
Distinct Immunophenotypic Features in Patients Affected by 22q11.2 Deletion Syndrome with Immune Dysregulation and Infectious Phenotype
 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Selection
2.2. Clinical Data Collection
2.3. Definition of the Study Subgroups
2.4. Flow Cytometric Analysis
2.5. Statistical Analysis
3. Results
3.1. Demographic and Clinical Features
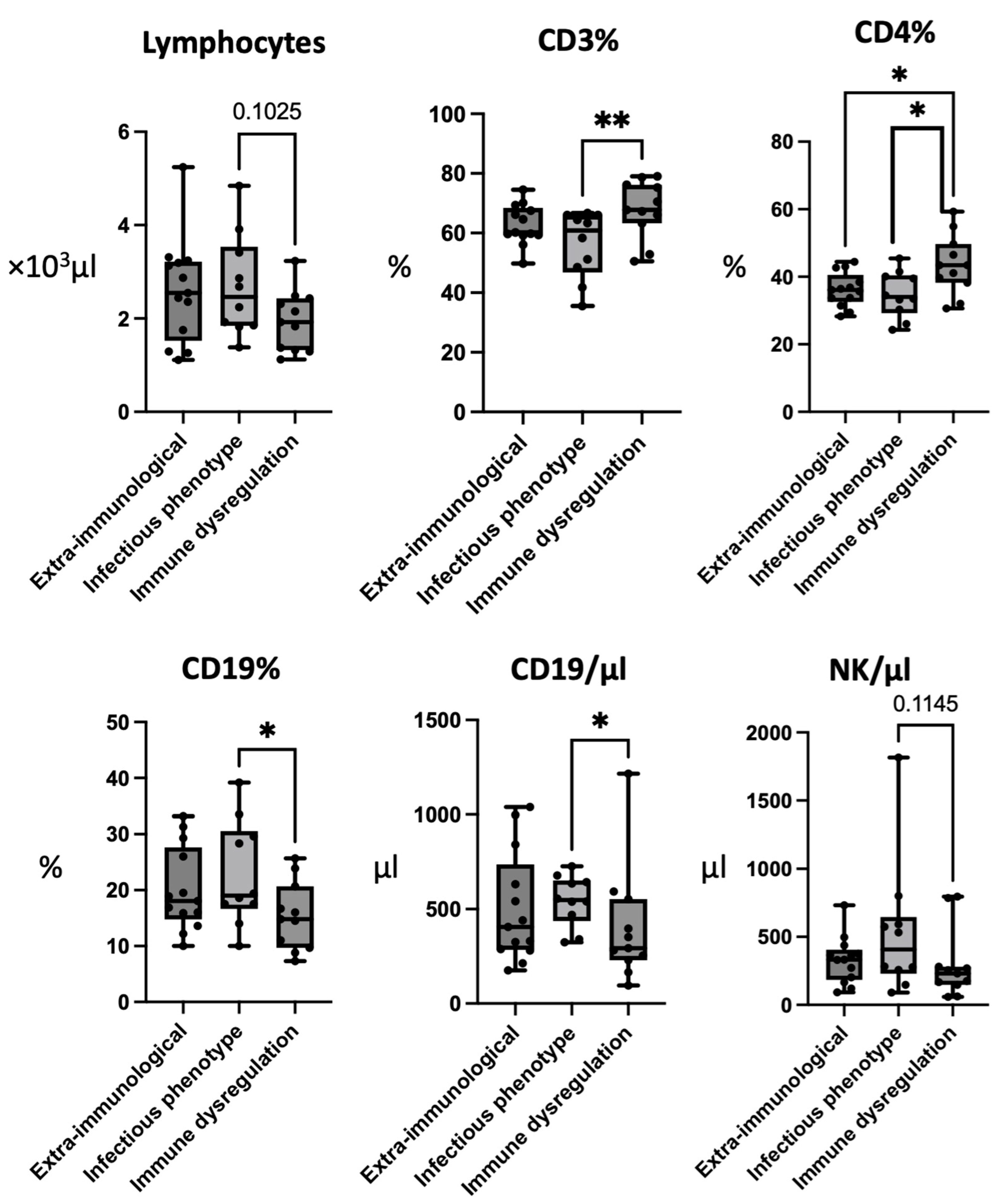
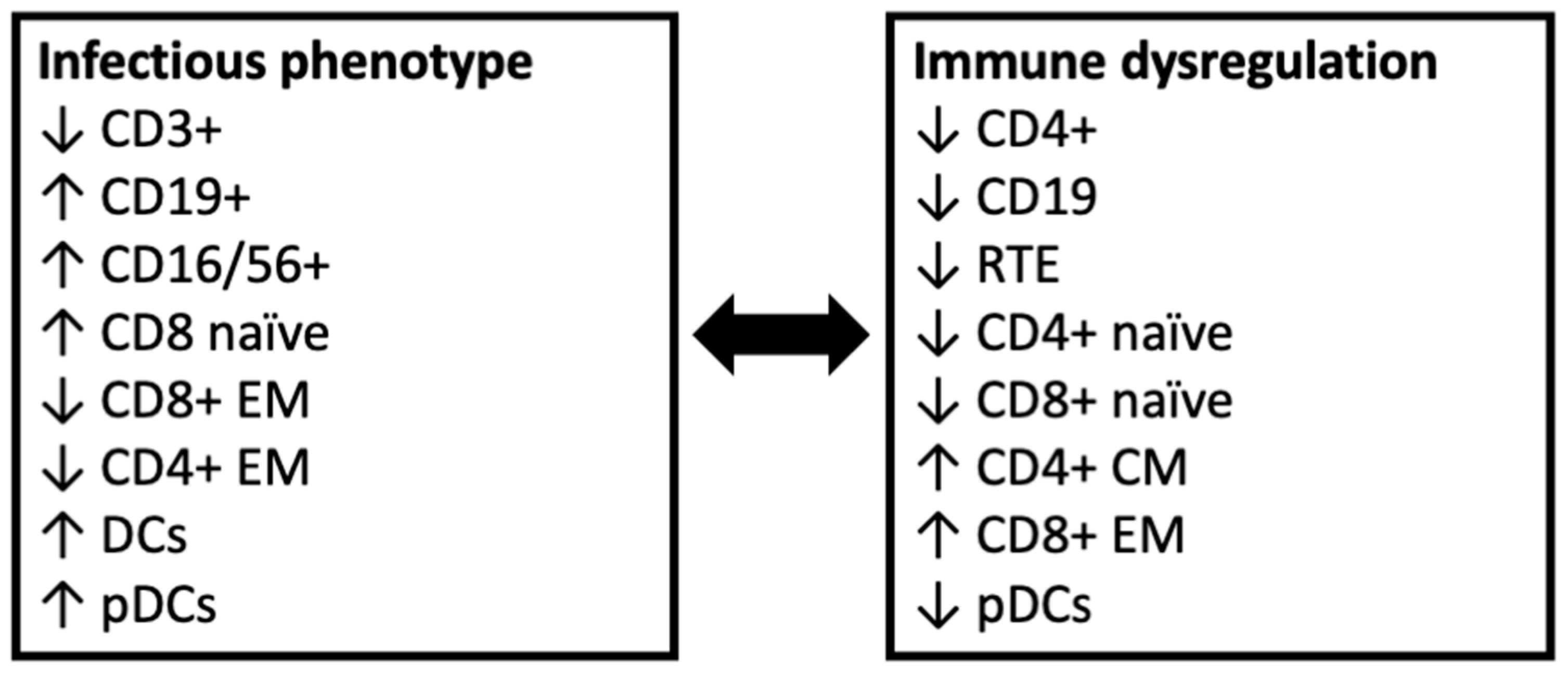
3.2. Patients with Infectious Phenotype Have Low CD3+ and CD4+ Cells and Elevated CD19+ Cells
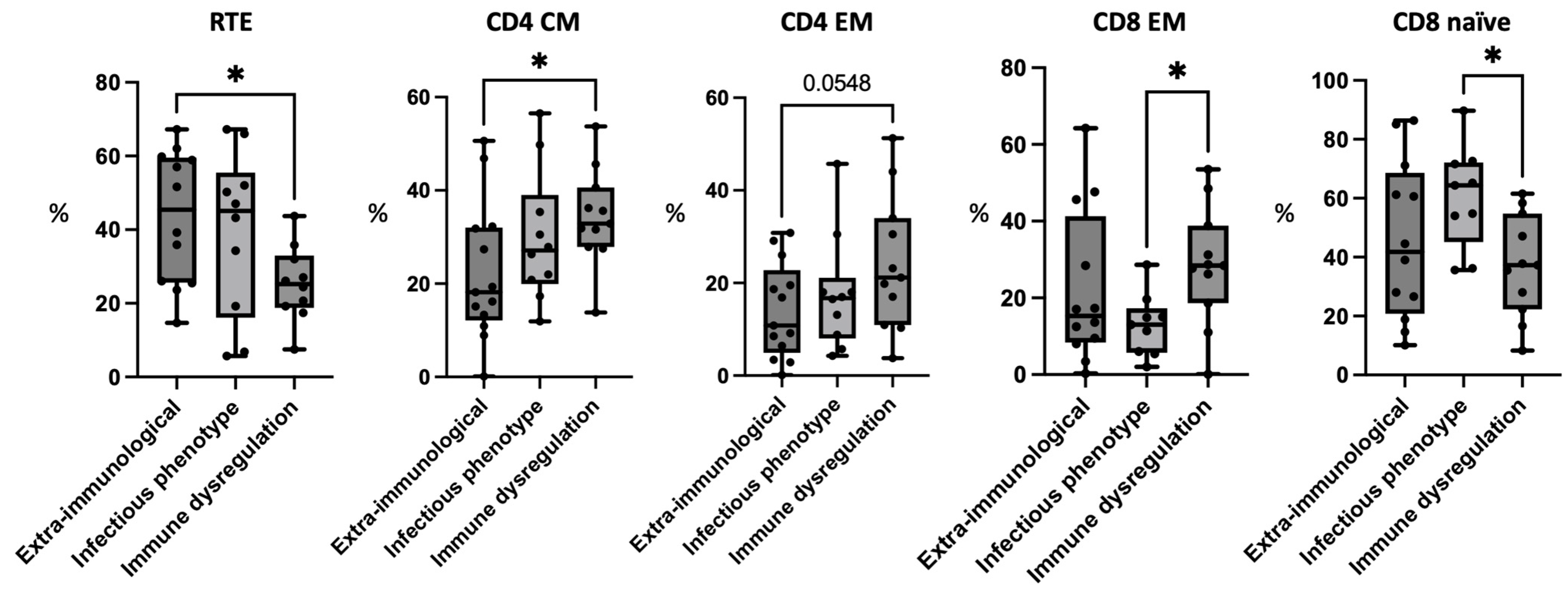
3.3. Patients with Immune Dysregulation Have Reduced Recent Thymic Emigrants
3.4. Patients with Immune Dysregulation Have Higher CD4+ Memory Cells
3.5. Patients with Immune Dysregulation Have Higher CD8+ Memory Cells and Patients with Infectious Phenotype Have High CD8+ Naïve Cells
3.6. Regulatory T Cells, Follicular T Cells, and B Cell Phenotyping
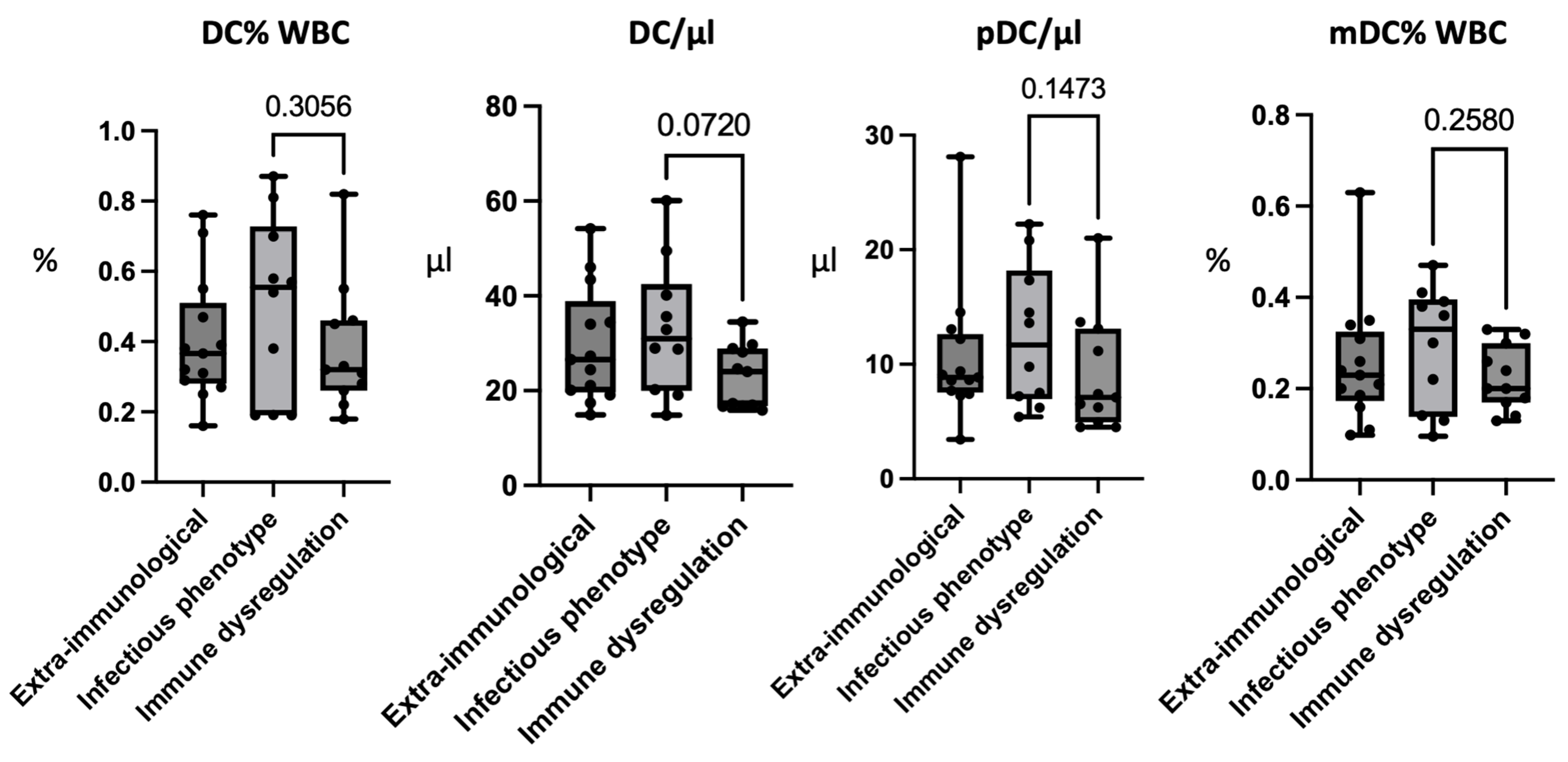
3.7. Patients with an Infectious Phenotype Have Higher Levels of Peripheral Dendritic Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Morrow, B.E.; McDonald-McGinn, D.M.; Emanuel, B.S.; Vermeesch, J.R.; Scambler, P.J. Molecular genetics of 22q11.2 deletion syndrome. Am. J. Med. Genet. Part A 2018, 176, 2070–2081. [Google Scholar] [CrossRef] [PubMed]
- Szczawińska-Popłonyk, A.; Schwartzmann, E.; Chmara, Z.; Głukowska, A.; Krysa, T.; Majchrzycki, M.; Olejnicki, M.; Ostrowska, P.; Babik, J. Chromosome 22q11.2 Deletion Syndrome: A Comprehensive Review of Molecular Genetics in the Context of Multidisciplinary Clinical Approach. Int. J. Mol. Sci. 2023, 24, 8317. [Google Scholar] [CrossRef] [PubMed]
- McDonald-McGinn, D.M.; Sullivan, K.E.; Marino, B.; Philip, N.; Swillen, A.; Vorstman, J.A.S.; Zackai, E.H.; Emanuel, B.S.; Vermeesch, J.R.; Morrow, B.E.; et al. 22q11.2 deletion syndrome. Nat. Rev. Dis. Primers 2015, 1, 15071. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, A.; Lioncino, M.; Maratea, A.; Passariello, A.; Fusco, A.; Fratta, F.; Monda, E.; Caiazza, M.; Signore, G.; Esposito, A.; et al. Clinical Manifestations of 22q11.2 Deletion Syndrome. Heart Fail. Clin. 2022, 18, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Mahé, P.; Nagot, N.; Portales, P.; Lozano, C.; Vincent, T.; Sarda, P.; Perez, M.; Amedro, P.; Marin, G.; Jeziorski, E. Risk factors of clinical dysimmune manifestations in a cohort of 86 children with 22q11.2 deletion syndrome: A retrospective study in France. Am. J. Med. Genet. Part A 2019, 179, 2207–2213. [Google Scholar] [CrossRef] [PubMed]
- Ricci, S.; Masini, M.; Valleriani, C.; Casini, A.; Cortimiglia, M.; Grisotto, L.; Canessa, C.; Indolfi, G.; Lippi, F.; Azzari, C. Reduced frequency of peripheral CD4 + CD45RA + CD31 + cells and autoimmunity phenomena in patients affected by Del22q11 syndrome. Clin. Immunol. 2018, 188, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Di Cesare, S.; Puliafito, P.; Ariganello, P.; Marcovecchio, G.E.; Mandolesi, M.; Capolino, R.; Digilio, M.C.; Aiuti, A.; Rossi, P.; Cancrini, C. Autoimmunity and regulatory T cells in 22q11.2 deletion syndrome patients. Pediatr. Allergy Immunol. 2015, 26, 591–594. [Google Scholar] [CrossRef]
- Chiappini, E.; Santamaria, F.; Marseglia, G.L.; Marchisio, P.; Galli, L.; Cutrera, R.; de Martino, M.; Antonini, S.; Becherucci, P.; Biasci, P.; et al. Prevention of recurrent respiratory infections: Inter-society Consensus. Ital. J. Pediatr. 2021, 47, 211. [Google Scholar] [CrossRef]
- Cai, T. Recurrent uncomplicated urinary tract infections: Definitions and risk factors. GMS Infect. Dis. 2021, 9, Doc03. [Google Scholar] [CrossRef]
- Eichenfield, L.F.; Tom, W.L.; Chamlin, S.L.; Feldman, S.R.; Hanifin, J.M.; Simpson, E.L.; Berger, T.G.; Bergman, J.N.; Cohen, D.E.; Cooper, K.D.; et al. Guidelines of care for the management of atopic dermatitis: Section 1. Diagnosis and assessment of atopic dermatitis. J. Am. Acad. Dermatol. 2014, 70, 338–351. [Google Scholar] [CrossRef]
- Orsini, G.; Legitimo, A.; Failli, A.; Massei, F.; Biver, P.; Consolini, R. Enumeration of human peripheral blood dendritic cells throughout the life. Int. Immunol. 2012, 24, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Marcovecchio, G.E.; Bortolomai, I.; Ferrua, F.; Fontana, E.; Imberti, L.; Conforti, E.; Amodio, D.; Bergante, S.; Macchiarulo, G.; D’Oria, V.; et al. Thymic Epithelium Abnormalities in DiGeorge and Down Syndrome Patients Contribute to Dysregulation in T Cell Development. Front. Immunol. 2019, 10, 447. [Google Scholar] [CrossRef] [PubMed]
- McLean-Tooke, A.; Barge, D.; Spickett, G.P.; Gennery, A.R. Flow cytometric analysis of TCR Vβ repertoire in patients with 22q11.2 deletion syndrome. Scand. J. Immunol. 2011, 73, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Gennery, A.R.; Barge, D.; O’Sullivan, J.J.; Flood, T.J.; Abinun, M.; Cant, A.J. Antibody deficiency and autoimmunity in 22q11.2 deletion syndrome. Arch. Dis. Child. 2002, 86, 422–425. [Google Scholar] [CrossRef] [PubMed]
- Derfalvi, B.; Maurer, K.; McGinn, D.M.M.; Zackai, E.; Meng, W.; Prak, E.T.L.; Sullivan, K.E. B cell development in chromosome 22q11.2 deletion syndrome. Clin. Immunol. 2016, 163, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Costagliola, G.; Cappelli, S.; Consolini, R. Autoimmunity in Primary Immunodeficiency Disorders: An Updated Review on Pathogenic and Clinical Implications. J. Clin. Med. 2021, 10, 4729. [Google Scholar] [CrossRef] [PubMed]
- Giardino, G.; Gallo, V.; Prencipe, R.; Gaudino, G.; Romano, R.; De Cataldis, M.; Lorello, P.; Palamaro, L.; Di Giacomo, C.; Capalbo, D.; et al. Unbalanced Immune System: Immunodeficiencies and Autoimmunity. Front. Pediatr. 2016, 4, 107. [Google Scholar] [CrossRef]
- Morsheimer, M.; Brown Whitehorn, T.F.; Heimall, J.; Sullivan, K.E. The immune deficiency of chromosome 22q11.2 deletion syndrome. Am. J. Med. Genet. Part A 2017, 173, 2366–2372. [Google Scholar] [CrossRef]
- Amaya-Uribe, L.; Rojas, M.; Azizi, G.; Anaya, J.-M.; Gershwin, M.E. Primary immunodeficiency and autoimmunity: A comprehensive review. J. Autoimmun. 2019, 99, 52–72. [Google Scholar] [CrossRef]
- Thalhammer, J.; Kindle, G.; Nieters, A.; Rusch, S.; Seppänen, M.R.; Fischer, A.; Grimbacher, B.; Edgar, D.; Buckland, M.; Mahlaoui, N.; et al. Initial presenting manifestations in 16,486 patients with inborn errors of immunity include infections and noninfectious manifestations. J. Allergy Clin. Immunol. 2021, 148, 1332–1341.e5. [Google Scholar] [CrossRef]
- Fischer, A.; Provot, J.; Jais, J.-P.; Alcais, A.; Mahlaoui, N.; Adoue, D.; Aladjidi, N.; Amoura, Z.; Arlet, P.; Armari-Alla, C.; et al. Autoimmune and inflammatory manifestations occur frequently in patients with primary immunodeficiencies. J. Allergy Clin. Immunol. 2017, 140, 1388–1393.e8. [Google Scholar] [CrossRef] [PubMed]
- McLean-Tooke, A.; Spickett, G.P.; Gennery, A.R. Immunodeficiency and autoimmunity in 22q11.2 deletion syndrome. Scand. J. Immunol. 2007, 66, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Tison, B.E.; Nicholas, S.K.; Abramson, S.L.; Hanson, I.C.; Paul, M.E.; Seeborg, F.O.; Shearer, W.T.; Perez, M.D.; Noroski, L.M.; Chinen, J. Autoimmunity in a cohort of 130 pediatric patients with partial DiGeorge syndrome. J. Allergy Clin. Immunol. 2011, 128, 1115–1117.e3. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, D.R.; Demirdag, Y.Y.; Marsh, R.A.; Sullivan, K.E.; Orange, J.S.; USIDNET Consortium. Relationship Between Severity of T Cell Lymphopenia and Immune Dysregulation in Patients with DiGeorge Syndrome (22q11.2 Deletions and/or Related TBX1 Mutations): A USIDNET Study. J. Clin. Immunol. 2021, 41, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Crowley, T.B.; Campbell, I.M.; Liebling, E.J.; Lambert, M.P.; Katz, L.E.L.; Heimall, J.; Bailey, A.; McGinn, D.E.; McGinn, D.M.M.; Sullivan, K.E. Distinct immune trajectories in patients with chromosome 22q11.2 deletion syndrome and immune-mediated diseases. J. Allergy Clin. Immunol. 2022, 149, 445–450. [Google Scholar] [CrossRef]
- Montin, D.; Marolda, A.; Licciardi, F.; Robasto, F.; Di Cesare, S.; Ricotti, E.; Ferro, F.; Scaioli, G.; Giancotta, C.; Amodio, D.; et al. Immunophenotype Anomalies Predict the Development of Autoimmune Cytopenia in 22q11.2 Deletion Syndrome. J. Allergy Clin. Immunol. Pract. 2019, 7, 2369–2376. [Google Scholar] [CrossRef]
- Giardino, G.; Radwan, N.; Koletsi, P.; Morrogh, D.M.; Adams, S.; Ip, W.; Worth, A.; Jones, A.; Meyer-Parsonson, I.; Gaspar, H.B.; et al. Clinical and immunological features in a cohort of patients with partial DiGeorge syndrome followed at a single center. Blood 2019, 133, 2586–2596. [Google Scholar] [CrossRef]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef]
- Amodio, G.; Gregori, S. Dendritic cells a double-edge sword in autoimmune responses. Front. Immunol. 2012, 3, 233. [Google Scholar] [CrossRef]
- Kassianos, A.J.; Hardy, M.Y.; Ju, X.; Vijayan, D.; Ding, Y.; Vulink, A.J.E.; McDonald, K.J.; Jongbloed, S.L.; Wadley, R.B.; Wells, C.; et al. Human CD1c (BDCA-1) + myeloid dendritic cells secrete IL-10 and display an immuno-regulatory phenotype and function in response to Escherichia coli. Eur. J. Immunol. 2012, 42, 1512–1522. [Google Scholar] [CrossRef]
- Macri, C.; Pang, E.S.; Patton, T.; O’Keeffe, M. Dendritic cell subsets. Semin. Cell Dev. Biol. 2018, 84, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Bigley, V.; Cytlak, U.; Collin, M. Human dendritic cell immunodeficiencies. Semin. Cell Dev. Biol. 2019, 86, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Mathan, T.S.M.M.; Figdor, C.G.; Buschow, S.I. Human plasmacytoid dendritic cells: From molecules to intercellular communication network. Front. Immunol. 2013, 4, 372. [Google Scholar] [CrossRef] [PubMed]
- Montoya, M.; Schiavoni, G.; Mattei, F.; Gresser, I.; Belardelli, F.; Borrow, P.; Tough, D.F. Type I interferons produced by dendritic cells promote their phenotypic and functional activation. Blood 2002, 99, 3263–3271. [Google Scholar] [CrossRef]
- Legitimo, A.; Bertini, V.; Costagliola, G.; Baroncelli, G.I.; Morganti, R.; Valetto, A.; Consolini, R. Vitamin D status and the immune assessment in 22q11.2 deletion syndrome. Clin. Exp. Immunol. 2020, 200, 272–286. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| G1 | G2 | G3 | |
|---|---|---|---|
| Age (years) | 13.61 ± 10.71 | 10.88 ± 9.31 | 18.35 ± 11.26 |
| Congenital heart disease requiring cardiosurgery | 3/13 | 3/10 | 4/11 |
| Hypoparathyroidism/hypocalcemia | 4/13 | 6/10 | 7/11 |
| Neuropsychiatric involvement | 8/13 | 8/10 | 7/11 |
| Velo-pharyngeal insufficiency or ear/nose abnormalities | 4/13 | 3/10 | 2/11 |
| Other relevant malformations (kidney, gastrointestinal, limbs) | 3/13 | 3/10 | 4/11 |
| Allergic disorders | 2/13 | 2/10 | 2/11 |
| Neoplasia | 0/13 | 0/10 | 1/11 |
| G1 | G2 | G3 | ||
|---|---|---|---|---|
| Lymphocytes | /μL | 2600 ± 1120 (1110–5240) | 2690 ± 1080 (1380–4840) | 1950 ± 650 (1120–3230) |
| CD3 | % | 62.81 ± 6.67 (49.8–74.5) | 56.42 ± 10.84 (41.8–66.7) | 67.96 ± 9.61 (50.5–78.7) |
| /μL | 1634.0 ± 717.52 (753–3102) | 1552.3 ± 708.96 (577–2608) | 1323.82 ± 512.67 (753–2552) | |
| CD4 | % | 36.16 ± 5.04 (28.3–44.4) | 32.91 ± 6.19 (24.3–41.5) | 43.53 ± 8.80 (32–59.3) |
| /μL | 954.01 ± 463.17 (326–1907) | 923.4 ± 474.49 (335–1621) | 851.0 ± 394.76 (486–1915) | |
| CD8 | % | 18.58 ± 4.84 (10.2–27) | 15.91 ± 5.29 (11–24.7) | 20.17 ± 5.66 (14.3–32.6) |
| /μL | 455.0 ± 211.32 (167–894) | 436.6 ± 232.72 (152–767) | 390.36 ± 163.62 (205–753) | |
| CD19 | % | 20.07 ± 7.49 (10–33.2) | 22.77 ± 9.35 (10–39.2) | 15.37± 6.09 (7.3–25.7) |
| /μL | 500.77 ± 292.66 (175–998) | 545.2 ± 135.36 (341–727) | 402.73 ± 308.26 (96–1215) | |
| CD16/56 | % | 13.98 ± 6.86 (5.2–30) | 18.06 ± 10.62 (6.6–37.5) | 15.41 ± 11.25 (2.5–32.3) |
| /μL | 329.69 ± 171.29 (92–732) | 537.2 ± 501.97 (91–1815) | 293.36 ± 256.92 (59–796) | |
| CD4 | ||||
| RTEs a | 43.45 ± 18.06 (14.7–67.2) | 39.16 ± 22.26 (5.7–67.2) | 25.39 ± 10.20 (7.5–43.7) | |
| Naïve a | 53.18 ± 25.3(20–87.6) | 50.21 ± 25.03 (10–80.5) | 38.45 ± 18.94 (12–69.5) | |
| TEMRA a | 10.4 ± 11.97(0.3–36.2) | 2.11 ± 2.19 (0.3–7.3) | 3.05 ± 4.62 (0.4–16) | |
| CM a | 22.38 ± 14.78 (0.1–50.6) | 29.84 ± 14.06 (11.9–56.5) | 34.3 ± 10.35 (13.8–53.7) | |
| EM a | 14.02 ± 10.27 (0.1–30.8) | 17.76 ± 12.36 (5.7–45.7) | 24.18 ± 14.59 (3.8–51.3) | |
| CD8 | ||||
| Naïve b | 45.51 ± 26.96 (10.1–86.4) | 60.45 ± 17.48 (35.6–89.7) | 37.05 ± 17.37 (8.3–61.5) | |
| TEMRA b | 19.69 ± 15.4 (5.1–56.5) | 14.39 ± 7.08 (6.7–30.3) | 19.6 ± 15.48 (0.1–54) | |
| CM b | 9.98 ± 7.73 (0.9–26.7) | 12.27 ± 8.78 (1.2–28.5) | 16.5 ± 10.02 (1.5–38.3) | |
| EM b | 22.28 ± 20.02 (0.3–64.2) | 12.88 ± 8.1 (2–28.6) | 28.43 ± 15.35 (0.1–53.5) | |
| Tregs a | 7.04 ± 2.69 (4.3–11.4) | 7.55 ± 1.69 (4.9–15) | 6.31 ± 3.21 (2.8–10.3) | |
| TfH c | 31.51 ± 8.1 (19.5–42.4) | 33.36 ± 6.89 (23.8–45.7) | 26.81 ± 5.57 (19.1–35.4) | |
| CD19 | ||||
| Naïve d | 79.91± 7.42 (66.6–92) | 78.32 ± 5.67 (69.5–88.2) | 76.74 ± 9.07 (60.1–88.2) | |
| Switched memory d | 6.64 ± 4.74 (2.3–15) | 7.58 ± 4.0 (1.1–14) | 6.05 ± 3.39 (2.5–13.74) | |
| Transitional d | 0.92 ± 0.57 (0.3–1.9) | 0.72 ± 0.75 (0.05–2.17) | 2.07 ± 3.76 (0.1–9.7) | |
| CD21low d | 6.16 ± 2.75 (3.4–10.3) | 8.04 ± 3.81 (3.4–14.2) | 5.71 ± 2.73 (2.3–10.5) | |
| DC e | 0.4 ± 0.18 (0.27–0.76) | 0.50 ± 0.26 (0.19–0.87) | 0.38 ± 0.18 (0.18–0.82) | |
| DC | /μL | 29.45 ± 12.2 (14.9–54.2) | 33.02 ± 14.01 (14.8–60.1) | 23.05 ± 6.63 (15.89–34.5) |
| pDC e | 0.15 ± 0.08 (0.06–0.37) | 0.21 ± 0.14 (0.05–0.48) | 0.16 ± 0.13 (0.05–0.25) | |
| pDC | /μL | 10.64 ± 5.96 (3.43–28.11) | 12.46 ± 6.17 (5.41–22.23) | 9.11 ± 5.14 (4.5–13.7) |
| mDC e | 0.26 ± 0.14 (0.098–0.63) | 0.29 ± 0.13 (0.095–0.41) | 0.22 ± 0.07 (0.13–0.33) | |
| mDC | /μL | 18.82 ± 8.89 (6.3–38.32) | 19.01 ± 10.99 (7.3–45.6) | 14.98 ± 4.48 (8.78–23.17) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costagliola, G.; Legitimo, A.; Bertini, V.; Alberio, A.M.Q.; Valetto, A.; Consolini, R. Distinct Immunophenotypic Features in Patients Affected by 22q11.2 Deletion Syndrome with Immune Dysregulation and Infectious Phenotype. J. Clin. Med. 2023, 12, 7579. https://doi.org/10.3390/jcm12247579
Costagliola G, Legitimo A, Bertini V, Alberio AMQ, Valetto A, Consolini R. Distinct Immunophenotypic Features in Patients Affected by 22q11.2 Deletion Syndrome with Immune Dysregulation and Infectious Phenotype. Journal of Clinical Medicine. 2023; 12(24):7579. https://doi.org/10.3390/jcm12247579
Chicago/Turabian StyleCostagliola, Giorgio, Annalisa Legitimo, Veronica Bertini, Antonino Maria Quintilio Alberio, Angelo Valetto, and Rita Consolini. 2023. "Distinct Immunophenotypic Features in Patients Affected by 22q11.2 Deletion Syndrome with Immune Dysregulation and Infectious Phenotype" Journal of Clinical Medicine 12, no. 24: 7579. https://doi.org/10.3390/jcm12247579
APA StyleCostagliola, G., Legitimo, A., Bertini, V., Alberio, A. M. Q., Valetto, A., & Consolini, R. (2023). Distinct Immunophenotypic Features in Patients Affected by 22q11.2 Deletion Syndrome with Immune Dysregulation and Infectious Phenotype. Journal of Clinical Medicine, 12(24), 7579. https://doi.org/10.3390/jcm12247579