
DR5 Up-Regulation Induced by Dichloroacetate Sensitizes Tumor Cells to Lipid Nanoparticles Decorated with TRAIL




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Cell Culture
2.2. Formation of Lipid Nanoparticles Anchored with Soluble Recombinant TRAIL
2.3. Cell Incubation with Drugs
2.4. Flow Cytometry Assays
2.5. Cell Growth Assay
2.6. Western-Blot Experiments
2.7. Statistical Analysis
3. Results
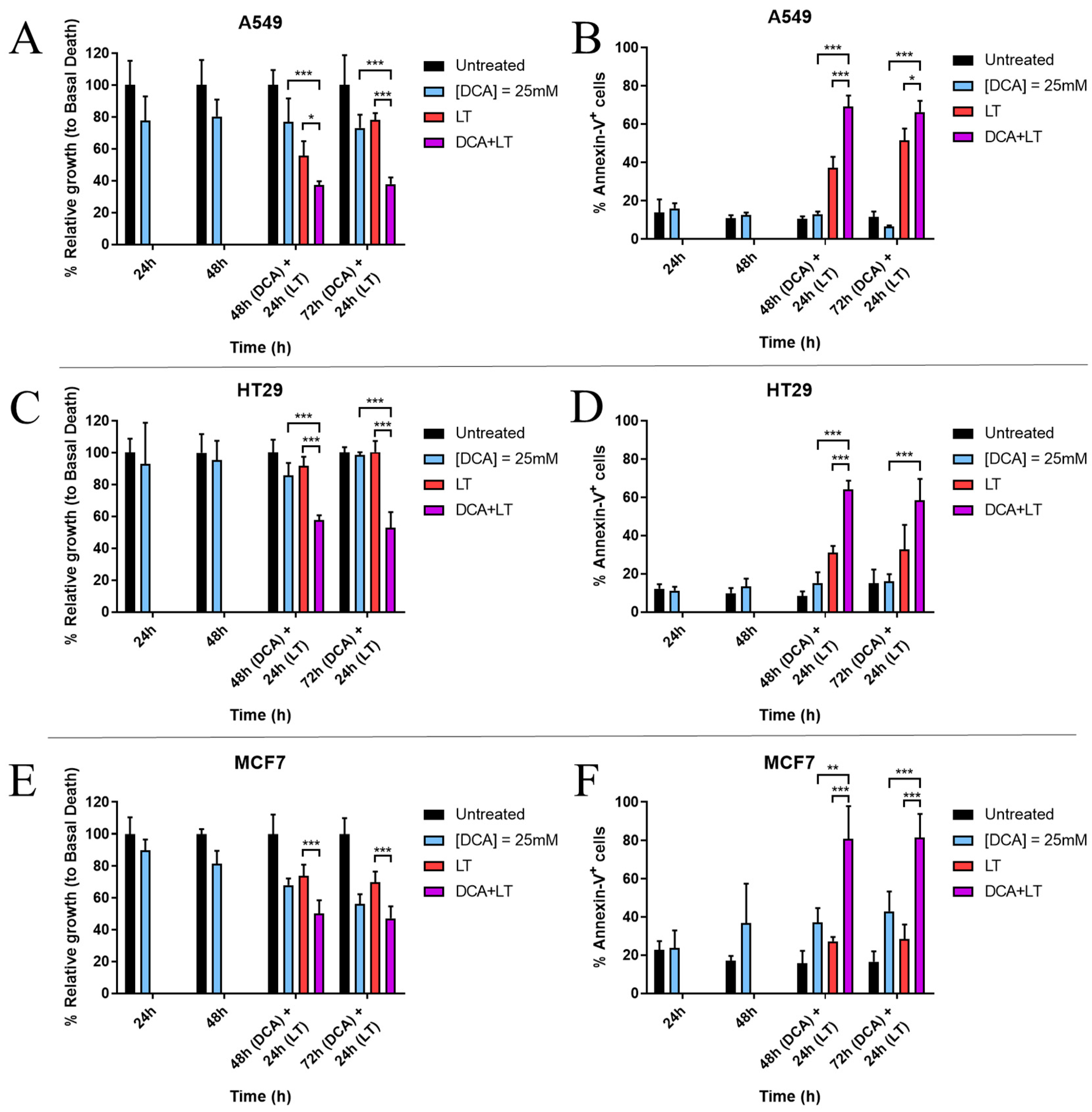
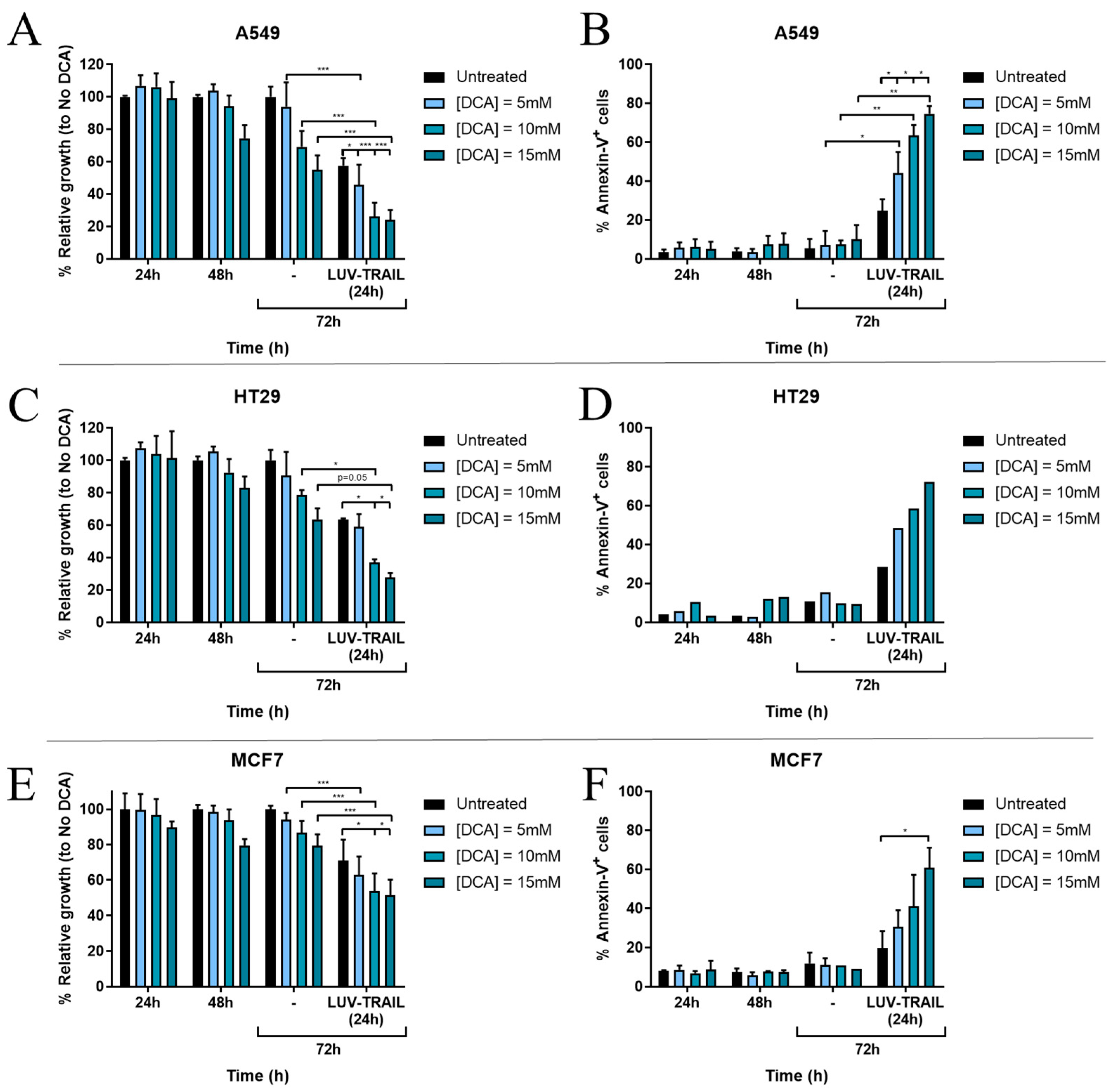
3.1. DCA Sensitizes Tumor Cells to LUV-TRAIL Treatment
3.2. DCA Synergizes with LUV-TRAIL at Lower Doses
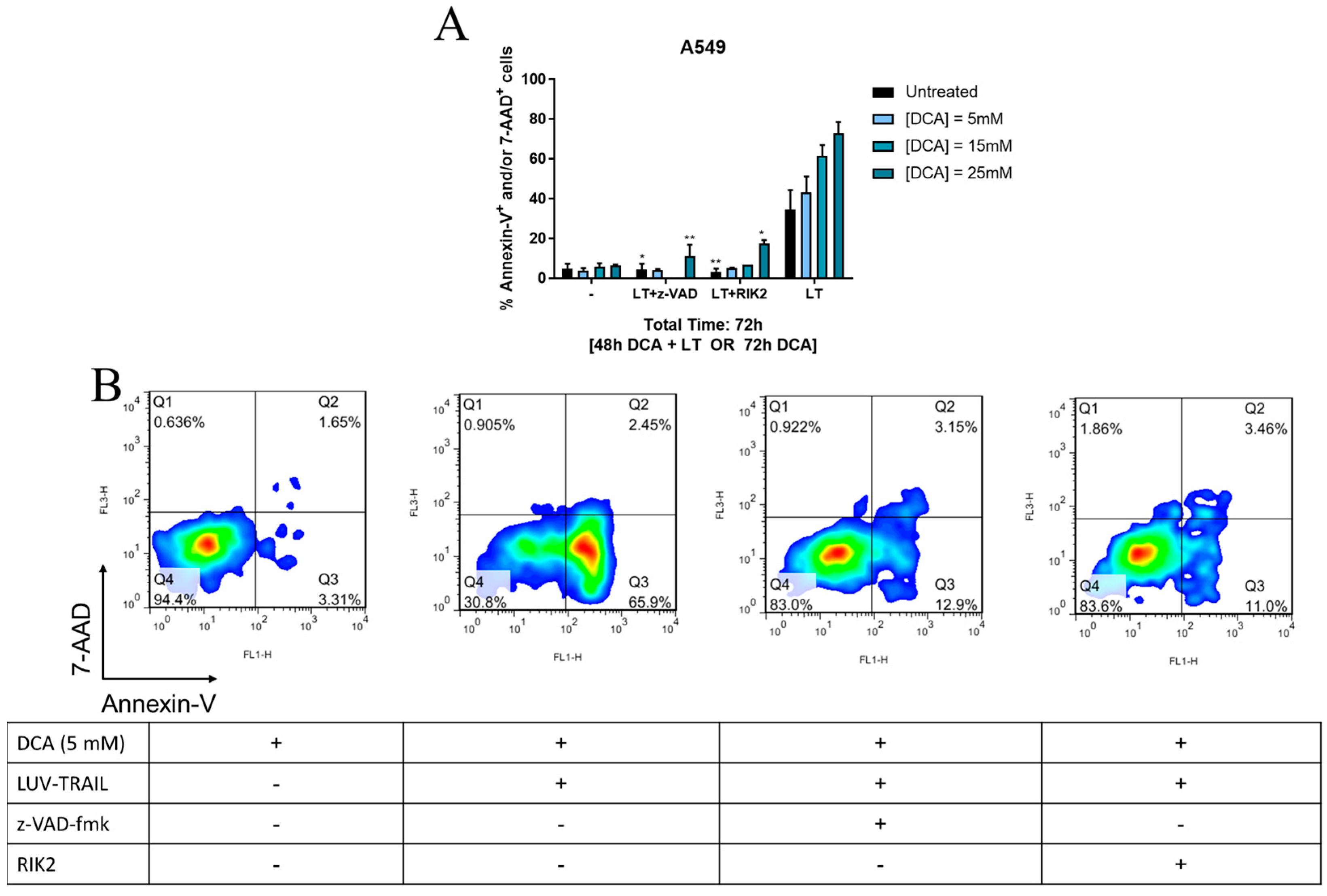
3.3. DCA + LUV-TRAIL Induce Apoptosis in A549 Cells
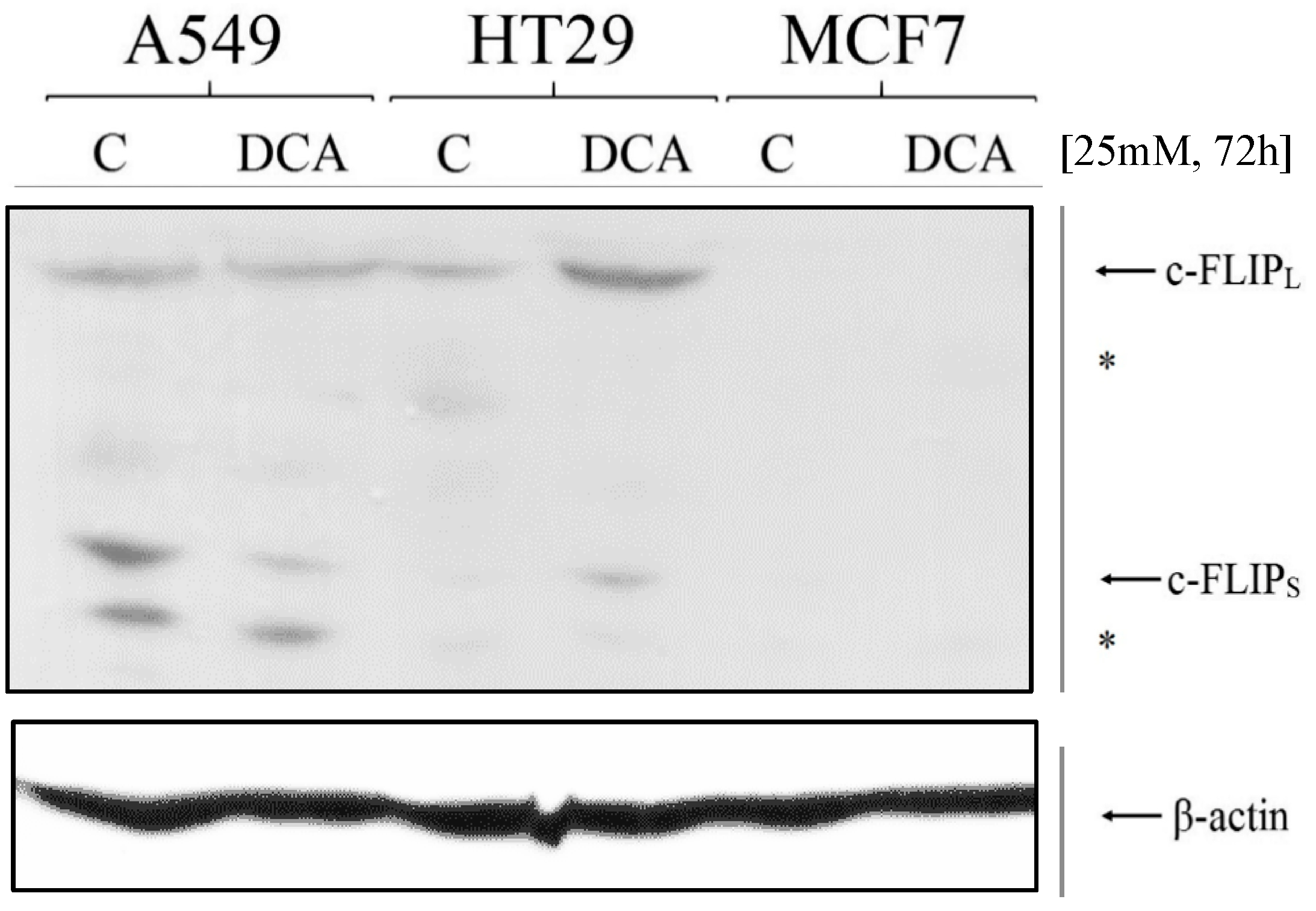
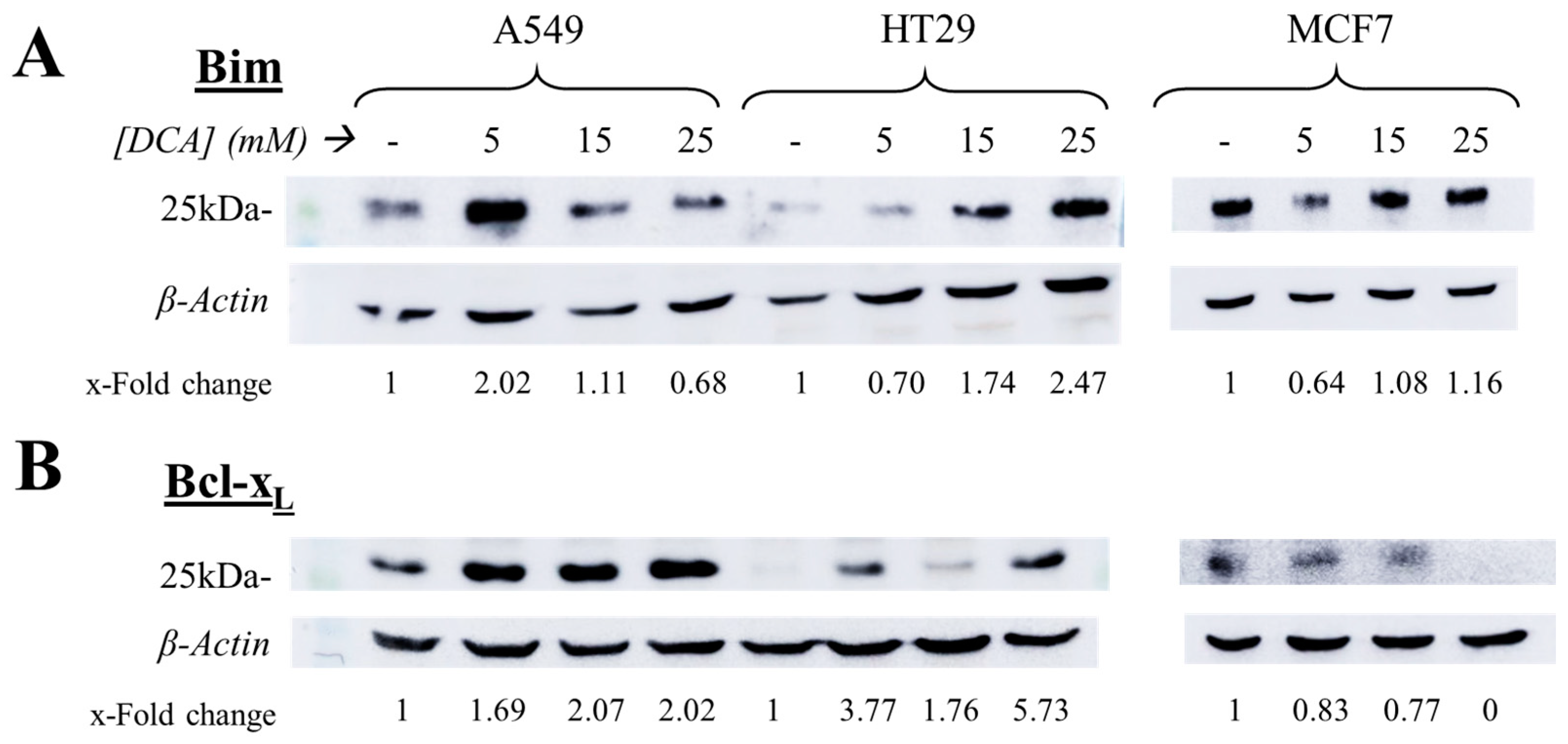
3.4. Expression of c-FLIP Is Altered in a Cell-Dependent Manner after DCA Exposure
3.5. The Intrinsic Apoptotic Pathway Is Altered in a Cell-Dependent Manner on Tumor Cells after DCA Exposure
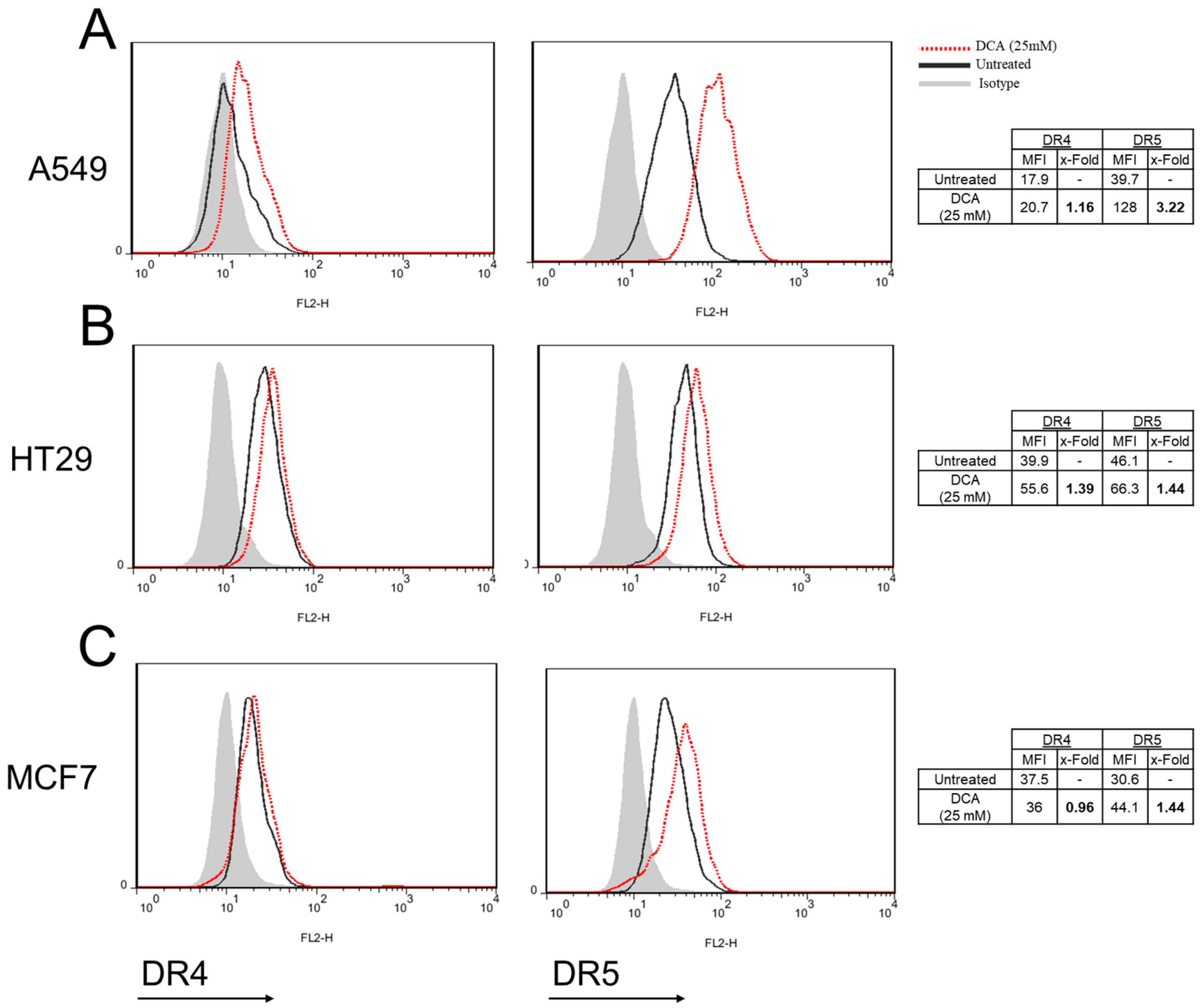
3.6. DCA Increases DR5 Surface Expression in Tumor Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- de Miguel, D.; Lemke, J.; Anel, A.; Walczak, H.; Martinez-Lostao, L. Onto better TRAILs for cancer treatment. Cell Death Differ. 2016, 23, 733–747. [Google Scholar] [CrossRef] [PubMed]
- Pitti, R.M.; Marsters, S.A.; Ruppert, S.; Donahue, C.J.; Moore, A.; Ashkenazi, A. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J. Biol. Chem. 1996, 271, 12687–12690. [Google Scholar] [CrossRef] [PubMed]
- Wiley, S.R.; Schooley, K.; Smolak, P.J.; Din, W.S.; Huang, C.P.; Nicholl, J.K.; Sutherland, G.R.; Smith, T.D.; Rauch, C.; Smith, C.A.; et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 1995, 3, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Jouan-Lanhouet, S.; Arshad, M.; Piquet-Pellorce, C.; Martin-Chouly, C.; Le Moigne-Muller, G.; Van Herreweghe, F.; Takahashi, N.; Sergent, O.; Lagadic-Gossmann, D.; Vandenabeele, P.; et al. TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ. 2012, 19, 2003–2014. [Google Scholar] [CrossRef]
- Lafont, E.; Kantari-Mimoun, C.; Draber, P.; De Miguel, D.; Hartwig, T.; Reichert, M.; Kupka, S.; Shimizu, Y.; Taraborrell, L.; Spit, M.; et al. The linear ubiquitin chain assembly complex regulates TRAIL-induced gene activation and cell death. EMBO J. 2017, 36, 1147–1166. [Google Scholar] [CrossRef] [PubMed]
- Almasan, A.; Ashkenazi, A. Apo2L/TRAIL: Apoptosis signaling, biology and potential for cance therapy. Cytokine Growth Factor Rev. 2003, 14, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.; Kim, M.; Choi, Y.; Sung, B.; Shin, N.; Shin, H.; Sung, Y.; Oh, B. 2.8 A resolution crystal structure of human TRAIL, a cytokine with selective antitumor activity. Immunity 1999, 11, 253–261. [Google Scholar] [CrossRef]
- Hymowitz, S.G.; Christinger, H.W.; Fuh, G.; Ultsch, M.; O’Connell, M.; Kelley, R.F.; Ashkenazi, A.; de Vos, A.M. Triggering cell death: The crystal structure of Apo2L/TRAIL in a complex with DR5. Mol. Cell 1999, 4, 563–571. [Google Scholar] [CrossRef]
- LeBlanc, H.N.; Ashkenazi, A. Apo2L/TRAIL and its death and decoy receptors. Cell Death Differ. 2003, 10, 66–75. [Google Scholar] [CrossRef]
- de Miguel, D.; Gallego-Lleyda, A.; Anel, A.; Martinez-Lostao, L. Liposome-bound TRAIL induces superior DR5 clustering and enhanced DISC recruitment in histiocytic lymphoma U937 cells. Leuk. Res. 2015, 39, 657–666. [Google Scholar] [CrossRef]
- van der Sloot, A.; Tur, V.; Szegezdi, E.; Mullally, M.; Cool, R.; Samali, A.; Serrano, L.; Quax, W. Designed tumor necrosis factor-related apoptosis-inducing ligand variants initiating apoptosis exclusively via the DR5 receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 8634–8639. [Google Scholar] [CrossRef] [PubMed]
- Bosque, A.; Pardo, J.; Martínez-Lorenzo, M.J.; Iturralde, M.; Marzo, I.; Piñeiro, A.; Alava, M.A.; Naval, J.; Anel, A. Down-regulation of normal human T cell blast activation: Roles of APO2L/TRAIL, FasL and c-FLIP, Bim or Bcl-x isoform expression. J. Leukoc. Biol. 2005, 77, 568–578. [Google Scholar] [CrossRef] [PubMed]
- Safa, A. Roles of c-FLIP in Apoptosis, Necroptosis, and Autophagy. J. Carcinog. Mutagen. 2013, 2013, 003. [Google Scholar] [CrossRef] [PubMed]
- Yamada, H.; Tada-Oikawa, S.; Uchida, A.; Kawanishi, S. TRAIL causes cleavage of bid by caspase-8 and loss of mitochondrial membrane potential resulting in apoptosis in BJAB cells. Biochem. Biophys. Res. Commun. 1999, 265, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Marzo, I.; Naval, J. Bcl-2 family members as molecular targets in cancer therapy. Biochem. Pharmacol. 2008, 76, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Walczak, H.; Miller, R.E.; Ariail, K.; Gliniak, B.; Griffith, T.S.; Kubin, M.; Chin, W.; Jones, J.; Woodward, A.; Le, T.; et al. Tumoricidal activity of TRAIL in vivo. Nat. Med. 1999, 5, 157–163. [Google Scholar] [CrossRef]
- Lemke, J.; von Karstedt, S.; Zinngrebe, J.; Walczak, H. Getting TRAIL back on track for cancer therapy. Cell Death Differ. 2014, 21, 1350–1364. [Google Scholar] [CrossRef]
- Chan, F.K.M. Three is better than one: Pre-ligand receptor assembly in the regulation of TNF receptor signaling. Cytokine 2007, 37, 101–107. [Google Scholar] [CrossRef]
- Anel, A.; Gallego-Lleyda, A.; de Miguel, D.; Naval, J.; Martinez-Lostao, L. Role of Exosomes in the Regulation of T-cell Mediated Immune Responses and in Autoimmune Disease. Cells 2019, 8, 154. [Google Scholar] [CrossRef]
- Martínez-Lorenzo, M.J.; Anel, A.; Gamen, S.; Monleón, I.; Lasierra, P.; Larrad, L.; Piñeiro, A.; Alava, M.A.; Naval, J. Activated human T cells release bioactive Fas ligand and APO2 ligand in microvesicles. J. Immunol. 1999, 163, 1274–1281. [Google Scholar] [CrossRef]
- de Miguel, D.; Basáñez, G.; Sánchez, D.; Galán, P.; Marzo, I.; Larrad, L.; Naval, J.; Pardo, J.; Anel, A.; Martínez-Lostao, L. Thethering Apo2L/TRAIL to liposomes overcomes chemoresistance of human hematological tumor cells. Mol. Pharm. 2013, 10, 893–904. [Google Scholar] [CrossRef] [PubMed]
- de Miguel, D.; Gallego-Lleyda, A.; Ayuso, J.; Pawlak, A.; Conde, B.; Ochoa, I.; Fernández, L.; Anel, A.; Martinez-Lostao, L. Improved Anti-Tumor Activity of Novel Highly Bioactive Liposome-Bound TRAIL in Breast Cancer Cells. Recent Pat. Anti-Cancer Drug Discov. 2016, 11, 197–214. [Google Scholar] [CrossRef] [PubMed]
- de Miguel, D.; Gallego-Lleyda, A.; Ayuso, J.; Pejenaute-Ochoa, D.; Jarauta, V.; Marzo, I.; Fernández, L.; Ochoa, I.; Conde, B.; Anel, A.; et al. High-order TRAIL oligomer formation in TRAIL-coated lipid nanoparticles enhances DR5 cross-linking and increases antitumour effect against colon cancer. Cancer Lett. 2016, 383, 250–260. [Google Scholar] [CrossRef] [PubMed]
- de Miguel, D.; Gallego-Lleyda, A.; Galan-Malo, P.; Rodriguez-Vigil, C.; Marzo, I.; Anel, A.; Martinez-Lostao, L. Immunotherapy with liposome-bound TRAIL overcomes partial protection to soluble TRAIL-induced apoptosis offered by down-regulation of Bim in leukemic cells. Clin. Transl. Oncol. 2015, 17, 657–667. [Google Scholar] [CrossRef] [PubMed]
- de Miguel, D.; Gallego-Lleyda, A.; Martinez-Ara, M.; Plou, J.; Anel, A.; Martinez-Lostao, L. Double-Edged Lipid Nanoparticles Combining Liposome-Bound TRAIL and Encapsulated Doxorubicin Showing an Extraordinary Synergistic Pro-Apoptotic Potential. Cancers 2019, 11, 1948. [Google Scholar] [CrossRef]
- Gallego-Lleyda, A.; De Miguel, D.; Anel, A.; Martinez-Lostao, L. Lipid Nanoparticles Decorated with TNF-Related Aptosis-Inducing Ligand (TRAIL) Are More Cytotoxic than Soluble Recombinant TRAIL in Sarcoma. Int. J. Mol. Sci. 2018, 19, 1449. [Google Scholar] [CrossRef] [PubMed]
- Balsas, P.; López-Royuela, N.; Galan-Malo, P.; Anel, A.; Marzo, I.; Naval, J. Cooperation between Apo2L/TRAIL and bortezomib in multiple myeloma apoptosis. Biochem. Pharmacol. 2009, 77, 804–812. [Google Scholar] [CrossRef]
- Bui, H.; Le, N.; Le, Q.; Kim, S.; Lee, S.; Kang, D. Synergistic apoptosis of human gastric cancer cells by bortezomib and TRAIL. Int. J. Mol. Sci. 2019, 16, 1412–1423. [Google Scholar] [CrossRef]
- Kretz, A.; Trauzold, A.; Hillenbrand, A.; Knippschild, U.; Henne-Bruns, D.; von Karstedt, S.; Lemke, J. TRAILblazing Strategies for Cancer Treatment. Cancers 2019, 30, 456. [Google Scholar] [CrossRef]
- Fulda, S. Smac Mimetics to Therapeutically Target IAP Proteins in Cancer. Int. Rev. Cell Mol. Biol. 2017, 330, 157–169. [Google Scholar]
- Mauro-Lizcano, M.; López-Rivas, A. Glutamine metabolism regulates FLIP expression and sensitivity to TRAIL in triple-negative breast cancer cells. Cell Death Dis. 2018, 9, 205. [Google Scholar] [CrossRef] [PubMed]
- Strekalova, E.; Malin, D.; Rajanala, H.; Cryns, V. Metformin sensitizes triple-negative breast cancer to proapoptotic TRAIL receptor agonists by suppressing XIAP expression. Breast Cancer Res. Treat. 2017, 163, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.; Deberardinis, R. Understanding the intersections between metabolism and cancer biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef]
- Bonnet, S.; Archer, S.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.; Lopaschuk, G.; Puttagunta, L.; Bonnet, S.; et al. A Mitochondria-K+ Channel Axis Is Suppressed in Cancer and Its Normalization Promotes Apoptosis and Inhibits Cancer Growth. Cancer Cell 2007, 11, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Chu, Q.; Sangha, R.; Spratlin, J.; Vos, L.; Mackey, J.; McEwan, A.; Venner, P.; Michelakis, E. A phase I open-labeled, single-arm, dose-escalation, study of dichloroacetate (DCA) in patients with advanced solid tumors. Investig. New Drugs 2015, 33, 603–610. [Google Scholar] [CrossRef]
- Dunbar, E.; Coats, B.; Shroads, A.; Langaee, T.; Lew, A.; Forder, J.; Shuster, J.; Wagner, D.; Stacpole, P. Phase 1 trial of dichloroacetate (DCA) in adults with recurrent brain tumors. Investig. New Drugs 2014, 32, 452–464. [Google Scholar] [CrossRef]
- Garon, E.; Christofk, H.; Hosmer, W.; Britten, C.; Bahng, A.; Crabtree, M.; Hong, C.; Kamranpour, N.; Pitts, S.; Kabbinavar, F.; et al. Dichloroacetate should be considered with platinum-based chemotherapy in hypoxic tumors rather than as a single agent in advanced non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 2014, 140, 443–452. [Google Scholar] [CrossRef]
- Marco-Brualla, J.; Al-Wasaby, S.; Soler, R.; Romanos, E.; Conde, B.; Justo-Méndez, R.; Enríquez, J.; Fernández-Silva, P.; Martínez-Lostao, L.; Villalba, M.; et al. Mutations in the ND2 subunit of mitochondrial complex I are sufficient to confer increased tumorigenic and metastatic potential to cancer cells. Cancers 2019, 11, 1027. [Google Scholar] [CrossRef]
- Stockwin, L.; Yu, S.; Borgel, S.; Hanckock, C.; Wolfe, T.; Phillips, L.; Hollingshead, M.; Newton, D. Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC. Int. J. Cancer 2010, 127, 2510–2519. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.; Park, J.; Kim, E.; Jang, H.; Kwon, M. Activation of mitochondrial oxidation by PDK2 inhibition reverses cisplatin resistance in head and neck cancer. Cancer Lett. 2016, 371, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Allende-Vega, N.; Krzywinska, E.; Orechioni, S.; López-Royuela, N.; Reggiani, F.; Talarico, G.; Rossi, J.; Rossignol, R.; Hicheri, Y.; Cartron, G.; et al. The presence of wild type p53 in hematological cancers improves the efficacy of combinational therapy targeting metabolism. Oncotarget 2015, 6, 19228–19245. [Google Scholar] [CrossRef] [PubMed]
- Voltan, R.; Rimondi, E.; Melloni, E.; Gilli, P.; Bertolasi, V.; Casciano, F.; Rigolin, G.; Zauli, G.; Secchiero, P. Metformin combined with sodium dichloroacetate promotes B leukemic cell death by suppressing anti-apoptotic protein Mcl-1. Oncotarget 2016, 7, 18965–18977. [Google Scholar] [CrossRef] [PubMed]
- Catalán, E.; Charni, S.; Jaime, P.; Aguiló, J.; Enríquez, J.; Naval, J.; Pardo, J.; Villalba, M.; Anel, A. MHC-I modulation due to changes in tumor cell metabolism regulates tumor sensitivity to CTL and NK cells. Oncoimmunology 2015, 4, e985924. [Google Scholar] [CrossRef]
- Belkahla, S.; Marco Brualla, J.; Fayd’herbe de Maudave, A.; Falvo, P.; Allende-Vega, N.; Constantinides, M.; Khan, A.; Coenon, L.; Alexia, C.; Mitola, G.; et al. The metabolism of cells regulates their sensitivity to NK cells depending on p53 status. Sci. Rep. 2022, 12, 3234. [Google Scholar] [CrossRef]
- Hong, S.; Kim, C.; An, S.; Kim, H.; Hwang, S.; Song, J.; Lee, J.; Hong, J.; Kim, J.; Noh, W.; et al. TRAIL restores DCA/metformin-mediated cell death in hypoxia. Biochem. Biophys. Res. Commun. 2016, 478, 1389–1395. [Google Scholar] [CrossRef]
- Martinez-Lostao, L.; García-Alvarez, F.; Basáñez, G.; Alegre-Aguarón, E.; Desportes, P.; Larrad, L.; Naval, J.; Martínez-Lorenzo, M.J.; Anel, A. Liposome-Bound APO2L/TRAIL Is an Effective Treatment in a Rabbit Model of Rheumatoid Arthritis. Arthrit. Rheum. 2010, 62, 2272–2282. [Google Scholar] [CrossRef]
- MacFarlane, M.; Ahmad, M.; Srinivasula, S.; Fernandes-Alnemri, T.; Cohen, G.; Alnemri, E. Identification and molecular cloning of two novel receptors for the cytotoxic ligand TRAIL. J. Biol. Chem. 1997, 272, 25417–25420. [Google Scholar] [CrossRef]
- Alley, M.C.; Scudiero, D.A.; Monks, A.; Hursey, M.L.; Czerwinski, M.J.; Fine, D.; Abbot, B.J.; Mayo, J.G.; Shoemaker, R.H.; Boyd, M.R. Sensibility of drug screening with panels of human tumor cell lines using microculture tetrazolium assay. Cancer Res. 1988, 46, 589–601. [Google Scholar]
- Kankotia, S.; Stacpoole, P. Dichloroacetate and cancer: New home for an orphan drug? Biochim. Biophys. Acta 2014, 1846, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Michelakis, E.D.; Sutendra, G.; Dromparis, P.; Webster, L.; Haromy, A.; Niven, E.; Maguire, C.; Gammer, T.L.; Mackey, J.R.; Fulton, D.; et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci. Transl. Med. 2010, 12, 31ra34. [Google Scholar] [CrossRef] [PubMed]
- Stacpoole, P.; Kerr, D.; Barnes, C.; Bunch, S.; Carney, P.; Fennell, E.; Felitsyn, N.; Gilmore, R.; Greer, M.; Henderson, G.; et al. Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatrics 2006, 117, 1519–1531. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Pinedo, C.; Ruiz-Ruiz, C.; Ruiz de Almodovar, C.; Palacios, C.; López-Rivas, A. Inhibition of glucose metabolism sensitizes tumor cells to death receptor-triggered apoptosis through enhancement of death-inducing signaling complex formation and apical procaspase-8 processing. J. Biol. Chem. 2003, 278, 12759–12768. [Google Scholar] [CrossRef]
- Tataranni, T.; Piccoli, C. Dichloroacetate (DCA) and Cancer: An Overview towards Clinical Applications. Oxidative Med. Cell. Longev. 2019, 2019, 8201079. [Google Scholar] [CrossRef]
- Snajdauf, M.; Havlova, K.; Vachtenheim, J.; Ozaniak, A.; Lischke, R.; Bartunkova, J.; Smrz, D.; Strizova, Z. The TRAIL in the Treatment of Human Cancer: An Update on Clinical Trials. Front. Mol. Biosci. 2021, 2021, 628332. [Google Scholar] [CrossRef]
- Wajant, H. Molecular Mode of Action of TRAIL Receptor Agonists-Common Principles and Their Translational Exploitation. Cancers 2019, 11, 954. [Google Scholar] [CrossRef]
- Cheah, C.; Belada, D.; Fanale, M.; Janikova, A.; Czucman, M.; Flinn, I.; Kapp, A.; Ashkenazi, A.; Kelley, S.; Bray, G.; et al. Dulanermin with rituximab in patients with relapsed indolent B-cell lymphoma: An open-label phase 1b/2 randomised study. Lancet Haeamtol. 2015, 2, e166–e174. [Google Scholar] [CrossRef]
- Soria, J.; Márk, Z.; Zatloukal, P.; Szima, B.; Albert, I.; Juhász, E.; Pujol, J.; Kozielski, J.; Baker, N.; Smethurst, D.; et al. Randomized phase II study of dulanermin in combination with paclitaxel, carboplatin, and bevacizumab in advanced non-small-cell lung cancer. J. Clin. Oncol. 2011, 29, 4442–4451. [Google Scholar] [CrossRef]
- Micheau, O.; Shirley, S.; Dufour, F. Death receptors as targets in cancer. Br. J. Pharmacol. 2013, 169, 1723–1744. [Google Scholar] [CrossRef]
- Ouyang, X.; Shi, M.; Jie, F.; Bai, Y.; Shen, P.; Yu, Z.; Wang, X.; Huang, C.; Tao, M.; Wang, Z.; et al. Phase III study of dulanermin (recombinant human tumor necrosis factor-related apoptosis-inducing ligand/Apo2 ligand) combined with vinorelbine and cisplatin in patients with advanced non-small-cell lung cancer. Investig. New Drugs 2018, 36, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Stacpoole, P. Therapeutic Targeting of the Pyruvate Dehydrogenase Complex/Pyruvate Dehydrogenase Kinase (PDC/PDK) Axis in Cancer. J. Natl. Cancer Inst. 2017, 109, 071. [Google Scholar] [CrossRef] [PubMed]
- Annibaldi, A.; Walczak, H. Death Receptors and Their Ligands in Inflammatory Disease and Cancer. Cold Spring Harb. Perspect. Biol. 2020, 12, a036384. [Google Scholar] [CrossRef]
- von Karstedt, S.; Montinaro, A.; Walczak, H. Exploring the TRAILs less travelled: TRAIL in cancer biology and therapy. Nat. Rev. Cancer 2017, 17, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Yacoub, S.; Shiverick, K.; Namiki, K.; Sakai, Y.; Porvasnik, S.; Urbanek, C.; Rosser, C. Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate 2008, 68, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Jiang, Y.; Yan, X.; Dai, X.; Huang, C.; Chen, L.; Li, T.; Zhang, Y.; Xiao, H.; Yang, M.; et al. Dichloroacetate enhances the anti-tumor effect of sorafenib via modulating the ROS-JNK-Mcl-1 pathway in liver cancer cells. Exp. Cell Res. 2021, 406, 112755. [Google Scholar] [CrossRef]
- Jones, R.; Plas, D.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.; Thompson, C. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef]
- Agnoletto, C.; Brunelli, L.; Melloni, E.; Pastorelli, R.; Casciano, F.; Rimondi, E.; Rigolin, G.; Cuneo, A.; Secchiero, P.; Zauli, G. The anti-leukemic activity of sodium dichloroacetate in p53mutated/null cells is mediated by a p53-independent ILF3/p21 pathway. Oncotarget 2014, 6, 2385–2396. [Google Scholar] [CrossRef]
- Agnoletto, C.; Melloni, E.; Casciano, F.; Rigolin, G.; Rimondi, E.; Celeghini, C.; Brunelli, L.; Cuneo, A.; Secchiero, P.; Zauli, G. Sodium dichloroacetate exhibits anti-leukemic activity in B-chronic lymphocytic leukemia (B-CLL) and synergizes with the p53 activator Nutlin-3. Oncotarget 2014, 5, 4347–4360. [Google Scholar] [CrossRef]
- Koit, A.; Timohhina, N.; Truu, L.; Chekulayev, V.; Gudlawar, S.; Shevchuk, I.; Lepik, K.; Mallo, L.; Kutner, R.; Valvere, V.; et al. Metabolic and OXPHOS Activities Quantified by Temporal ex vivo Analysis Display Patient-Specific Metabolic Vulnerabilities in Human Breast Cancers. Front. Oncol. 2020, 10, 1053. [Google Scholar] [CrossRef]
- Owens, K.; Kulawiec, M.; Desouki, M.; Vanniarajan, A.; Singh, K. Impaired OXPHOS complex III in breast cancer. PLoS ONE 2011, 6, e23846. [Google Scholar] [CrossRef] [PubMed]
- James, M.; Stacpoole, P. Pharmacogenetic considerations with dichloroacetate dosing. Pharmacogenomics 2016, 17, 743–753. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marco-Brualla, J.; de Miguel, D.; Martínez-Lostao, L.; Anel, A. DR5 Up-Regulation Induced by Dichloroacetate Sensitizes Tumor Cells to Lipid Nanoparticles Decorated with TRAIL. J. Clin. Med. 2023, 12, 608. https://doi.org/10.3390/jcm12020608
Marco-Brualla J, de Miguel D, Martínez-Lostao L, Anel A. DR5 Up-Regulation Induced by Dichloroacetate Sensitizes Tumor Cells to Lipid Nanoparticles Decorated with TRAIL. Journal of Clinical Medicine. 2023; 12(2):608. https://doi.org/10.3390/jcm12020608
Chicago/Turabian StyleMarco-Brualla, Joaquín, Diego de Miguel, Luis Martínez-Lostao, and Alberto Anel. 2023. "DR5 Up-Regulation Induced by Dichloroacetate Sensitizes Tumor Cells to Lipid Nanoparticles Decorated with TRAIL" Journal of Clinical Medicine 12, no. 2: 608. https://doi.org/10.3390/jcm12020608
APA StyleMarco-Brualla, J., de Miguel, D., Martínez-Lostao, L., & Anel, A. (2023). DR5 Up-Regulation Induced by Dichloroacetate Sensitizes Tumor Cells to Lipid Nanoparticles Decorated with TRAIL. Journal of Clinical Medicine, 12(2), 608. https://doi.org/10.3390/jcm12020608