
Challenges and Opportunities for Immunotherapeutic Intervention against Myeloid Immunosuppression in Glioblastoma

Abstract
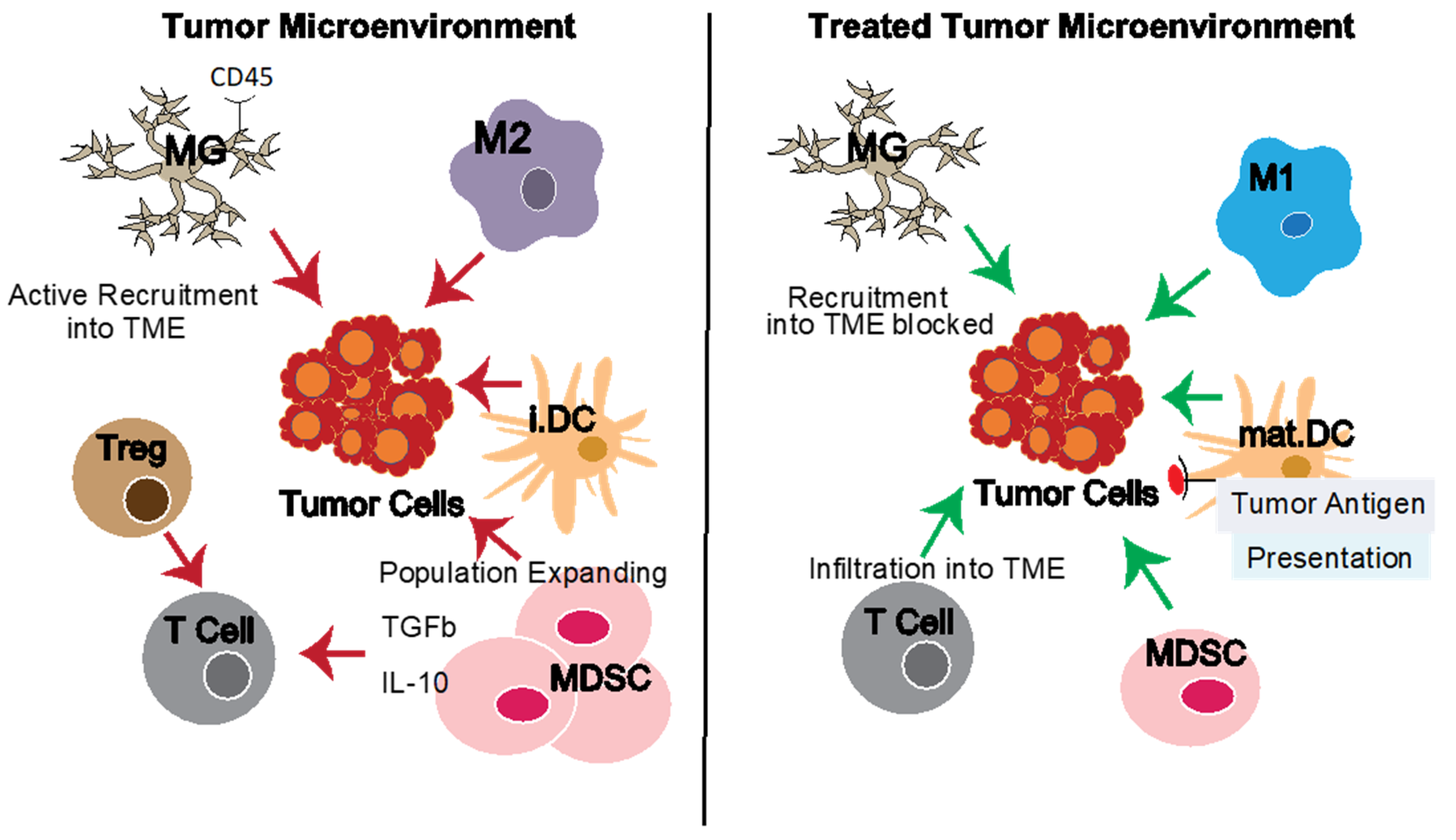
:1. Introduction
2. Defects in Myeloid Cell Populations in GBM
3. Targeting Myeloid Cell Populations in GBM
3.1. Microglia Targeting in GBM
3.2. Macrophage Re-Polarization and Re-Direction in GBM
3.3. Myeloid-Derived Suppressor Cell (MDSC) Targeting in GBM
3.4. Dendritic Cell (DC) Targeting in GBM
4. Further Molecular Targets and Myeloid Cell Relevance
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
References
- Chang, R.B.; Beatty, G.L. The interplay between innate and adaptive immunity in cancer shapes the productivity of cancer immunosurveillance. J. Leukoc. Biol. 2020, 108, 363–376. [Google Scholar] [CrossRef]
- Magaña-Maldonado, R.; Chávez-Cortez, E.G.; Olascoaga-Arellano, N.K.; López-Mejía, M.; Maldonado-Leal, F.M.; Sotelo, J.; Pineda, B. Immunological Evasion in Glioblastoma. BioMed Res. Int. 2016, 2016, 7487313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomaszewski, W.; Sanchez-Perez, L.; Gajewski, T.F.; Sampson, J.H. Brain Tumor Microenvironment and Host State: Implications for Immunotherapy. Clin. Cancer Res. 2019, 25, 4202–4210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, A.S.; Routkevitch, D.; Jackson, C.; Lim, M. Targeting Myeloid Cells in Combination Treatments for Glioma and Other Tumors. Front. Immunol. 2019, 10, 1715. [Google Scholar] [CrossRef] [Green Version]
- Locarno, C.V.; Simonelli, M.; Carenza, C.; Capucetti, A.; Stanzani, E.; Lorenzi, E.; Persico, P.; Bella, S.D.; Passoni, L.; Mavilio, D.; et al. Role of myeloid cells in the immunosuppressive microenvironment in gliomas. Immunobiology 2020, 225, 151853. [Google Scholar] [CrossRef]
- Pombo Antunes, A.R.; Scheyltjens, I.; Duerinck, J.; Neyns, B.; Movahedi, K.; van Ginderachter, J.A. Understanding the glioblastoma immune microenvironment as basis for the development of new immunotherapeutic strategies. eLife 2020, 9, e52176. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, M.M.; Sankey, E.W.; Ryan, K.J.; Chongsathidkiet, P.; Lorrey, S.J.; Wilkinson, D.S.; Fecci, P.E. Immune suppression in gliomas. J. Neuro-Oncol. 2021, 151, 3–12. [Google Scholar] [CrossRef]
- Daubon, T.; Hemadou, A.; Romero Garmendia, I.; Saleh, M. Glioblastoma Immune Landscape and the Potential of New Immunotherapies. Front. Immunol. 2020, 11, 2495. [Google Scholar] [CrossRef]
- Pires-Afonso, Y.; Niclou, S.P.; Michelucci, A. Revealing and Harnessing Tumour-Associated Microglia/Macrophage Heterogeneity in Glioblastoma. Int. J. Mol. Sci. 2020, 21, 689. [Google Scholar] [CrossRef] [Green Version]
- Gutmann, D.H.; Kettenmann, H. Microglia/Brain Macrophages as Central Drivers of Brain Tumor Pathobiology. Neuron 2019, 104, 442–449. [Google Scholar] [CrossRef]
- dello Russo, C.; Lisi, L.; Tentori, L.; Navarra, P.; Graziani, G.; Combs, C.K. Exploiting Microglial Functions for the Treatment of Glioblastoma. Curr. Cancer Drug Targets 2017, 17, 267–281. [Google Scholar] [CrossRef] [PubMed]
- Arcuri, C.; Fioretti, B.; Bianchi, R.; Mecca, C.; Tubaro, C.; Beccari, T.; Franciolini, F.; Giambanco, I.; Donato, R. Microglia-glioma cross-talk: A two way approach to new strategies against glioma. Front. Biosci. (Landmark Ed.) 2017, 22, 268–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, A. Microglia and brain macrophages: An update. Neuropathology 2017, 37, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Catalano, M.; D’Alessandro, G.; Trettel, F.; Limatola, C. Role of Infiltrating Microglia/Macrophages in Glioma. Adv. Exp. Med. Biol. 2020, 1202, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, R.K.; Singh, S.; Gupta, C.L.; Bajpai, P. Microglial TLR9: Plausible Novel Target for Therapeutic Regime Against Glioblastoma Multiforme. Cell. Mol. Neurobiol. 2021, 41, 1391–1393. [Google Scholar] [CrossRef]
- Mignogna, C.; Signorelli, F.; Vismara, M.F.M.; Zeppa, P.; Camastra, C.; Barni, T.; Donato, G.; Vito, A.D. A reappraisal of macrophage polarization in glioblastoma: Histopathological and immunohistochemical findings and review of the literature. Pathol. Res. Pract. 2016, 212, 491–499. [Google Scholar] [CrossRef]
- Vetsika, E.-K.; Koukos, A.; Kotsakis, A. Myeloid-Derived Suppressor Cells: Major Figures that Shape the Immunosuppressive and Angiogenic Network in Cancer. Cells 2019, 8, 1647. [Google Scholar] [CrossRef] [Green Version]
- Dysthe, M.; Parihar, R. Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1224, 117–140. [Google Scholar] [CrossRef]
- Richard, S.A. Explicating the Pivotal Pathogenic, Diagnostic, and Therapeutic Biomarker Potentials of Myeloid-Derived Suppressor Cells in Glioblastoma. Dis. Mark. 2020, 2020, 8844313. [Google Scholar] [CrossRef]
- Siew, J.J.; Chern, Y. Microglial Lectins in Health and Neurological Diseases. Front. Mol. Neurosci. 2018, 11, 158. Available online: https://www.frontiersin.org/article/10.3389/fnmol.2018.00158 (accessed on 7 February 2022). [CrossRef]
- Wang, W.-Y.; Tan, M.-S.; Yu, J.-T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Chakraborty, K.; Ray, P. Immunosuppressive MDSCs induced by TLR signaling during infection and role in resolution of inflammation. Front. Cell. Infect. Microbiol. 2013, 3, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, R.L.; Klemm, F.; Akkari, L.; Pyonteck, S.M.; Sevenich, L.; Quail, D.F.; Dhara, S.; Simpson, K.; Gardner, E.E.; lacobuzio-Donahue, C.A.; et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep. 2016, 17, 2445–2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, A.; Brandenburg, S.; Turkowski, K.; Müller, S.; Vajkoczy, P. Resident microglia, and not peripheral macrophages, are the main source of brain tumor mononuclear cells. Int. J. Cancer 2015, 137, 278–288. [Google Scholar] [CrossRef]
- Martins, T.A.; Schmassmann, P.; Shekarian, T.; Boulay, J.L.; Ritz, M.F.; Zanganeh, S.; Berg, J.V.; Hutter, G. Microglia-Centered Combinatorial Strategies Against Glioblastoma. Front. Immunol. 2020, 11, 2359. [Google Scholar] [CrossRef]
- Cheng, L.; Wang, Y.; Huang, L. Exosomes from M1-Polarized Macrophages Potentiate the Cancer Vaccine by Creating a Pro-inflammatory Microenvironment in the Lymph Node. Mol. Ther. 2017, 25, 1665–1675. [Google Scholar] [CrossRef] [Green Version]
- Prosniak, M.; Harshyne, L.A.; Andrews, D.W.; Kenyon, L.C.; Bedelbaeva, K.; Apanasovich, T.V.; Heber-Katz, E.; Curtis, M.T.; Cotzia, P.; Hooper, D.C. Glioma grade is associated with the accumulation and activity of cells bearing M2 monocyte markers. Clin. Cancer Res. 2013, 19, 3776–3786. [Google Scholar] [CrossRef] [Green Version]
- Harshyne, L.A.; Nasca, B.J.; Kenyon, L.C.; Andrews, D.W.; Hooper, D.C. Serum exosomes and cytokines promote a T-helper cell type 2 environment in the peripheral blood of glioblastoma patients. Neuro-Oncol. 2016, 18, 206–215. [Google Scholar] [CrossRef] [Green Version]
- Baig, M.S.; Roy, A.; Rajpoot, S.; Liu, D.; Savai, R.; Banerjee, S.; Kawada, M.; Faisal, S.M.; Saluja, R.; Saqib, U.; et al. Tumor-derived exosomes in the regulation of macrophage polarization. Inflamm. Res. 2020, 69, 435–451. [Google Scholar] [CrossRef]
- Wang, Q.; Yu, C. Identification of biomarkers associated with extracellular vesicles based on an integrative pan-cancer bioinformatics analysis. Med. Oncol. 2020, 37, 79. [Google Scholar] [CrossRef]
- Zheng, Z.; Zhang, J.; Jiang, J.; He, Y.; Zhang, W.; Mo, X.; Kang, X.; Xu, Q.; Wang, B.; Huang, Y. Remodeling tumor immune microenvironment (TIME) for glioma therapy using multi-targeting liposomal codelivery. J. Immunother. Cancer 2020, 8, e000207. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Wucherpfennig, K.; Chiocca, E.A. Immunotherapy for glioblastoma: On the sidelines or in the game? Discov. Med. 2017, 24, 201–208. [Google Scholar] [PubMed]
- Lorenzo-Sanz, L.; Muñoz, P. Tumor-Infiltrating Immunosuppressive Cells in Cancer-Cell Plasticity, Tumor Progression and Therapy Response. Cancer Microenviron. 2019, 12, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Chaib, M.; Chauhan, S.C.; Makowski, L. Friend or Foe? Recent Strategies to Target Myeloid Cells in Cancer. Front. Cell Dev. Biol. 2020, 8, 351. [Google Scholar] [CrossRef]
- Ngiow, S.F.; Young, A. Re-education of the Tumor Microenvironment With Targeted Therapies and Immunotherapies. Front. Immunol. 2020, 11, 1633. Available online: https://www.frontiersin.org/article/10.3389/fimmu.2020.01633 (accessed on 7 February 2022). [CrossRef]
- Miyazaki, T.; Ishikawa, E.; Sugii, N.; Matsuda, M. Therapeutic Strategies for Overcoming Immunotherapy Resistance Mediated by Immunosuppressive Factors of the Glioblastoma Microenvironment. Cancers 2020, 12, 1960. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeori, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [Green Version]
- Fraternale, A.; Brundu, S.; Magnani, M. Polarization and Repolarization of Macrophages. J. Clin. Cell. Immunol. 2015, 6, 2. [Google Scholar] [CrossRef] [Green Version]
- Hörhold, F.; Eisel, D.; Oswald, M.; Kolte, A.; Röll, D.; Osen, W.; Eichmuller, S.B.; Konig, R. Reprogramming of macrophages employing gene regulatory and metabolic network models. PLoS Comput. Biol. 2020, 16, e1007657. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, R.K.; Singh, S.; Gupta, C.L.; Pandey, P.; Singh, V.K.; Sayyed, U.; Shekh, R.; Bajpai, P. Repolarization of glioblastoma macrophage cells using non-agonistic Dectin-1 ligand encapsulating TLR-9 agonist: Plausible role in regenerative medicine against brain tumor. Int. J. Neurosci. 2021, 131, 591–598. [Google Scholar] [CrossRef]
- Xiong, Z.; Ampudia Mesias, E.; Pluhar, G.E.; Rathe, S.K.; Largaespada, D.A.; Sham, Y.Y.; Moertel, C.L.; Olin, M.R. CD200 Checkpoint Reversal: A Novel Approach to Immunotherapy. Clin. Cancer Res. 2020, 26, 232–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salacz, M.E.; Kast, R.E.; Saki, N.; Brüning, A.; Karpel-Massler, G.; Halatsch, M.E. Toward a noncytotoxic glioblastoma therapy: Blocking MCP-1 with the MTZ Regimen. Onco Targets Ther. 2016, 9, 2535–2545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kast, R.E.; Hill, Q.A.; Wion, D.; Mellstedt, H.; Focosi, D.; Karpel-Massler, G.; Heiland, T.; Halatsch, M.-E. Glioblastoma-synthesized G-CSF and GM-CSF contribute to growth and immunosuppression: Potential therapeutic benefit from dapsone, fenofibrate, and ribavirin. Tumor Biol. 2017, 39, 1010428317699797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voth, B.L.; Pelargos, P.E.; Barnette, N.E.; Bhatt, N.S.; Chen, C.H.J.; Lagman, C.; Chung, L.K.; Nguyen, T.; Sheppard, J.P.; Romiyo, P.; et al. Intratumor injection of CCL21-coupled vault nanoparticles is associated with reduction in tumor volume in an in vivo model of glioma. J. Neuro-Oncol. 2020, 147, 599–605. [Google Scholar] [CrossRef]
- Li, C.; Deng, H.; Zhou, Y.; Ye, Y.; Zhao, S.; Liang, S.; Cai, S.; Lin, J.; Tang, Y.; Wu, Y. Expression and clinical significance of CXC chemokines in the glioblastoma microenvironment. Life Sci. 2020, 261, 118486. [Google Scholar] [CrossRef]
- Huang, M.-N.; Nicholson, L.T.; Batich, K.A.; Swartz, A.M.; Kopin, D.; Wellford, S.; Prabhakar, V.K.; Woroniecka, K.; Nair, S.K.; Fecci, P.E.; et al. Antigen-loaded monocyte administration induces potent therapeutic antitumor T cell responses. J. Clin. Investig. 2020, 130, 774–788. [Google Scholar] [CrossRef]
- Spadaro, O.; Camell, C.D.; Bosurgi, L.; Nguyen, K.Y.; Youm, Y.H.; Rothlin, C.V.; Dixit, V.D. IGF1 Shapes Macrophage Activation in Response to Immunometabolic Challenge. Cell Rep. 2017, 19, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Morin-Brureau, M.; Hooper, K.M.; Prosniak, M.; Sauma, S.; Harshyne, L.A.; Andrews, D.W.; Hooper, D.C. Enhancement of glioma-specific immunity in mice by “NOBEL”, an insulin-like growth factor 1 receptor antisense oligodeoxynucleotide. Cancer Immunol. Immunother. 2015, 64, 447–457. [Google Scholar] [CrossRef]
- Harshyne, L.A.; Hooper, K.M.; Andrews, E.G.; Nasca, B.J.; Kenyon, L.C.; Andrews, D.W.; Hooper, D.C. Glioblastoma exosomes and IGF-1R/AS-ODN are immunogenic stimuli in a translational research immunotherapy paradigm. Cancer Immunol. Immunother. 2015, 64, 299–309. [Google Scholar] [CrossRef]
- Andrews, D.W.; Resnicoff, M.; Flanders, A.E.; Kenyon, L.; Curtis, M.; Merli, G.; Baserga, R.; Iliakis, G.; Aiken, R.D. Results of a pilot study involving the use of an antisense oligodeoxynucleotide directed against the insulin-like growth factor type I receptor in malignant astrocytomas. J. Clin. Oncol. 2001, 19, 2189–2200. [Google Scholar] [CrossRef]
- Andrews, D.W.; Judy, K.D.; Scott, C.B.; Garcia, S.; Harshyne, L.A.; Kenyon, L.; Talekar, K.; Flanders, A.; Atsina, K.-B.; Kim, L.; et al. Phase 1b Clinical Trial of IGV-001 for Patients with Newly Diagnosed Glioblastoma. Clin. Cancer Res. 2021, 27, 1912–1922. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zeng, S.; Gong, Z.; Xu, Z. Clinical implication of cellular vaccine in glioma: Current advances and future prospects. J. Exp. Clin. Cancer Res. 2020, 39, 257. [Google Scholar] [CrossRef]
- Guedan, S.; Ruella, M.; June, C.H. Emerging Cellular Therapies for Cancer. Annu. Rev. Immunol. 2019, 37, 145–171. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.D.; Maus, M.; June, C.H.; Sampson, J.H. Immunotherapy for Glioblastoma: Adoptive T-cell Strategies. Clin. Cancer Res. 2019, 25, 2042–2048. [Google Scholar] [CrossRef] [PubMed]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Tian, L.; Dai, X.; Yu, H.; Wang, J.; Lei, A.; Zhu, M.; Xu, J.; Zhao, W.; Zhu, Y.; et al. Induced Pluripotent Stem Cell-derived CAR-Macrophage Cells with Antigen-dependent Anti-Cancer Cell Functions for Liquid and Solid Tumors. BioRxiv 2020. [Google Scholar] [CrossRef]
- Anderson, N.R.; Minutolo, N.G.; Gill, S.; Klichinsky, M. Macrophage-based approaches for cancer immunotherapy. Cancer Res. 2021, 81, 1201–1208. [Google Scholar] [CrossRef]
- Sumida, K.; Wakita, D.; Narita, Y.; Masuko, K.; Terada, S.; Watanabe, K.; Satoh, T.; Kitamura, H.; Nishimura, T. Anti-IL-6 receptor mAb eliminates myeloid-derived suppressor cells and inhibits tumor growth by enhancing T-cell responses. Eur. J. Immunol. 2012, 42, 2060–2072. [Google Scholar] [CrossRef]
- Condamine, T.; Kumar, V.; Ramachandran, I.R.; Youn, J.I.; Celis, E.; Finnberg, N.; EI-Deiry, W.S.; Winograd, R.; Vonderheide, R.H.; English, N.R.; et al. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J. Clin. Investig. 2014, 124, 2626–2639. [Google Scholar] [CrossRef] [Green Version]
- Mi, Y.; Guo, N.; Luan, J.; Cheng, J.; Hu, Z.; Jiang, P.; Jin, W.; Gao, X. The Emerging Role of Myeloid-Derived Suppressor Cells in the Glioma Immune Suppressive Microenvironment. Front. Immunol. 2020, 11, 737. [Google Scholar] [CrossRef]
- Zhu, X.; Fujita, M.; Snyder, L.A.; Okada, H. Systemic delivery of neutralizing antibody targeting CCL2 for glioma therapy. J. Neuro-Oncol. 2011, 104, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Pan, R.-Y.; Chung, W.-H.; Chu, M.-T.; Chen, S.J.; Chen, H.C.; Zheng, L.; Hung, S.I. Recent Development and Clinical Application of Cancer Vaccine: Targeting Neoantigens. J. Immunol. Res. 2018, 2018, 4325874. [Google Scholar] [CrossRef] [PubMed]
- Buchroithner, J.; Erhart, F.; Pichler, J.; Widhalm, G.; Preusser, M.; Stockhammer, G.; Nowosielski, M.; Iglseder, S.; Freyschlag, C.F.; Oberndorfer, S.; et al. Audencel Immunotherapy Based on Dendritic Cells Has No Effect on Overall and Progression-Free Survival in Newly Diagnosed Glioblastoma: A Phase II Randomized Trial. Cancers 2018, 10, 372. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Shi, Z.; Jiang, J. Cyclooxygenase-2 in glioblastoma multiforme. Drug Discov. Today 2017, 22, 148–156. [Google Scholar] [CrossRef] [Green Version]
- Kast, R.E. Adding high-dose celecoxib to increase effectiveness of standard glioblastoma chemoirradiation. Ann. Pharm. Fr. 2021, 79, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Privorotskiy, A.; Bhavsar, S.P.; Lang, F.F.; Hu, J.; Cata, J.P. Impact of anesthesia and analgesia techniques on glioblastoma progression. A narrative review. Neurooncol. Adv. 2020, 2, vdaa123. [Google Scholar] [CrossRef] [PubMed]
- Benveniste, E.N.; Nguyen, V.T.; Wesemann, D.R. Molecular regulation of CD40 gene expression in macrophages and microglia. Brain Behav. Immun. 2004, 18, 7–12. [Google Scholar] [CrossRef]
- Werner, J.M.; Kuhl, S.; Ulrich, K.; Krischek, B.; Stavrinou, P.; Goldbrunner, R.; Timmer, M. Expression of CD40 Correlates Negatively with Overall and Progression-Free Survival of Low- and High-Grade Gliomas. World Neurosurg. 2019, 130, e17–e25. [Google Scholar] [CrossRef]
- Expression of CD40 in Cancer. Available online: https://www.proteinatlas.org/ENSG00000101017-CD40/pathology (accessed on 7 February 2022).
- Berberich, A.; Bartels, F.; Tang, Z.; Knoll, M.; Pusch, S.; Hucke, N.; Kessler, T.; Dong, Z.; Wiestler, B.; Winkler, F.; et al. LAPTM5-CD40 Crosstalk in Glioblastoma Invasion and Temozolomide Resistance. Front. Oncol. 2020, 10, 747. [Google Scholar] [CrossRef]
- Chonan, M.; Saito, R.; Shoji, T.; Shibahara, I.; Kanamori, M.; Sonoda, Y.; Watanabe, M.; Kikuchi, T.; Ishii, N.; Tominaga, T. CD40/CD40L expression correlates with the survival of patients with glioblastomas and an augmentation in CD40 signaling enhances the efficacy of vaccinations against glioma models. Neuro Oncol. 2015, 17, 1453–1462. [Google Scholar] [CrossRef] [Green Version]
- Hussain, S.F.; Yang, D.; Suki, D.; Aldape, K.; Grimm, E.; Heimberger, A.B. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro Oncol. 2006, 8, 261–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Huang, J.; Gong, W.; Zhang, L.; Yu, P.; Wang, J.M. CD40/CD40L dyad in the inflammatory and immune responses in the central nervous system. Cell Mol. Immunol. 2006, 3, 163–169. [Google Scholar] [PubMed]
- Shoji, T.; Saito, R.; Chonan, M.; Shibahara, I.; Sato, A.; Kanamori, M.; Sonoda, Y.; Kondo, T.; Ishii, N.; Tominaga, T. Local convection-enhanced delivery of an anti-CD40 agonistic monoclonal antibody induces antitumor effects in mouse glioma models. Neuro Oncol. 2016, 18, 1120–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lookian, P.P.; Zhao, D.; Medina, R.; Wang, H.; Zenka, J.; Gilbert, M.R.; Pacak, K.; Zhuang, Z. Mannan-BAM, TLR Ligands, Anti-CD40 Antibody (MBTA) Vaccine Immunotherapy: A Review of Current Evidence and Applications in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 3455. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; He, Z.; Duan, H.; Zhang, D.; Li, J.; Yang, H.; Dorsey, J.F.; Zou, W.; Nabavizadeh, S.A.; Bagley, S.J.; et al. Synergistic immunotherapy of glioblastoma by dual targeting of IL-6 and CD40. Nat. Commun. 2021, 12, 3424. [Google Scholar] [CrossRef]
- Lee-Chang, C.; Miska, J.; Hou, D.; Rashidi, A.; Zhang, P.; Burga, R.A.; Jusué-Torres, I.; Xiao, T.; Arrieta, V.A.; Zhang, D.Y.; et al. Activation of 4-1BBL+ B cells with CD40 agonism and IFNγ elicits potent immunity against glioblastoma. J. Exp. Med. 2021, 218, e20200913. [Google Scholar] [CrossRef]
- Genoud, V.; Espinoza, F.I.; Marinari, E.; Rochemont, V.; Dietrich, P.Y.; McSheehy, P.; Bachmann, F.; Lane, H.A.; Walker, P.R. Treating ICB-resistant glioma with anti-CD40 and mitotic spindle checkpoint controller BAL101553 (lisavanbulin). JCI Insight 2021, 6, e142980. [Google Scholar] [CrossRef]
- Nguyen, V.T.; Benveniste, E.N. Involvement of STAT-1 and its family members in interferon-γ induction of CD40 transcription in microglia/macrophages. J. Biol. Chem. 2000, 275, 23674–23684. [Google Scholar] [CrossRef] [Green Version]
- Qin, H.; Wilson, C.A.; Lee, S.J.; Zhao, X.; Benveniste, E.N. LPS induces CD40 gene expression through the activation of NF-κB and STAT-1α in macrophages and microglia. Blood 2005, 106, 3114–3122. [Google Scholar] [CrossRef] [Green Version]
- Yeung, Y.T.; McDonald, K.L.; Grewal, T.; Munoz, L. Interleukins in glioblastoma pathophysiology: Implications for therapy. Br. J. Pharmacol. 2013, 168, 591–606. [Google Scholar] [CrossRef] [Green Version]
- Hori, T.; Sasayama, T.; Tanaka, K.; Koma, Y.I.; Nishihara, M.; Tanaka, H.; Nakamizo, S.; Nagashima, H.; Maeyama, M.; Fujita, Y.; et al. Tumor-associated macrophage related interleukin-6 in cerebrospinal fluid as a prognostic marker for glioblastoma. J. Clin. Neurosci. 2019, 68, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Lamano, J.B.; Lamano, J.B.; Li, Y.D.; DiDomenico, J.D.; Choy, W.; Veliceasa, D.; Oyon, D.E.; Fakurnejad, S.; Ampie, L.; Kesavabhotla, K.; et al. Glioblastoma-Derived IL6 Induces Immunosuppressive Peripheral Myeloid Cell PD-L1 and Promotes Tumor Growth. Clin. Cancer Res. 2019, 25, 3643–3657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holst, C.B.; Christensen, I.J.; Vitting-Seerup, K.; Skjøth-Rasmussen, J.; Hamerlik, P.; Poulsen, H.S.; Johansen, J.S. Plasma IL-8 and ICOSLG as prognostic biomarkers in glioblastoma. Neurooncol. Adv. 2021, 3, vdab072. [Google Scholar] [CrossRef]
- Chiorean, R.; Berindan-Neagoe, I.; Braicu, C.; Florian, I.S.; Leucuta, D.; Crisan, D.; Cernea, V. Quantitative expression of serum biomarkers involved in angiogenesis and inflammation, in patients with glioblastoma multiforme: Correlations with clinical data. Cancer Biomark. 2014, 14, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Andersen, B.M.; Faust Akl, C.; Wheeler, M.A.; Chiocca, E.A.; Reardon, D.A.; Quintana, F.J. Glial and myeloid heterogeneity in the brain tumour microenvironment. Nat. Rev. Cancer 2021, 21, 786–802. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Cell Type | Markers: | Refs. | ||||
|---|---|---|---|---|---|---|
| CD14/CD16/CD68/… | Lectins … | TLRs | FcRs | Cytokines/Chemokines | ||
| Microglia | CD40/TMEM119 … | Galectins, SigLecs, MBLs | +(1–9) | + | CCL5, IL-1, IL-6, TGFβ, TNFα | [15,20,21] |
| Monocytes/ Macrophages | CD14/CD16 CD68/CD163 | ScavR, MannoseR | 1,2,4,−8 | CD16 | IL-1, IL-6, IL-8, IL-10, IFNγ, TNFα | [1,2,3,16] |
| MDSCs | CD11b/GR-1/CD33 | Galectins | 4 | − | IL-10, TGFβ | [17,18,22] |
| DCs | CD1c, CD11s | SigLecs | 9 | +/− | IL-10, IL-12 | [1,2,3] |
| Cell Type | Activities: | Refs. | ||||
|---|---|---|---|---|---|---|
| Phagocytosis | Cytokines/Chemokines | Immuno-suppression | TLRs | APC | ||
| Microglia | + | + | + | + | + | [12,13,23,24] |
| Monocytes/Macrophages | ++ | ++ | +/− | + | + | [1,2,3,23] |
| MDSCs | − | + | ++ | +/− | − | [17,18,19] |
| DCs | +/− | + | +/− | ++ | ++ | [1,2,3] |
| Cell Type | Targets | Refs. | |||||
|---|---|---|---|---|---|---|---|
| CSFs | IGF1 | Cytokines/Chemokines | TLRs: TLR9 … | Exosomes | CARs | ||
| Microglia | + | + | IL-6 | +/− | + | − | [12,13,20,21,22,24,25,26] |
| Monocytes/Macrophages | ++ | ++ | IL-6, IL-8, IL-10 | + | ++ | + | [16,26,27,28] |
| MDSCs | + | +/− | IL-10, TGFβ | +/− | +/− | − | [17,18,19,22] |
| DCs | − | +/− | IL-12/-18 | ++ | +/− | − | [1,2,3] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Exley, M.A.; Garcia, S.; Zellander, A.; Zilberberg, J.; Andrews, D.W. Challenges and Opportunities for Immunotherapeutic Intervention against Myeloid Immunosuppression in Glioblastoma. J. Clin. Med. 2022, 11, 1069. https://doi.org/10.3390/jcm11041069
Exley MA, Garcia S, Zellander A, Zilberberg J, Andrews DW. Challenges and Opportunities for Immunotherapeutic Intervention against Myeloid Immunosuppression in Glioblastoma. Journal of Clinical Medicine. 2022; 11(4):1069. https://doi.org/10.3390/jcm11041069
Chicago/Turabian StyleExley, Mark A., Samantha Garcia, Amelia Zellander, Jenny Zilberberg, and David W. Andrews. 2022. "Challenges and Opportunities for Immunotherapeutic Intervention against Myeloid Immunosuppression in Glioblastoma" Journal of Clinical Medicine 11, no. 4: 1069. https://doi.org/10.3390/jcm11041069
APA StyleExley, M. A., Garcia, S., Zellander, A., Zilberberg, J., & Andrews, D. W. (2022). Challenges and Opportunities for Immunotherapeutic Intervention against Myeloid Immunosuppression in Glioblastoma. Journal of Clinical Medicine, 11(4), 1069. https://doi.org/10.3390/jcm11041069