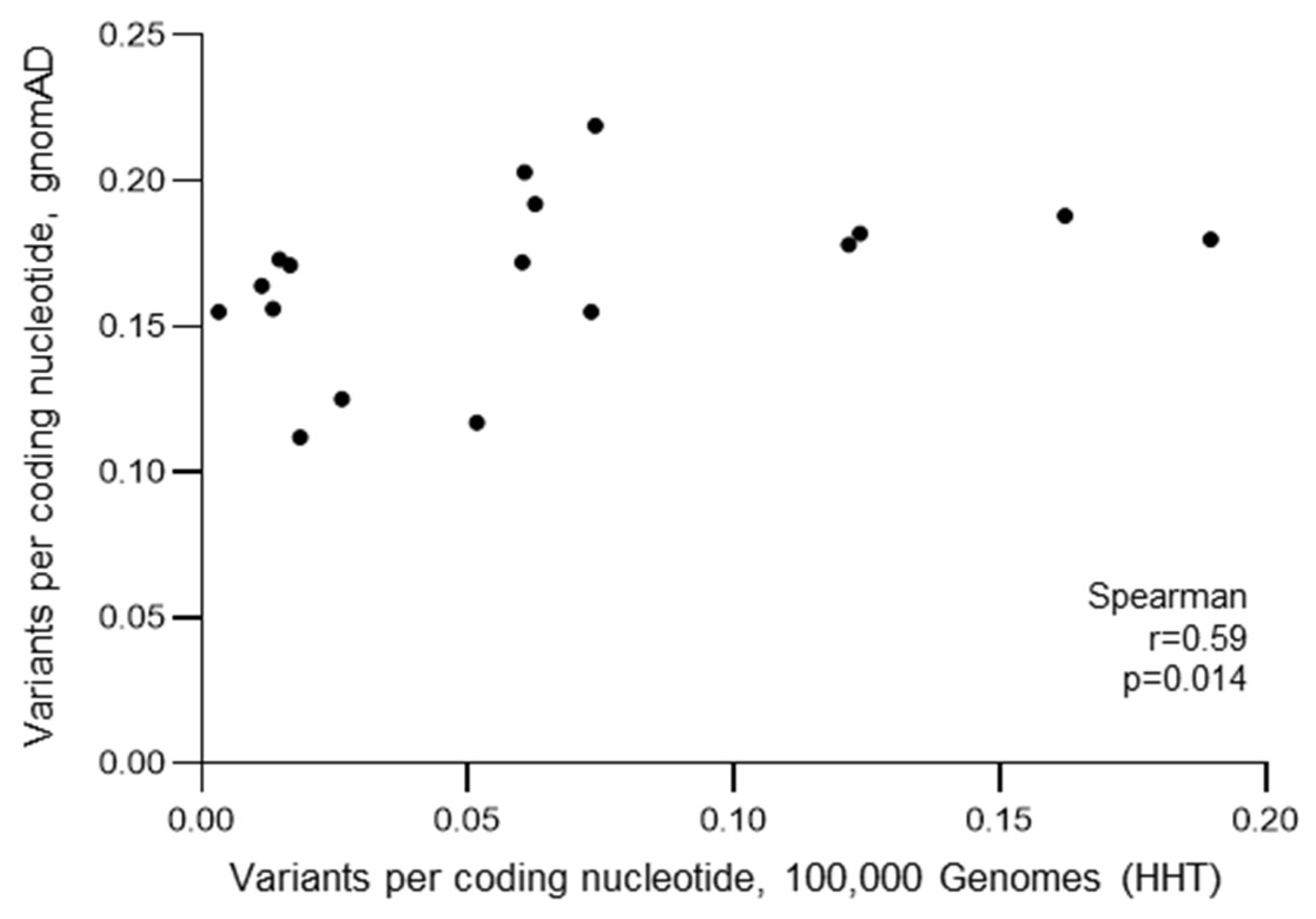
3.3. General Population Variant Burdens
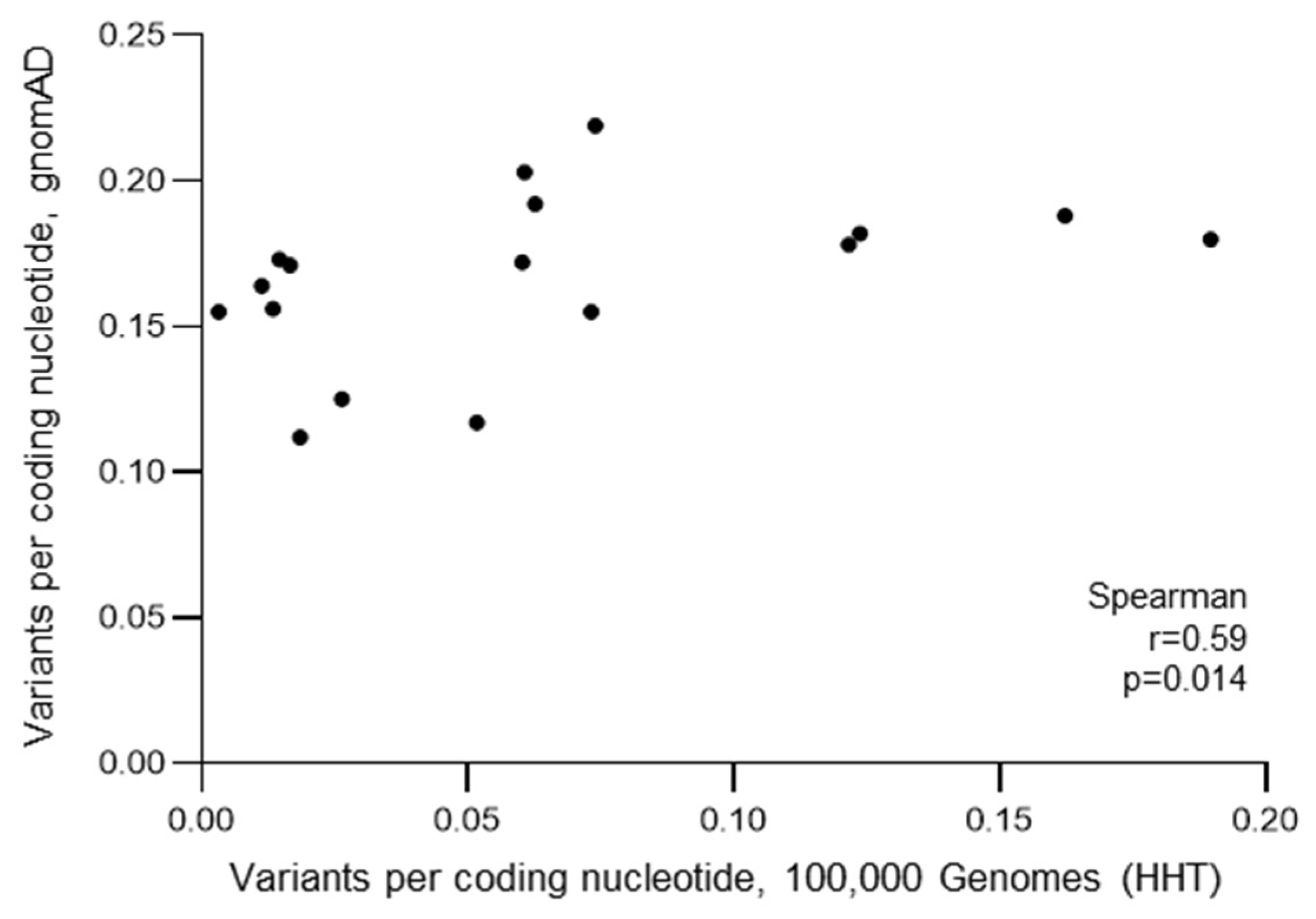
3.4. HHT Population Variant Burdens
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- McAllister, K.A.; Grogg, K.M.; Johnson, D.W.; Gallione, C.J.; Baldwin, M.A.; Jackson, C.E.; Helmbold, E.A.; Markel, D.S.; McKinnon, W.C.; Murrel, J.; et al. Endoglin, a TGF-β binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat. Genet. 1994, 8, 345–351. [Google Scholar] [CrossRef]
- Johnson, D.W.; Berg, J.N.; Baldwin, M.A.; Gallione, C.J.; Marondel, I.; Yoon, S.-J.; Stenzel, T.T.; Speer, M.; Pericak-Vance, M.A.; Diamond, A.; et al. Mutations in the activin receptor–like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat. Genet. 1996, 13, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Gallione, C.J.; Repetto, G.M.; Legius, E.; Rustgi, A.K.; Schelley, S.L.; Tejpar, S.; Mitchell, G.; Drouin, E.; Westermann, C.J.; Marchuk, D.A. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet 2004, 363, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Shovlin, C.L.; Simeoni, I.; Downes, K.; Frazer, Z.C.; Megy, K.; Bernabeu-Herrero, M.E.; Shurr, A.; Brimley, J.; Patel, D.; Kell, L.; et al. Mutational and phenotypic characterization of hereditary hemorrhagic telangiectasia. Blood 2020, 136, 1907–1918. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Kai, Z.; Murphy, D.; Li, D.; Patel, D.; Bielowka, A.M.; Bernabeu-Herrero, M.E.; Abdulmogith, A.; Mumford, A.D.; Westbury, S.K.; et al. Functional filter for whole-genome sequencing data identifies HHT and stress-associated non-coding SMAD4 polyadenylation site variants >5 kb from coding DNA. Am. J. Hum. Genet. 2023, 110, 1903–1918. [Google Scholar] [CrossRef] [PubMed]
- VASCERN Orphanet Definition of Hereditary Hemorrhagic Telangiectasia. 2019. Available online: https://www.orpha.net/consor/www/cgi-bin/OC_Exp.php?lng=EN&Expert=774 (accessed on 11 December 2023).
- Faughnan, M.E.; Mager, J.J.; Hetts, S.W.; Palda, V.A.; Lang-Robertson, K.; Buscarini, E.; Deslandres, E.; Kasthuri, R.S.; Lausman, A.; Poetker, D.; et al. Second International Guidelines for the Diagnosis and Management of Hereditary Hemorrhagic Telangiectasia. Ann. Intern. Med. 2020, 173, 989–1001. [Google Scholar] [CrossRef] [PubMed]
- Shovlin, C.L.; Buscarini, E.; Sabbà, C.; Mager, H.J.; Kjeldsen, A.D.; Pagella, F.; Sure, U.; Ugolini, S.; Torring, P.M.; Suppressa, P.; et al. The European Rare Disease Network for HHT Frameworks for management of hereditary haemorrhagic telangiectasia in general and speciality care. Eur. J. Med. Genet. 2022, 65, 104370. [Google Scholar] [CrossRef] [PubMed]
- Danesino, C.; Cantarini, C.; Olivieri, C. Hereditary Hemorrhagic Telangiectasia in Pediatric Age: Focus on Genetics and Diagnosis. Pediatr. Rep. 2023, 15, 129–142. [Google Scholar] [CrossRef]
- Shovlin, C.L.; Guttmacher, A.E.; Buscarini, E.; Faughnan, M.E.; Hyland, R.H.; Westermann, C.J.; Kjeldsen, A.D.; Plauchu, H. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am. J. Med. Genet. 2000, 91, 66–67. [Google Scholar] [CrossRef]
- Faughnan, M.E.; Palda, V.A.; Garcia-Tsao, G.; Geisthoff, U.W.; McDonald, J.; Proctor, D.D.; Spears, J.; Brown, D.H.; Buscarini, E.; Chesnutt, M.S.; et al. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J. Med. Genet. 2011, 48, 73–87. [Google Scholar] [CrossRef]
- Van Gent, M.W.; Velthuis, S.; Post, M.C.; Snijder, R.J.; Westermann, C.J.; Letteboer, T.G.; Mager, J.J. Hereditary hemorrhagic telangiectasia: How accurate are the clinical criteria? Am. J. Med. Genet. A 2013, 161, 461–466. [Google Scholar] [CrossRef]
- Pollak, M.; Gatt, D.; Shaw, M.; Hewko, S.L.; Lamanna, A.; Santos, S.; Ratjen, F. Longitudinal Assessment of Curaçao Criteria in Children with Hereditary Hemorrhagic Telangiectasia. J. Pediatr. 2023, 263, 113665. [Google Scholar] [CrossRef]
- Pahl, K.S.; Choudhury, A.; Wusik, K.; Hammill, A.; White, A.; Henderson, K.; Pollak, J.; Kasthuri, R.S. Applicability of the Curaçao Criteria for the Diagnosis of Hereditary Hemorrhagic Telangiectasia in the Pediatric Population. J. Pediatr. 2018, 197, 207–213. [Google Scholar] [CrossRef]
- Gonzalez, C.D.; Cipriano, S.D.; Topham, C.A.; Stevenson, D.A.; Whitehead, K.J.; Vanderhooft, S.; Presson, A.P.; McDonald, J. Localization and age distribution of telangiectases in children and adolescents with hereditary hemorrhagic telangiectasia: A retrospective cohort study. J. Am. Acad. Dermatol. 2019, 81, 950–955. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.; Kornish, J.; Stevenson, D.A.; Hanson-Kahn, A.; Balch, H.; James, J.; Naik, H.; Whitehead, K.J. Frequency of epistaxis and telangiectasia in patients with hereditary hemorrhagic telangiectasia (HHT) in comparison with the general population: Curaçao diagnostic criteria revisited. Genet Med. 2023, 25, 100865. [Google Scholar] [CrossRef] [PubMed]
- Shovlin, C.L.; Almaghlouth, F.I.; Alsafi, A.; Coote, N.; Rennie, C.; Wallace, G.M.; Govani, F.S.; Research Consortium, G.E. Updates on diagnostic criteria for hereditary haemorrhagic telangiectasia in the light of whole genome sequencing of ‘gene-negative’ individuals recruited to the 100,000 Genomes Project. J. Med. Genet. 2023, 16, jmg-2023-109195. Available online: https://jmg.bmj.com/content/early/2023/08/15/jmg-2023-109195 (accessed on 11 December 2023).
- Baysal, M.; Demir, S.; Ümit, E.G.; Gürkan, H.; Baş, V.; Karaman Gülsaran, S.; Demirci, U.; Kırkızlar, H.O.; Demir, A.M. Genetic Diagnosis of Hereditary Hemorrhagic Telangiectasia: Four Novel Pathogenic Variations in Turkish Patients. Balkan Med. J. 2019, 37, 43–46. [Google Scholar] [PubMed]
- Zhao, Y.; Zhang, Y.; Wang, X.; Zhang, L. Variant analysis in Chinese families with hereditary hemorrhagic telangiectasia. Mol. Genet. Genomic. Med. 2019, 7, e893. [Google Scholar] [CrossRef] [PubMed]
- Koenighofer, M.; Parzefall, T.; Frohne, A.; Allen, M.; Unterberger, U.; Laccone, F.; Schoefer, C.; Frei, K.; Lucas, T. Spectrum of Novel Hereditary Hemorrhagic Telangiectasia Variants in an Austrian Patient Cohort. Clin. Exp. Otorhinolaryngol. 2019, 12, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Mutize, T.T.; Seedat, R.Y.; Ploos van Amstel, J.K.; Mager, J.J.; Brown, S.C.; Gebremariam, F.; Coetzee, M.J. The clinical and genetic features of hereditary haemorrhagic telangiectasia (HHT) in central South Africa-three novel pathogenic variants. Mol. Biol. Rep. 2020, 47, 9967–9972. [Google Scholar] [CrossRef]
- Gil, R.; Añón, S.; Salazar-Mendiguchía, J.; Riera-Mestre, A. RiHHTa Investigators of the Rare Diseases Working Group from the Spanish Society of Internal Medicine. Current HHT genetic overview in Spain and its phenotypic correlation: Data from RiHHTa registry. Orphanet J. Rare Dis. 2020, 15, 138. [Google Scholar]
- Kitayama, K.; Ishiguro, T.; Komiyama, M.; Morisaki, T.; Morisaki, H.; Minase, G.; Hamanaka, K.; Miyatake, S.; Matsumoto, N.; Kato, M.; et al. Mutational and clinical spectrum of Japanese patients with hereditary hemorrhagic telangiectasia. BMC Med. Genomics. 2021, 14, 288. [Google Scholar] [CrossRef]
- Major, T.; Bereczky, Z.; Gindele, R.; Balogh, G.; Rácz, B.; Bora, L.; Kézsmárki, Z.; Brúgós, B.; Pfliegler, G. Current Status of Clinical and Genetic Screening of Hereditary Hemorrhagic Telangiectasia Families in Hungary. J. Clin. Med. 2021, 10, 3774. [Google Scholar] [CrossRef]
- Kim, B.G.; Jung, J.H.; Kim, M.J.; Moon, E.H.; Oh, J.H.; Park, J.W.; Cha, H.E.; Kim, J.H.; Kim, Y.J.; Chung, J.W.; et al. Genetic Variants and Clinical Phenotypes in Korean Patients with Hereditary Hemorrhagic Telangiectasia. Clin. Exp. Otorhinolaryngol. 2021, 14, 399–406. [Google Scholar] [CrossRef]
- Heald, B.; Rigelsky, C.; Moran, R.; LaGuardia, L.; O’Malley, M.; Burke, C.A.; Zahka, K. Prevalence of thoracic aortopathy in patients with juvenile Polyposis Syndrome-Hereditary Hemorrhagic Telangiectasia due to SMAD4. Am. J. Med. Genet. A 2015, 167, 1758–1762. [Google Scholar] [CrossRef] [PubMed]
- Vorselaars, V.M.M.; Diederik, A.; Prabhudesai, V.; Velthuis, S.; Vos, J.A.; Snijder, R.J.; Westermann, C.J.J.; Mulder, B.J.; Ploos van Amstel, J.K.; Mager, J.J.; et al. SMAD4 gene mutation increases the risk of aortic dilation in patients with hereditary haemorrhagic telangiectasia. Int. J. Cardiol. 2017, 245, 114–118. [Google Scholar] [CrossRef] [PubMed]
- McDonald, N.M.; Ramos, G.P.; Sweetser, S. SMAD4 mutation and the combined juvenile polyposis and hereditary hemorrhage telangiectasia syndrome: A single center experience. Int. J. Color. Dis. 2020, 35, 1963–1965. [Google Scholar] [CrossRef] [PubMed]
- Jelsig, A.M.; Kjeldsen, A.; Christensen, L.L.; Bertelsen, B.; Karstensen, J.G.; Brusgaard, K.; Torring, P.M. Hereditary haemorrhagic telangiectasia in Danish patients with pathogenic variants in SMAD4: A nationwide study. J. Med. Genet. 2023, 60, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Boland, C.R.; Idos, G.E.; Durno, C.; Giardiello, F.M.; Anderson, J.C.; Burke, C.A.; Dominitz, J.A.; Gross, S.; Gupta, S.; Jacobson, B.C.; et al. Diagnosis and management of cancer risk in the gastrointestinal hamartomatous polyposis syndromes: Recommendations from the U.S. Multi-Society Task Force on Colorectal Cancer. Gastrointest Endosc. 2022, 95, 1025–1047. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Umeno, J.; Jimbo, K.; Arai, M.; Iwama, I.; Kashida, H.; Kudo, T.; Koizumi, K.; Sato, Y.; Sekine, S.; et al. Clinical Guidelines for Diagnosis and Management of Juvenile Polyposis Syndrome in Children and Adults-Secondary Publication. J. Anus Rectum Colon 2023, 7, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Kjeldsen, A.D.; Kjeldsen, J. Gastrointestinal bleeding in patients with hereditary hemorrhagic telangiectasia. Am. J. Gastroenterol. 2000, 95, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Finnamore, H.; Le Couteur, J.; Hickson, M.; Busbridge, M.; Whelan, K.; Shovlin, C.L. Hemorrhage-adjusted iron requirements, hematinics and hepcidin define hereditary hemorrhagic telangiectasia as a model of hemorrhagic iron deficiency. PLoS ONE 2013, 8, e76516. [Google Scholar] [CrossRef] [PubMed]
- Cherif, H.; Karlsson, T. Combination treatment with an erythropoiesis-stimulating agent and intravenous iron alleviates anaemia in patients with hereditary haemorrhagic telangiectasia. Ups J. Med. Sci. 2014, 119, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Thielemans, L.; Layton, D.M.; Shovlin, C.L. Low serum haptoglobin and blood films suggest intravascular hemolysis contributes to severe anemia in hereditary hemorrhagic telangiectasia. Haematologica 2019, 104, e127–e130. [Google Scholar] [CrossRef] [PubMed]
- Zarka, J.; Jeong, K.; Yabes, J.G.; Ragni, M.V. Prevalence and risk factors for bleeding in hereditary hemorrhagic telangiectasia: A National Inpatient Sample study. Blood Adv. 2023, 7, 5843–5850. [Google Scholar] [CrossRef]
- Hvelplund, T.; Lange, B.; Bird, S.D.; Korsholm, M.; Kjeldsen, A.D. A retrospective cohort study on European Reference Network for Rare Vascular Diseases 5 outcome measures for Hereditary Haemorrhagic Telangiectasia in Denmark. Orphanet J. Rare Dis. 2022, 17, 8. [Google Scholar] [CrossRef]
- Livesey, J.A.; Manning, R.A.; Meek, J.H.; Jackson, J.E.; Kulinskaya, E.; Laffan, M.A.; Shovlin, C.L. Low serum iron levels are associated with elevated plasma levels of coagulation factor VIII and pulmonary emboli/deep venous thromboses in replicate cohorts of patients with hereditary haemorrhagic telangiectasia. Thorax 2012, 67, 328–333. [Google Scholar] [CrossRef]
- Riera-Mestre, A.; Mora-Luján, J.M.; Trujillo-Santos, J.; Del Toro, J.; Nieto, J.A.; Pedrajas, J.M.; López-Reyes, R.; Soler, S.; Ballaz, A.; Cerdà, P.; et al. Natural history of patients with venous thromboembolism and hereditary hemorrhagic telangiectasia. Findings from the RIETE registry. Orphanet J. Rare Dis. 2019, 14, 196. [Google Scholar] [CrossRef]
- Tentoni, N.; Lapidus, M.I.; Peuchot, V.A.; Vazquez, F.J.; Serra, M.M. Bleeding events during anticoagulation in patients with hereditary hemorrhagic telangiectasia. Thromb. Res. 2021, 197, 109–111. [Google Scholar] [CrossRef]
- Penaloza, A.; Vekemans, M.C.; Lambert, C.; Hermans, C. Deep vein thrombosis induced by thalidomide to control epistaxis secondary to hereditary haemorrhagic telangiectasia. Blood Coagul. Fibrinolysis 2011, 22, 616–618. [Google Scholar] [CrossRef]
- Maestraggi, Q.; Bouattour, M.; Toquet, S.; Jaussaud, R.; Kianmanesh, R.; Durand, F.; Servettaz, A. Bevacizumab to Treat Cholangiopathy in Hereditary Hemorrhagic Telangiectasia: Be Cautious: A Case Report. Medicine 2015, 94, e1966. [Google Scholar] [CrossRef] [PubMed]
- Buscarini, E.; Leandro, G.; Conte, D.; Danesino, C.; Daina, E.; Manfredi, G.; Zambelli, A. Natural history and outcome of hepatic vascular malformations in a large cohort of patients with hereditary hemorrhagic teleangiectasia. Dig Dis Sci. 2011, 56, 2166–2178. [Google Scholar] [CrossRef]
- Velthuis, S.; Swaans, M.J.; Mager, J.J.; Rensing, B.J.; Boersma, L.V.; Post, M.C. Left atrial appendage closure for stroke prevention in patients with atrial fibrillation and hereditary hemorrhagic telangiectasia. Case Rep. Cardiol. 2012, 2012, 646505. [Google Scholar] [CrossRef] [PubMed]
- Pepe, M.; Suppressa, P.; Giuliano, A.F.; Nestola, P.L.; Bortone, A.S.; DECillis, E.; Acquaviva, T.; Forleo, C.; Moscarelli, M.; Lenato, G.M.; et al. Safety of reduced or absent antithrombotic therapy after left atrial appendage closure in patients affected by hereditary hemorrhagic telangiectasia and atrial fibrillation. Minerva Cardiol. Angiol. 2022, 70, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Cepas-Guillen, P.L.; López-Mínguez, J.R.; García, J.C.N.; Nombela-Franco, L.; Benito-González, T.; Cruz-González, I.; Freixa, X. Left Atrial Appendage Occlusion in Hereditary Haemorrhagic Telangiectasia Patients (Rendu Osler Syndrome) with Non-Valvular Atrial Fibrillation: Prevention of Cardioembolic Events While Avoiding the Long-Term Risks of Oral Anticoagulation. Cardiovasc. Revasc. Med. 2022, 43, 140–142. [Google Scholar] [CrossRef] [PubMed]
- Ducloy-Bouthors, A.S.; Baldini, A.; Abdul-Kadir, R.; Nizard, J. ESA VTE Guidelines Task Force. European guidelines on perioperative venous thromboembolism prophylaxis: Surgery during pregnancy and the immediate postpartum period. Eur. J. Anaesthesiol. 2018, 35, 130–133. [Google Scholar] [CrossRef]
- Spyropoulos, A.C.; Levy, J.H.; Ageno, W.; Connors, J.M.; Hunt, B.J.; Iba, T.; Levi, M.; Samama, C.M.; Thachil, J.; Giannis, D.; et al. Scientific and Standardization Committee communication: Clinical guidance on the diagnosis, prevention, and treatment of venous thromboembolism in hospitalized patients with COVID-19. J. Thromb. Haemost. 2020, 18, 1859–1865. [Google Scholar] [CrossRef]
- Plauchu, H.; de Chadarévian, J.P.; Bideau, A.; Robert, J.M. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am. J. Med. Genet. 1989, 32, 291–297. [Google Scholar] [CrossRef]
- Kjeldsen, A.D.; Vase, P.; Green, A. Hereditary haemorrhagic telangiectasia: A population-based study of prevalence and mortality in Danish patients. J. Intern. Med. 1999, 245, 31–39. [Google Scholar] [CrossRef]
- Dakeishi, M.; Shioya, T.; Wada, Y.; Shindo, T.; Otaka, K.; Manabe, M.; Nozaki, J.; Inoue, S.; Koizumi, A. Genetic epidemiology of hereditary hemorrhagic telangiectasia in a local community in the northern part of Japan. Hum. Mutat. 2002, 19, 140–148. [Google Scholar] [CrossRef]
- Donaldson, J.W.; McKeever, T.M.; Hall, I.P.; Hubbard, R.B.; Fogarty, A.W. The UK prevalence of hereditary haemorrhagic telangiectasia and its association with sex, socioeconomic status and region of residence: A population-based study. Thorax 2014, 69, 161–167. [Google Scholar] [CrossRef]
- Du, P.; Bergamasco, A.; Moride, Y.; Truong Berthoz, F.; Özen, G.; Tzivelekis, S. Von Willebrand Disease Epidemiology, Burden of Illness and Management: A Systematic Review. J. Blood Med. 2023, 14, 189–208. [Google Scholar] [CrossRef] [PubMed]
- Iorio, A.; Stonebraker, J.S.; Chambost, H.; Makris, M.; Coffin, D.; Herr, C.; Germini, F. Data and Demographics Committee of the World Federation of Hemophilia. Establishing the Prevalence and Prevalence at Birth of Hemophilia in Males: A Meta-analytic Approach Using National Registries. Ann. Intern. Med. 2019, 171, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Shovlin, C.L.; Millar, C.M.; Droege, F.; Kjeldsen, A.; Manfredi, G.; Suppressa, P.; Ugolini, S.; Coote, N.; Fialla, A.D.; Geisthoff, U.; et al. Safety of direct oral anticoagulants in patients with hereditary hemorrhagic telangiectasia. Orphanet J. Rare Dis. 2019, 14, 210. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.P.; Shehata, N.; Faughnan, M.E. Hereditary hemorrhagic telangiectasia patients can tolerate anticoagulation. Ann. Hematol. 2012, 91, 1959–1968. [Google Scholar] [CrossRef] [PubMed]
- Devlin, H.L.; Hosman, A.E.; Shovlin, C.L. Antiplatelet and anticoagulant agents in hereditary hemorrhagic telangiectasia. N. Engl. J. Med. 2013, 368, 876–878. [Google Scholar] [CrossRef] [PubMed]
- Gaetani, E.; Agostini, F.; Giarretta, I.; Porfidia, A.; Di Martino, L.; Gasbarrini, A.; Pola, R.; on behalf of the Multidisciplinary Gemelli Hospital Group for HHT. Antithrombotic Therapy in Hereditary Hemorrhagic Telangiectasia: Real-World Data from the Gemelli Hospital HHT Registry. J. Clin. Med. 2020, 9, 1699. [Google Scholar] [CrossRef] [PubMed]
- Virk, Z.M.; Zhang, E.; Rodriguez-Lopez, J.; Witkin, A.; Wong, A.K.; Luther, J.; Lin, A.E.; Ning, M.; Grabowski, E.; Holbrook, E.H.; et al. Safety, tolerability, and effectiveness of anticoagulation and antiplatelet therapy in hereditary hemorrhagic telangiectasia. J. Thromb. Haemostasis. 2023, 21, 26–36. [Google Scholar] [CrossRef]
- Grobost, V.; Hammi, S.; Pereira, B.; Guilhem, A.; Duffau, P.; Seguier, J.; Parrot, A.; Gautier, G.; Alric, L.; Kerjouan, M.; et al. Antiplatelet and anticoagulant therapies in hereditary hemorrhagic telangiectasia: A large French cohort study (RETROPLACOTEL). Thromb. Res. 2023, 229, 107–113. [Google Scholar] [CrossRef]
- Serra, M.M.; Elizondo, C.M.; Alonso, M.; Peuchot, V.; Vazquez, F.J. Incidence of Thromboembolic Disease in Hereditary Hemorrhagic Telangiectasia (Osler Weber Rendu Syndrome) Abstract. Blood 2018, 132 (Suppl. S1), 4967. [Google Scholar] [CrossRef]
- Zhang, E.; Virk, Z.M.; Rodriguez-Lopez, J.; Al-Samkari, H. Anticoagulation and antiplatelet therapy in hereditary hemorrhagic telangiectasia: A scoping review. Thromb. Res. 2023, 226, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Joyce, K.E.; Onabanjo, E.; Brownlow, S.; Nur, F.; Olupona, K.; Fakayode, K.; Sroya, M.; Thomas, G.A.; Ferguson, T.; Redhead, J.; et al. Whole genome sequences discriminate hereditary hemorrhagic telangiectasia phenotypes by non-HHT deleterious DNA variation. Blood Adv. 2022, 6, 3956–3969. [Google Scholar] [CrossRef] [PubMed]
- Egido-Turrión, C.; Rossi, E.; Ollauri-Ibáñez, C.; Pérez-García, M.L.; Sevilla, M.A.; Bastida, J.M.; González-Porras, J.R.; Rodríguez-Barbero, A.; Bernabeu, C.; Lopez-Novoa, J.M.; et al. Functional Alterations Involved in Increased Bleeding in Hereditary Hemorrhagic Telangiectasia Mouse Models. Front. Med. 2022, 9, 871903. [Google Scholar] [CrossRef] [PubMed]
- Pirmohammed, M.; O’Donoghue, D.; Turner, R.; Magavern, E.; Roebuck, D.; Ross, P.; Keavney, B.; Shovlin, C.; Popoola, J.; Nasser, S.; et al. Personalised Prescribing. Using Pharmacogenomics to Improve Patient Outcomes. A Report from the Royal College of Physicians and British Pharmacological Society Joint Working Party. Available online: https://www.rcp.ac.uk/projects/outputs/personalised-prescribing-using-pharmacogenomics-improve-patient-outcomes (accessed on 24 March 2022).
- Franconi, F.; Campesi, I. Pharmacogenomics, pharmacokinetics and pharmacodynamics: Interaction with biological differences between men and women. Br. J. Pharmacol. 2014, 171, 580–594. [Google Scholar] [CrossRef] [PubMed]
- Kabbani, D.; Akika, R.; Wahid, A.; Daly, A.K.; Cascorbi, I.; Zgheib, N.K. Pharmacogenomics in practice: A review and implementation guide. Front Pharmacol. 2023, 14, 1189976. [Google Scholar] [CrossRef]
- Swen, J.J.; van der Wouden, C.H.; Manson, L.E.; Abdullah-Koolmees, H.; Blagec, K.; Blagus, T.; Böhringer, S.; Cambon-Thomsen, A.; Cecchin, E.; Cheung, K.C.; et al. A 12-gene pharmacogenetic panel to prevent adverse drug reactions: An open-label, multicentre, controlled, cluster-randomised crossover implementation study. Lancet 2023, 401, 347–356, Erratum in Lancet 2023, 402, 692. [Google Scholar] [CrossRef]
- European Medicines Agency. Good Pharmacogenomic Practice-Scientific Guideline. Available online: https://www.ema.europa.eu/en/good-pharmacogenomic-practice-scientific-guideline (accessed on 19 March 2018).
- NHS England. Community Pharmacy Oral Anticoagulant Safety Audit 2021/22. Available online: https://www.england.nhs.uk/long-read/community-pharmacy-oral-anticoagulant-safety-audit-2021-22/ (accessed on 27 March 2023).
- Beunk, L.; Nijenhuis, M.; Soree, B.; de Boer-Veger, N.J.; Buunk, A.M.; Guchelaar, H.J.; Houwink, E.J.F.; Risselada, A.; Rongen, G.A.P.J.M.; van Schaik, R.H.N.; et al. Dutch Pharmacogenetics Working Group (DPWG) guideline for the gene-drug interaction between CYP2D6, CYP3A4 and CYP1A2 and antipsychotics. Eur. J. Hum. Genet. 2023; epub ahead of print. [Google Scholar] [CrossRef]
- Relling, M.V.; Klein, T.E. CPIC: Clinical Pharmacogenetics Implementation Consortium of the Pharmacogenomics Research Network. Clin. Pharmacol. Ther. 2011, 89, 464–467. [Google Scholar] [CrossRef]
- Hewett, M.; Oliver, D.E.; Rubin, D.L.; Easton, K.L.; Stuart, J.M.; Altman, R.B.; Klein, T.E. PharmGKB: The Pharmacogenetics Knowledge Base. Nucleic Acids Res. 2002, 30, 163–165. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information, U.S. National Library of Medicine: Genome Reference Consortium Human Build 38. 2023. Available online: https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_000001405.26/ (accessed on 11 December 2023).
- Nassar, L.R.; Barber, G.P.; Benet-Pagès, A.; Casper, J.; Clawson, H.; Diekhans, M.; Fischer, C.; Gonzalez, J.N.; Hinrichs, A.S.; Lee, B.T.; et al. The UCSC Genome Browser database: 2023 update. Nucleic Acids Res. 2023, 51, D1188–D1195. [Google Scholar] [CrossRef]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Farrell, C.M.; Feldgarden, M.; Fine, A.M.; Funk, K.; et al. Database resources of the National Center for Biotechnology Information in 2023. Nucleic Acids Res. 2023, 51, D29–D38. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- The National Genomics Research and Healthcare Knowledgebase v5; Genomics England: London, UK, 2019. Available online: https://figshare.com/articles/dataset/GenomicEnglandProtocol_pdf/4530893 (accessed on 11 December 2023).
- Morales, J.; Pujar, S.; Loveland, J.E.; Astashyn, A.; Bennett, R.; Berry, A.; Cox, E.; Davidson, C.; Ermolaeva, O.; Farrell, C.M.; et al. A joint NCBI and EMBL-EBI transcript set for clinical genomics and research. Nature 2022, 604, 310–315. [Google Scholar] [CrossRef]
- Shovlin, C.L.; Hughes, J.M.; Tuddenham, E.G.; Temperley, I.; Perembelon, Y.F.; Scott, J.; Seidman, C.E.; Seidman, J.G. A gene for hereditary haemorrhagic telangiectasia maps to chromosome 9q3. Nat. Genet. 1994, 6, 205–209. [Google Scholar] [CrossRef] [PubMed]
- American Society of Hematology. To Test or Not to Test: Is Pharmacogenomic Testing for Warfarin Valuable? Available online: https://ashpublications.org/ashclinicalnews/news/2776/To-Test-or-Not-to-Test-Is-Pharmacogenomic-Testing (accessed on 30 December 2021).
- Thomas, O.; Lybeck, E.; Strandberg, K.; Tynngård, N.; Schött, U. Monitoring low molecular weight heparins at therapeutic levels: Dose-responses of, and correlations and differences between aPTT, anti-factor Xa and thrombin generation assays. PLoS ONE 2015, 10, e0116835. [Google Scholar] [CrossRef] [PubMed]
- Crowther, M.A.; Ginsberg, J.B.; Kearon, C.; Harrison, L.; Johnson, J.; Massicotte, M.P.; Hirsh, J. A randomized trial comparing 5-mg and 10-mg warfarin loading doses. Arch Intern Med. 1999, 159, 46–48. [Google Scholar] [CrossRef] [PubMed]
- Tait, R.C.; Sefcick, A. A warfarin induction regimen for out-patient anticoagulation in patients with atrial fibrillation. Br. J. Haematol. 1998, 101, 450–454. [Google Scholar] [CrossRef]
- Flockhart, D.A.; O’Kane, D.; Williams, M.S.; Watson, M.S.; Flockhart, D.A.; Gage, B.; Gandolfi, R.; King, R.; Lyon, E.; Nussbaum, R.; et al. Pharmacogenetic testing of CYP2C9 and VKORC1 alleles for warfarin. Genet Med. 2008, 10, 139–150. [Google Scholar] [CrossRef]
- Johnson, J.A.; Gong, L.; Whirl-Carrillo, M.; Gage, B.F.; Scott, S.A.; Stein, C.M.; Anderson, J.L.; Kimmel, S.E.; Lee, M.T.; Pirmohamed, M.; et al. Clinical Pharmacogenetics Implementation Consortium Guidelines for CYP2C9 and VKORC1 genotypes and warfarin dosing. Clin. Pharmacol. Ther. 2011, 90, 625–629. [Google Scholar] [CrossRef]
- Kim, S.; Yun, Y.M.; Chae, H.J.; Cho, H.J.; Ji, M.; Kim, I.S.; Wee, K.A.; Lee, W.; Song, S.H.; Woo, H.I.; et al. Clinical Pharmacogenetic Testing and Application: Laboratory Medicine Clinical Practice Guidelines. Ann. Lab. Med. 2017, 37, 180–193. [Google Scholar] [CrossRef]
- Pratt, V.M.; Cavallari, L.H.; Del Tredici, A.L.; Hachad, H.; Ji, Y.; Kalman, L.V.; Ly, R.C.; Moyer, A.M.; Scott, S.A.; Whirl-Carrillo, M.; et al. Recommendations for Clinical Warfarin Genotyping Allele Selection: A Report of the Association for Molecular Pathology and the College of American Pathologists. J. Mol. Diagn. 2020, 22, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Giugliano, R.P.; Morrow, D.A.; Patel, M.R.; Wallentin, L.; Alexander, J.H.; Cecilia Bahit, M.; Benz, A.P.; Bohula, E.A.; Chao, T.F.; Dyal, L.; et al. Direct Oral Anticoagulants Versus Warfarin in Patients with Atrial Fibrillation: Patient-Level Network Meta-Analyses of Randomized Clinical Trials With Interaction Testing by Age and Sex. Circulation 2022, 145, 242–255. [Google Scholar]
- National Institute for Health and Care Ecxcellence. Oral Anticoagulants. Available online: https://cks.nice.org.uk/topics/deep-vein-thrombosis/prescribing-information/oral-anticoagulants/ (accessed on 11 December 2023).
- Ho, K.H.; Van Hove, M.; Leng, G. Trends in anticoagulant prescribing: A review of local policies in English primary care. BMC Health Serv Res. 2020, 20, 279. [Google Scholar] [CrossRef] [PubMed]
- Afzal, S.; Zaidi, S.T.R.; Merchant, H.A.; Babar, Z.U.D.; Hasan, S.S. Prescribing trends of oral anticoagulants in England over the last decade: A focus on new and old drugs and adverse events reporting. J. Thromb. Thrombolysis. 2021, 52, 646–653. [Google Scholar] [CrossRef]
- PharmGKB Clinical Annotation Levels of Evidence. 2023. Available online: https://www.pharmgkb.org/page/clinAnnLevels (accessed on 11 December 2023).
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Raymond, J.; Imbert, L.; Cousin, T.; Duflot, T.; Varin, R.; Wils, J.; Lamoureux, F. Pharmacogenetics of Direct Oral Anticoagulants: A Systematic Review. J. Pers. Med. 2021, 11, 37. [Google Scholar] [CrossRef]
- NHS England. National Genomic Test Directory. Available online: https://www.england.nhs.uk/publication/national-genomic-test-directories/ (accessed on 20 September 2023).
- NHS England. Accelerating Genomic Medicine in the NHS. Available online: https://www.england.nhs.uk/long-read/accelerating-genomic-medicine-in-the-nhs/ (accessed on 31 October 2022).
- NHS North West Genomic Medicine Service Alliance. Spotlight: PROGRESS Project. 2023. Available online: https://www.nw-gmsa.nhs.uk/about-us/our-projects/spotlight#:~:text=PROGRESS%20is%20the%20first%20study,being%20prescribed%20a%20new%20medicine (accessed on 11 December 2023).
- Connelly, D. Pharmacogenomic Testing Pilot to Start in General Practice from June 2023. The Pharmaceutical Journal. 18 May 2023. Available online: https://pharmaceutical-journal.com/article/news/pharmacogenomic-testing-pilot-to-start-in-general-practice-from-june-2023 (accessed on 11 December 2023).
- Nakagawa, T.; Kishino, S.; Itoh, S.; Sugawara, M.; Miyazaki, K. Differential binding of disopyramide and warfarin enantiomers to human alpha(1)-acid glycoprotein variants. Br. J. Clin. Pharmacol. 2003, 56, 664–669. [Google Scholar] [CrossRef]
- Otagiri, M.; Maruyama, T.; Imai, T.; Suenaga, A.; Imamura, Y. A comparative study of the interaction of warfarin with human α1-acid glycoprotein and human albumin. J. Pharm. Pharmacol. 1987, 39, 416–420. [Google Scholar] [CrossRef]
- Wadelius, M.; Pirmohamed, M. Pharmacogenetics of warfarin: Current status and future challenges. Pharmacogenomics J. 2007, 7, 99–111. [Google Scholar] [CrossRef]
- Wadelius, M.; Sörlin, K.; Wallerman, O.; Karlsson, J.; Yue, Q.Y.; Magnusson, P.K.; Wadelius, C.; Melhus, H. Warfarin sensitivity related to CYP2C9, CYP3A5, ABCB1 (MDR1) and other factors. Pharmacogenomics J. 2003, 4, 40–48. [Google Scholar] [CrossRef]
- Assenat, E.; Gerbal-Chaloin, S.; Larrey, D.; Saric, J.; Fabre, J.M.; Maurel, P.; Vilarem, M.J.; Pascussi, J.M. Interleukin 1beta inhibits CAR-induced expression of hepatic genes involved in drug and bilirubin clearance. Hepatology 2004, 40, 951–960. [Google Scholar] [CrossRef]
- Lehmann, J.M.; McKee, D.D.; Watson, M.A.; Willson, T.M.; Moore, J.T.; Kliewer, S.A. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J. Clin. Investig. 1998, 102, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ferguson, S.S.; Negishi, M.; Goldstein, J.A. Induction of human CYP2C9 by rifampicin, hyperforin, and phenobarbital is mediated by the pregnane X receptor. J. Pharmacol. Exp. Ther. 2004, 308, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Jonas, D.E.; McLeod, H.L. Genetic and clinical factors relating to warfarin dosing. Trends Pharmacol. Sci. 2009, 30, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Moualla, H.; Garcia, D. Vitamin K Antagonists—Current Concepts and Challenges. Thromb Res. 2011, 128, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Kohlmeier, M.; Salomon, A.; Saupe, J.; Shearer, M.J. Transport of vitamin K to bone in humans. J. Nutr. 1996, 126, 1192S–1196S. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.Y.; Sun, X.; Wadelius, M.; Huang, L.; Peng, C.; Ma, W.L.; Yang, G.P. Influence of APOE Gene Polymorphism on Interindividual and Interethnic Warfarin Dosage Requirement: A Systematic Review and Meta-Analysis. Cardiovasc. Ther. 2016, 34, 297–307. [Google Scholar] [CrossRef]
- Tian, L.; Xiao, P.; Zhou, B.; Chen, Y.; Kang, L.; Wang, Q.; Lin, J.; Son, M.; Wu, Q. Influence of NQO1 Polymorphisms on Warfarin Maintenance Dose: A Systematic Review and Meta-Analysis (rs1800566 and rs10517). Cardiovasc Ther. 2021, 2021, 5534946. [Google Scholar] [CrossRef] [PubMed]
- Mishima, E.; Ito, J.; Wu, Z.; Nakamura, T.; Wahida, A.; Doll, S.; Tonnus, W.; Nepachalovich, P.; Eggenhofer, E.; Aldrovandi, M.; et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature 2022, 608, 778–783. [Google Scholar] [CrossRef]
- McDonald, M.G.; Rieder, M.J.; Nakano, M.; Hsia, C.K.; Rettie, A.E. CYP4F2 Is a Vitamin K1 Oxidase: An Explanation for Altered Warfarin Dose in Carriers of the V433M Variant. Mol. Pharmacol. 2009, 75, 1337–1346. [Google Scholar] [CrossRef]
- Danziger, J. Vitamin K-dependent proteins, warfarin, and vascular calcification. Clin. J. Am. Soc. Nephrol. 2008, 3, 1504–1510. [Google Scholar] [CrossRef] [PubMed]
- Vecsler, M.; Loebstein, R.; Almog, S.; Kurnik, D.; Goldman, B.; Halkin, H.; Gak, E. Combined genetic profiles of components and regulators of the vitamin K-dependent γ-carboxylation system affect individual sensitivity to warfarin. Thromb Haemost. 2006, 95, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Loebstein, R.; Vecsler, M.; Kurnik, D.; Austerweil, N.; Gak, E.; Halkin, H.; Almog, S. Common Genetic Variants of Microsomal Epoxide Hydrolase Affect Warfarin Dose Requirements Beyond the Effect of Cytochrome P450 2C9. Clin. Pharmacol. Ther. 2005, 77, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Larsen, T.B.; Lassen, J.F.; Dahler-Eriksen, B.S.; Petersen, P.H.; Brandslund, I. Effect of anticoagulant therapy on the hypercoagulable state in patients carrying the factor V Arg506Gln mutation. Thromb Res. 1998, 92, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Franchini, M.; Liumbruno, G.M.; Bonfanti, C.; Lippi, G. The evolution of anticoagulant therapy. Blood Transfus. 2016, 14, 175–184. [Google Scholar] [PubMed]
- Oduah, E.I.; Linhardt, R.J.; Sharfstein, S.T. Heparin: Past, Present, and Future. Pharmaceuticals 2016, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Alridha, A.M.A.; Al-Gburi, K.M.; Abbood, S.K. A review of pharmacogenetics of anticoagulant therapy: Heparins, rivaroxaban, apixaban, and dabigatran. Med. J. Babylon. 2023, 19, 332–340. [Google Scholar] [CrossRef]
- Vandell, A.G.; Lee, J.; Shi, M.; Rubets, I.; Brown, K.S.; Walker, J.R. An integrated pharmacokinetic/pharmacogenomic analysis of ABCB1 and SLCO1B1 polymorphisms on edoxaban exposure. Pharmacogenomics J. 2016, 18, 153–159. [Google Scholar] [CrossRef]
- Shnayder, N.A.; Petrova, M.M.; Shesternya, P.A.; Savinova, A.V.; Bochanova, E.N.; Zimnitskaya, O.V.; Pozhilenkova, E.A.; Nasyrova, R.F. Using Pharmacogenetics of Direct Oral Anticoagulants to Predict Changes in Their Pharmacokinetics and the Risk of Adverse Drug Reactions. Biomedicines 2021, 9, 451. [Google Scholar] [CrossRef]
- Merali, Z.; Ross, S.; Paré, G. The pharmacogenetics of carboxylesterases: CES1 and CES2 genetic variants and their clinical effect. Drug Metabol. Drug Interact. 2014, 29, 143–151. [Google Scholar] [CrossRef]
- Tseng, A.S.; Patel, R.D.; Quist, H.E.; Kekic, A.; Maddux, J.T.; Grilli, C.B.; Shamoun, F.E. Clinical Review of the Pharmacogenomics of Direct Oral Anticoagulants. Cardiovasc Drugs Ther. 2018, 32, 121–126. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCarley, S.C.; Murphy, D.A.; Thompson, J.; Shovlin, C.L. Pharmacogenomic Considerations for Anticoagulant Prescription in Patients with Hereditary Haemorrhagic Telangiectasia. J. Clin. Med. 2023, 12, 7710. https://doi.org/10.3390/jcm12247710
McCarley SC, Murphy DA, Thompson J, Shovlin CL. Pharmacogenomic Considerations for Anticoagulant Prescription in Patients with Hereditary Haemorrhagic Telangiectasia. Journal of Clinical Medicine. 2023; 12(24):7710. https://doi.org/10.3390/jcm12247710
Chicago/Turabian StyleMcCarley, Sarah C., Daniel A. Murphy, Jack Thompson, and Claire L. Shovlin. 2023. "Pharmacogenomic Considerations for Anticoagulant Prescription in Patients with Hereditary Haemorrhagic Telangiectasia" Journal of Clinical Medicine 12, no. 24: 7710. https://doi.org/10.3390/jcm12247710
APA StyleMcCarley, S. C., Murphy, D. A., Thompson, J., & Shovlin, C. L. (2023). Pharmacogenomic Considerations for Anticoagulant Prescription in Patients with Hereditary Haemorrhagic Telangiectasia. Journal of Clinical Medicine, 12(24), 7710. https://doi.org/10.3390/jcm12247710