
Clinical and Genetic Characteristics of Finnish Patients with Autosomal Recessive and Dominant Non-Syndromic Hearing Loss Due to Pathogenic TMC1 Variants

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Recruitment
2.2. Familial and Clinical Examination
2.3. Genetic Testing and Data Analysis
3. Results
3.1. Clinical Characteristics of the Patients
3.2. Genetic Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smith, R.J.H.; Bale, J.F.; White, K.R. Sensorineural Hearing Loss in Children. Lancet 2005, 365, 879–890. [Google Scholar] [CrossRef]
- Koffler, T.; Ushakov, K.; Avraham, K.B. Genetics of Hearing Loss: Syndromic. Otolaryngol. Clin. N. Am. 2015, 48, 1041–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, M.; Bitner-Glindzicz, M. Genetic Investigations in Childhood Deafness. Arch. Dis Child. 2015, 100, 271–278. [Google Scholar] [CrossRef]
- Shearer, A.E.; Hildebrand, M.S.; Smith, R.J. Hereditary Hearing Loss and Deafness Overview. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Pan, B.; Géléoc, G.S.; Asai, Y.; Horwitz, G.C.; Kurima, K.; Ishikawa, K.; Kawashima, Y.; Griffith, A.J.; Holt, J.R. TMC1 and TMC2 Are Components of the Mechanotransduction Channel in Hair Cells of the Mammalian Inner Ear. Neuron 2013, 79, 504–515. [Google Scholar] [CrossRef] [Green Version]
- Kawashima, Y.; Géléoc, G.S.G.; Kurima, K.; Labay, V.; Lelli, A.; Asai, Y.; Makishima, T.; Wu, D.K.; Della Santina, C.C.; Holt, J.R.; et al. Mechanotransduction in Mouse Inner Ear Hair Cells Requires Transmembrane Channel-like Genes. J. Clin. Investig. 2011, 121, 4796–4809. [Google Scholar] [CrossRef]
- Kurima, K.; Ebrahim, S.; Pan, B.; Sedlacek, M.; Sengupta, P.; Millis, B.A.; Cui, R.; Nakanishi, H.; Fujikawa, T.; Kawashima, Y.; et al. TMC1 and TMC2 Localize at the Site of Mechanotransduction in Mammalian Inner Ear Hair Cell Stereocilia. Cell Rep. 2015, 12, 1606–1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, B.; Akyuz, N.; Liu, X.-P.; Asai, Y.; Nist-Lund, C.; Kurima, K.; Derfler, B.H.; György, B.; Limapichat, W.; Walujkar, S.; et al. TMC1 Forms the Pore of Mechanosensory Transduction Channels in Vertebrate Inner Ear Hair Cells. Neuron 2018, 99, 736–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurima, K.; Peters, L.M.; Yang, Y.; Riazuddin, S.; Ahmed, Z.M.; Naz, S.; Arnaud, D.; Drury, S.; Mo, J.; Makishima, T.; et al. Dominant and Recessive Deafness Caused by Mutations of a Novel Gene, TMC1, Required for Cochlear Hair-Cell Function. Nat. Genet. 2002, 30, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Sirmaci, A.; Duman, D.; Oztürkmen-Akay, H.; Erbek, S.; Incesulu, A.; Oztürk-Hişmi, B.; Arici, Z.S.; Yüksel-Konuk, E.B.; Taşir-Yilmaz, S.; Tokgöz-Yilmaz, S.; et al. Mutations in TMC1 Contribute Significantly to Nonsyndromic Autosomal Recessive Sensorineural Hearing Loss: A Report of Five Novel Mutations. Int. J. Pediatr. Otorhinolaryngol. 2009, 73, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Sommen, M.; Schrauwen, I.; Vandeweyer, G.; Boeckx, N.; Corneveaux, J.J.; van den Ende, J.; Boudewyns, A.; De Leenheer, E.; Janssens, S.; Claes, K.; et al. DNA Diagnostics of Hereditary Hearing Loss: A Targeted Resequencing Approach Combined with a Mutation Classification System. Hum. Mutat. 2016, 37, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Zazo Seco, C.; Wesdorp, M.; Feenstra, I.; Pfundt, R.; Hehir-Kwa, J.Y.; Lelieveld, S.H.; Castelein, S.; Gilissen, C.; de Wijs, I.J.; Admiraal, R.J.; et al. The Diagnostic Yield of Whole-Exome Sequencing Targeting a Gene Panel for Hearing Impairment in The Netherlands. Eur. J. Hum. Genet. 2017, 25, 308–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bademci, G.; Foster, J.; Mahdieh, N.; Bonyadi, M.; Duman, D.; Cengiz, F.B.; Menendez, I.; Diaz-Horta, O.; Shirkavand, A.; Zeinali, S.; et al. Comprehensive Analysis via Exome Sequencing Uncovers Genetic Etiology in Autosomal Recessive Nonsyndromic Deafness in a Large Multiethnic Cohort. Genet. Med. 2016, 18, 364–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Heer, A.-M.R.; Collin, R.W.J.; Huygen, P.L.M.; Schraders, M.; Oostrik, J.; Rouwette, M.; Kunst, H.P.M.; Kremer, H.; Cremers, C.W.R.J. Progressive Sensorineural Hearing Loss and Normal Vestibular Function in a Dutch DFNB7/11 Family with a Novel Mutation in TMC1. Audiol. Neurootol. 2011, 16, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Schrauwen, I.; Sommen, M.; Corneveaux, J.J.; Reiman, R.A.; Hackett, N.J.; Claes, C.; Claes, K.; Bitner-Glindzicz, M.; Coucke, P.; Van Camp, G.; et al. A Sensitive and Specific Diagnostic Test for Hearing Loss Using a Microdroplet PCR-Based Approach and next Generation Sequencing. Am. J. Med. Genet. A 2013, 161A, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Imtiaz, A.; Maqsood, A.; Rehman, A.U.; Morell, R.J.; Holt, J.R.; Friedman, T.B.; Naz, S. Recessive Mutations of TMC1 Associated with Moderate to Severe Hearing Loss. Neurogenetics 2016, 17, 115–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishio, S.-Y.; Usami, S.-I. Prevalence and Clinical Features of Autosomal Dominant and Recessive TMC1-Associated Hearing Loss. Hum. Genet. 2021. [Google Scholar] [CrossRef]
- Ramzan, K.; Al-Owain, M.; Al-Numair, N.S.; Afzal, S.; Al-Ageel, S.; Al-Amer, S.; Al-Baik, L.; Al-Otaibi, G.F.; Hashem, A.; Al-Mashharawi, E.; et al. Identification of TMC1 as a Relatively Common Cause for Nonsyndromic Hearing Loss in the Saudi Population. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2020, 183, 172–180. [Google Scholar] [CrossRef]
- Wang, H.; Wu, K.; Guan, J.; Yang, J.; Xie, L.; Xiong, F.; Lan, L.; Wang, D.; Wang, Q. Identification of Four TMC1 Variations in Different Chinese Families with Hereditary Hearing Loss. Mol. Genet. Genom. Med. 2018, 6, 504–513. [Google Scholar] [CrossRef]
- Makishima, T.; Kurima, K.; Brewer, C.C.; Griffith, A.J. Early Onset and Rapid Progression of Dominant Nonsyndromic DFNA36 Hearing Loss. Otol. Neurotol. 2004, 25, 714–719. [Google Scholar] [CrossRef]
- Gallo, S.; Trevisi, P.; Rigon, C.; Caserta, E.; Seif Ali, D.; Bovo, R.; Martini, A.; Cassina, M. Auditory Outcome after Cochlear Implantation in Children with DFNB7/11 Caused by Pathogenic Variants in TMC1 Gene. Audiol. Neurootol. 2021, 26, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-Y.; Oh, D.-Y.; Han, J.H.; Kim, M.Y.; Kim, B.; Kim, B.J.; Song, J.-J.; Koo, J.-W.; Lee, J.H.; Oh, S.H.; et al. Flexible Real-Time Polymerase Chain Reaction-Based Platforms for Detecting Deafness Mutations in Koreans: A Proposed Guideline for the Etiologic Diagnosis of Auditory Neuropathy Spectrum Disorder. Diagnostics 2020, 10, 672. [Google Scholar] [CrossRef]
- Willberg, T.; Sivonen, V.; Linder, P.; Dietz, A. Comparing the Speech Perception of Cochlear Implant Users with Three Different Finnish Speech Intelligibility Tests in Noise. J. Clin. Med. 2021, 10, 3666. [Google Scholar] [CrossRef]
- Black, J.; Hickson, L.; Black, B. Defining and Evaluating Success in Paediatric Cochlear Implantation—An Exploratory Study. Int. J. Pediatr. Otorhinolaryngol. 2012, 76, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Holden, L.K.; Finley, C.C.; Firszt, J.B.; Holden, T.A.; Brenner, C.; Potts, L.G.; Gotter, B.D.; Vanderhoof, S.S.; Mispagel, K.; Heydebrand, G.; et al. Factors Affecting Open-Set Word Recognition in Adults with Cochlear Implants. Ear Hear. 2013, 34, 342–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, E.E.; Dornhoffer, J.R.; Loftus, C.; Nguyen, S.A.; Meyer, T.A.; Dubno, J.R.; McRackan, T.R. Association of Patient-Related Factors with Adult Cochlear Implant Speech Recognition Outcomes: A Meta-Analysis. JAMA Otolaryngol. Head Neck Surg. 2020, 146, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Hilgert, N.; Monahan, K.; Kurima, K.; Li, C.; Friedman, R.A.; Griffith, A.J.; Van Camp, G. Amino Acid 572 in TMC1: Hot Spot or Critical Functional Residue for Dominant Mutations Causing Hearing Impairment. J. Hum. Genet. 2009, 54, 188–190. [Google Scholar] [CrossRef]
- Wei, Q.; Zhu, H.; Qian, X.; Chen, Z.; Yao, J.; Lu, Y.; Cao, X.; Xing, G. Targeted Genomic Capture and Massively Parallel Sequencing to Identify Novel Variants Causing Chinese Hereditary Hearing Loss. J. Transl. Med. 2014, 12, 311. [Google Scholar] [CrossRef] [Green Version]
- Beurg, M.; Schimmenti, L.A.; Koleilat, A.; Amr, S.S.; Oza, A.; Barlow, A.J.; Ballesteros, A.; Fettiplace, R. New Tmc1 Deafness Mutations Impact Mechanotransduction in Auditory Hair Cells. J. Neurosci. 2021, 41, 4378–4391. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
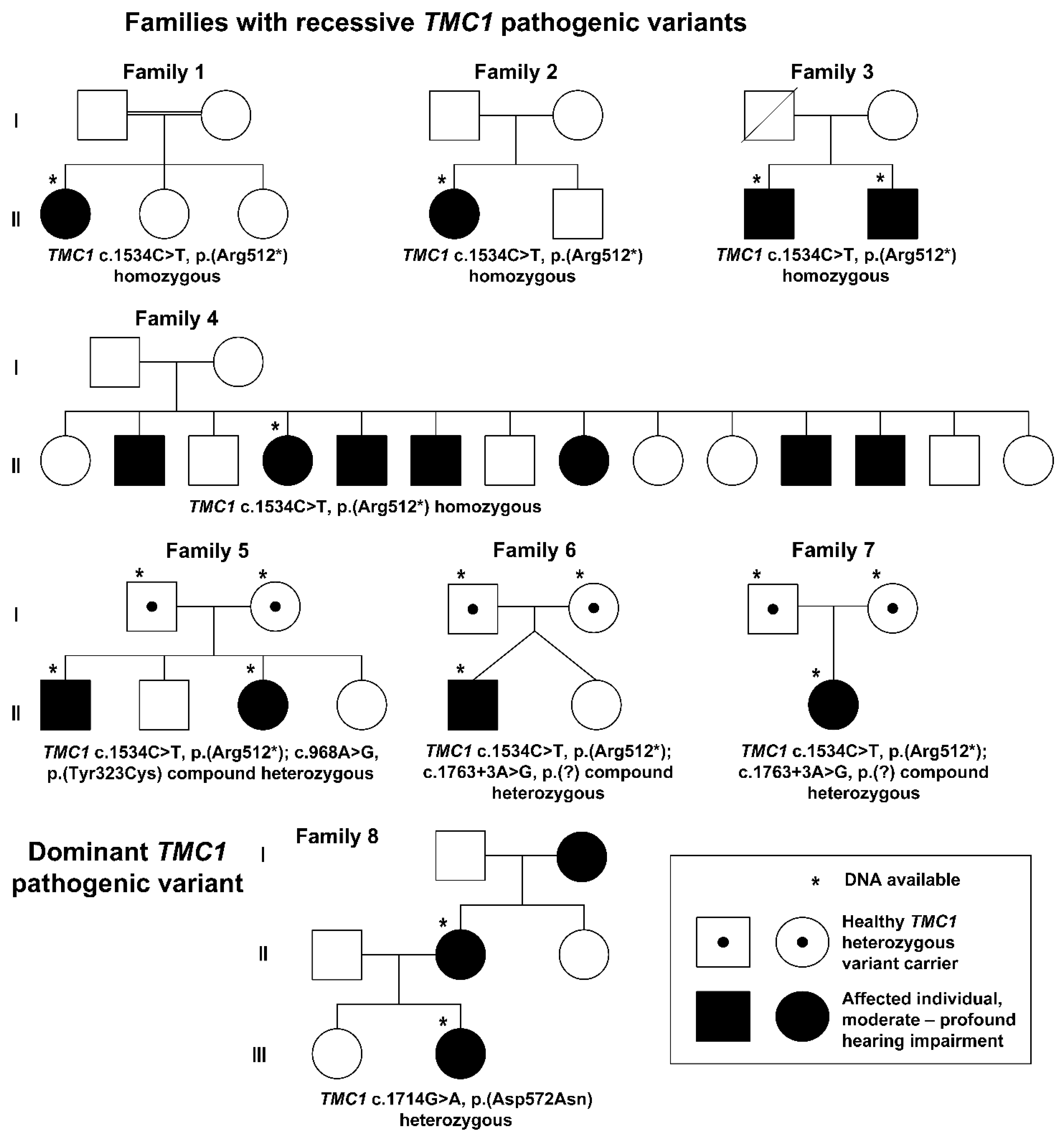
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Patient 7 | Patient 8 | Patient 9 | Patient 10 | Patient 11 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Family | 1 | 2 | 3 | 3 | 4 | 5 | 5 | 6 | 7 | 8 | 8 |
| Current age | 21 | 5 | 42 | 46 | 53 | 23 | 18 | 7 | 0 | 57 | 21 |
| Sex | F | F | M | M | F | M | F | M | F | F | F |
| Genotype | c.1534C>T, p.(Arg512*), homozygous | c.1534C>T, p.(Arg512*), homozygous | c.1534C>T, p.(Arg512*), homozygous | c.1534C>T, p.(Arg512*), homozygous | c.1534C>T, p.(Arg512*), homozygous | c.1534C>T, p.(Arg512*) and c.968A>G, p.(Tyr323Cys) | c.1534C>T, p.(Arg512*) and c.968A>G, p.(Tyr323Cys) | c.1534C>T, p.(Arg512*) and c.1763+3A>G | c.1534C>T, p.(Arg512*) and c.1763+3A>G | c.1714G>A, p.(Asp572Asn) heterozygous | c.1714G>A, p.(Asp572Asn) heterozygous |
| Newborn screening | Abnormal | Abnormal | NA | NA | NA | NA | Abnormal | Abnormal | Abnormal | NA | NA |
| OAE | No response | No response | NA | NA | NA | No response | No response | No response | No response | NA | NA |
| TEOAE | NA | No response | NA | NA | NA | NA | NA | NA | No response | NA | NA |
| AABR | NA | No response | NA | NA | NA | NA | NA | NA | No response | NA | NA |
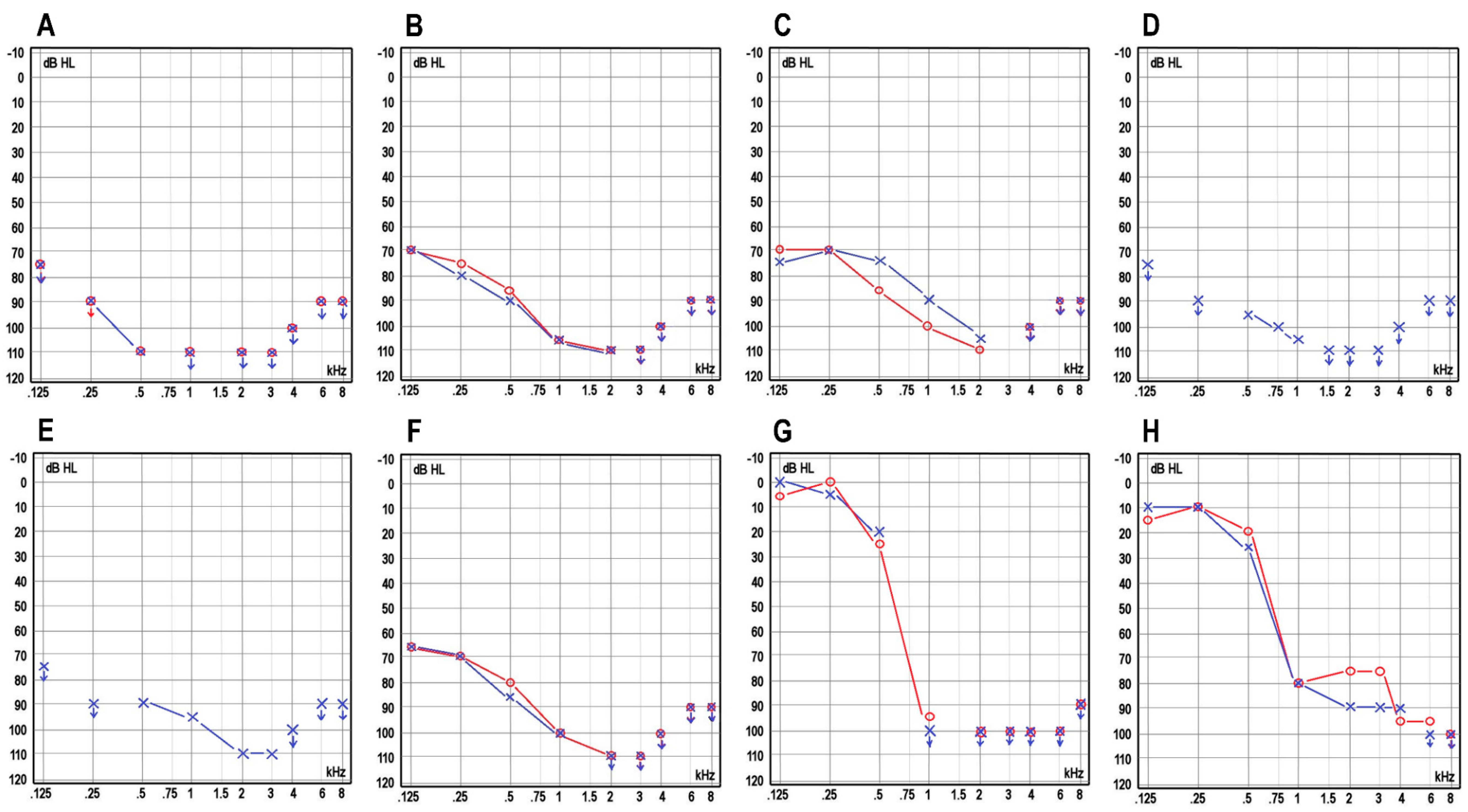
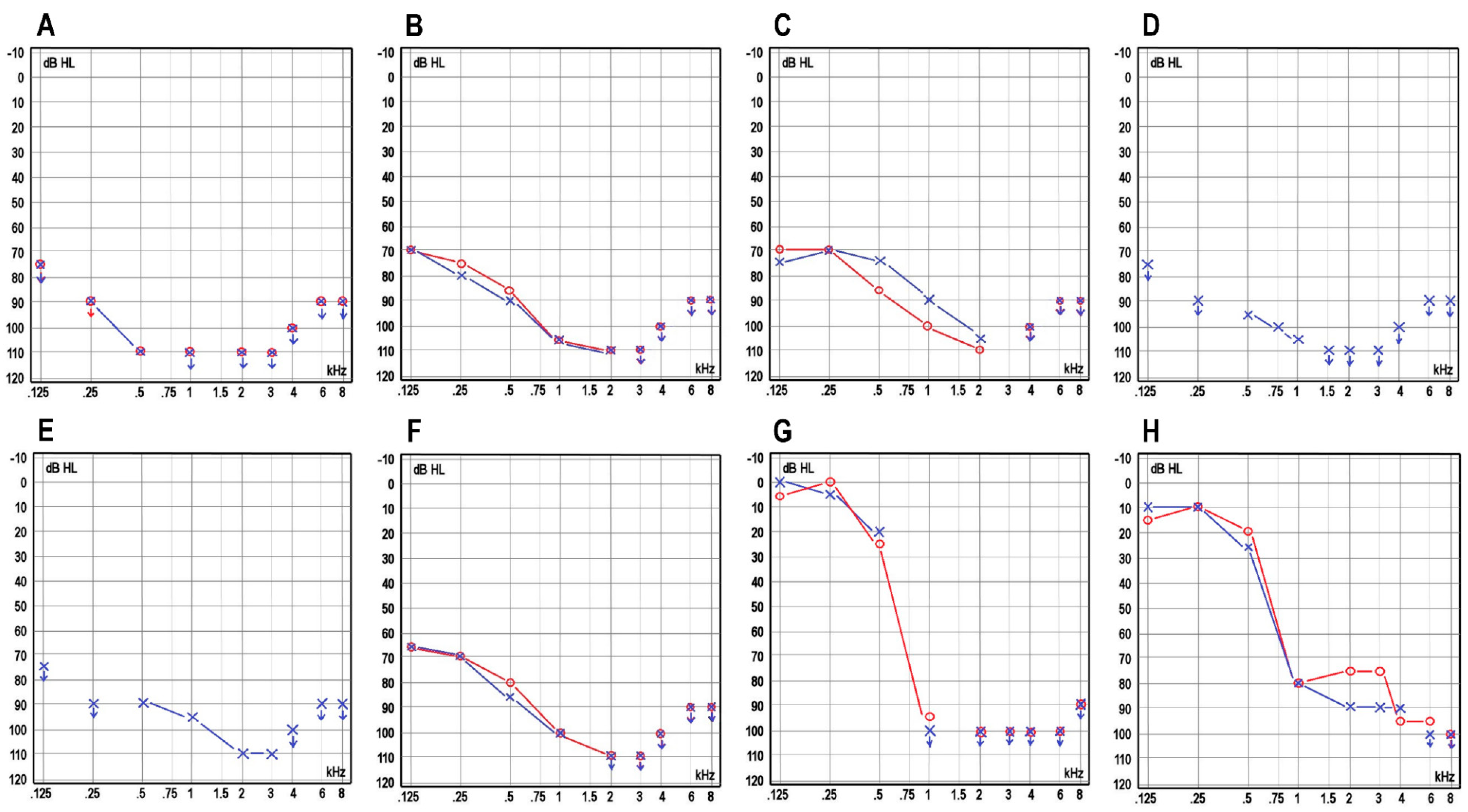
| ABR (dB) | No response | l.dx: no response/l.sin: 90 (4 kHz) and click 85 | NA | NA | NA | No response | No response | No response | Cochlear microphonics at 85, no neural responses at 85 | NA | NA |
| Speech-in-noise test | SRS +10 dB SNR l.a. 99%; SRT l.dx. −8.0 dB SNR, SRT l.sin −8.6 dB SNR | SRT l.dx −5,9 dB SNR, SRT l.sin −4.9 dB SNR, SRT l.a. −6.6 dB SNR | NA | NA | NA | SRS +10 dB SNR l.a. 75%, SRT l.dx −2.3 dB SNR, SRT l.a. −3.4 dB SNR | SRS +10 dB SNR l.a. 100%, SRT l.dx −7.6 dB SNR, SRT l.a. −6.6/−7.6 dB SNR | SRT l.dx −6.3 dB SNR, SRT l.sin −4.9 dB SNR, SRT l.a. −6.2 dB SNR, | NA | SRS +10 dB SNR l.a. 94%; SRT l.sin. −1.3 dB SNR, SRT l.dx. −2.0 dB SNR, SRT l.a. −2.8 dB SNR | SRS +10 dB SNR l.a. 88%, SRT l.a. +0.4 dB SNR |
| Age at diagnosis | 7 m | 2 m | From birth | 1 y 8 m | From birth | From birth | 1 m | 8 m | From birth | 7 y | 7 y |
| Severity | Profound | Profound | Profound | Profound | Profound | Profound | Profound | Profound | Profound | Profound | Severe |
| Progression | No | No | No | No | No | No | No | No | No | Yes | Yes |
| Hearing aids (from the age) | Yes (7 m) | Yes (4 m) | No | No | Yes (1.5 y) | Yes (1 y 4 m) | Yes (3 m) | Yes (9 m) | Yes (4 m) | No | Yes |
| Cochlear implant (from the age) | Yes (1 y 5 m) (right) and 13 y (left) | Yes (1 y) | No | No | No | Yes (2 y 1 m (right) and 20 y 9 m (left) | Yes (1 y (right) and 15 y 5 m (left) | Yes (1 y 5 m (right) and 1y6m (left) | Planned (8–9 m) | Yes (41 y (left) and 51 y (right) | No |
| Balance problems | No | No | No | No | No | Yes | No | No | NA | No | No |
| Language perception | Good progress with CI use | Good progress with CI use | Sign language | Sign language | Sign language | Good progress with CI use | Good progress with CI use | Good progress with CI use | NA | Normal spoken language | Normal spoken language |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kraatari-Tiri, M.; Haanpää, M.K.; Willberg, T.; Pohjola, P.; Keski-Filppula, R.; Kuismin, O.; Moilanen, J.S.; Häkli, S.; Rahikkala, E. Clinical and Genetic Characteristics of Finnish Patients with Autosomal Recessive and Dominant Non-Syndromic Hearing Loss Due to Pathogenic TMC1 Variants. J. Clin. Med. 2022, 11, 1837. https://doi.org/10.3390/jcm11071837
Kraatari-Tiri M, Haanpää MK, Willberg T, Pohjola P, Keski-Filppula R, Kuismin O, Moilanen JS, Häkli S, Rahikkala E. Clinical and Genetic Characteristics of Finnish Patients with Autosomal Recessive and Dominant Non-Syndromic Hearing Loss Due to Pathogenic TMC1 Variants. Journal of Clinical Medicine. 2022; 11(7):1837. https://doi.org/10.3390/jcm11071837
Chicago/Turabian StyleKraatari-Tiri, Minna, Maria K. Haanpää, Tytti Willberg, Pia Pohjola, Riikka Keski-Filppula, Outi Kuismin, Jukka S. Moilanen, Sanna Häkli, and Elisa Rahikkala. 2022. "Clinical and Genetic Characteristics of Finnish Patients with Autosomal Recessive and Dominant Non-Syndromic Hearing Loss Due to Pathogenic TMC1 Variants" Journal of Clinical Medicine 11, no. 7: 1837. https://doi.org/10.3390/jcm11071837
APA StyleKraatari-Tiri, M., Haanpää, M. K., Willberg, T., Pohjola, P., Keski-Filppula, R., Kuismin, O., Moilanen, J. S., Häkli, S., & Rahikkala, E. (2022). Clinical and Genetic Characteristics of Finnish Patients with Autosomal Recessive and Dominant Non-Syndromic Hearing Loss Due to Pathogenic TMC1 Variants. Journal of Clinical Medicine, 11(7), 1837. https://doi.org/10.3390/jcm11071837