Abstract
Hyperphenylalaninemia (HPA), the most common amino acid metabolism disorder, is caused by defects in enzymes involved in phenylalanine metabolism, with the consequent accumulation of phenylalanine and its secondary metabolites in body fluids and tissues. Clinical manifestations of HPA include mental retardation, and its early diagnosis with timely treatment can improve the prognosis of affected patients. Due to the genetic complexity and heterogeneity of HPA, high-throughput molecular technologies, such as next-generation sequencing (NGS), are becoming indispensable tools to fully characterize the etiology, helping clinicians to promptly identify the exact patients’ genotype and determine the appropriate treatment. In this review, after a brief overview of the key enzymes involved in phenylalanine metabolism, we represent the wide spectrum of genes and their variants associated with HPA and discuss the utility of genomic testing for improved diagnosis and clinical management of HPA.
1. Introduction
A burden of phenylalanine (Phe) in the blood and other tissues is the hallmark of hyperphenylalaninemia (HPA), the most common inborn error of amino acid metabolism, with an incidence that varies widely among ethnic and geographical regions around the world [1,2]. HPA is often the result of genetic alterations in the phenylalanine hydroxylase (PAH) gene, encoding an enzyme catalyzing the conversion of L-Phe to L-Tyrosine (Tyr), but it may also derive from defects in genes encoding enzymes involved in the biosynthesis or regeneration of the cofactor tetrahydrobiopterin (BH4) [3]. Although HPA is primarily characterized by progressive mental retardation, distinctive genotypes associated with HPA have different effects on the severity and prognosis of the disease and the response of patients to therapy [4,5]. To avoid irreversible damage to the nervous system, it is essential to perform an early and accurate diagnosis and begin the appropriate treatment in a timely manner.
To date, many countries in the world have implemented newborn screening (NBS) programs that allow the diagnosis of HPA and elicit a prompt therapy, which is often based on a diet throughout life [6,7]. However, traditional differential diagnosis methods are time consuming and are nowadays inadequate to capture the extensive genetic heterogeneity of HPA. In this context, high-throughput technologies, such as multiplex ligation-dependent probe amplification, DNA microarray and next-generation sequencing (NGS), allow the simultaneous analysis of multiple genetic variants associated with this heterogeneous disorder and, thus, optimize patient care and management [8,9,10,11,12,13].
In this review, after a brief overview of the key enzymes involved in Phe metabolism, we represent the wide spectrum of genes and their variants associated with HPA and discuss the utility of genomic testing for improved diagnosis and clinical management of HPA. To introduce the readers to genomic testing, we exemplify the workflow and illustrate time and cost of targeted NGS applied to HPA.
2. Enzymes Involved in Phe Metabolism
To better highlight the effects of enzymatic deficiencies on Phe metabolism, all the enzymes directly or indirectly involved in Phe catabolism are illustrated in Figure 1 [14]. Phe is an essential amino acid exclusively obtained by the diet or by endogenous proteolysis. Following cellular exogenous uptake through specific transporters, Phe is converted in L-Tyr by PAH, an enzyme mainly expressed in the liver and kidney, which represents the rate-limiting step in Phe catabolism [15,16,17]. This conversion is dependent on tetrahydrobiopterin (BH4), as a cofactor, molecular oxygen and iron [18]. When PAH is nonfunctional, Phe accumulates in the body and is converted by different enzymes into phenylpyruvic acid, a substance that is normally produced only in small quantities. Individuals with mutations of PAH excrete in the urine large quantities of phenylpyruvic acid along with Phe, a condition known as phenylketonuria (PKU) [19].
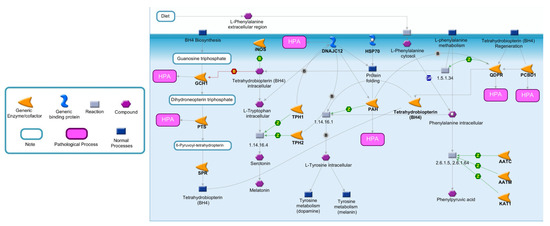
Figure 1.
Metabolic pathway of Phe and BH4. Phe metabolism is crucial for protein synthesis, as well as for the synthesis of Tyr and its derivatives. The major catabolic pathway involves Phe hydroxylation to Tyr by PAH. In one of the minor pathways, Phe may undergo conversion to Phenylpyruvic acid. An essential cofactor and regulator of PAH is BH4, whose biosynthesis and regeneration depend by different enzymes. Abbreviations: AATC, Aspartate aminotransferase, cytoplasmic; AATM, Aspartate aminotransferase, mitochondrial; DCoH, Pterin-4-alpha-carbinolamine dehydratase; DHPR, Dihydropteridine reductase; DNAJC12, DNAJ homolog subfamily C member 12; GCH1, Guanosine triphosphate (GTP) cyclohydrolase 1; KAT1, Kynurenine-oxoglutarate transaminase 1; PAH, Phenylalanine-4-hydroxylase; PTS, 6-pyruvoyl tetrahydrobiopterin synthase; SPR, Sepiapterin reductase; TPH1, Tryptophan 5-hydroxylase 1; TPH2, Tryptophan 5-hydroxylase 2; PCBD1, Pterin-4a-carbinolamine dehydratase; EC number 1.5.1.34: Dihydrobiopterin + NADH = NAD+ + Tetrahydrobiopterin; EC number 1.14.16.1: O2 + Tetrahydrobiopterin + S-Methylcysteine = S-Methylcysteine-sulfoxide + H2O + Dihydrobiopterin; EC number: 1.14.16.4 L-Tryptophan + O2 + Tetrahydrobiopterin = 5-Hydroxyl-L-Tryptophan+4alpha-Hydroxytetrahydrobiopterin; EC number 2.6.1.5-2.6.1.64: L-Phenylalanine + 2-Oxoglutaric acid = L-Glutamic acid + Phenylpyruvic acid.
PAH activity depends on BH4, a cofactor also involved in the hydroxylation of intracellular tryptophan (a precursor of serotonin and melatonin) and Tyr (a precursor of dopamine and melanin), as well as the synthesis of nitric oxide synthase and the cleavage of lipid ethers into glycerol and the corresponding aldehyde [20,21]. As illustrated in Figure 1, under normal conditions, the de novo biosynthesis of BH4 from guanosine triphospate (GTP) is catalyzed by three enzymes: GTP cyclohydrolase I (GCH1), 6-pyruvoyl-tetrahydropterin synthase (PTS) and sepiapterin reductase (SPR) [18,22,23]. An alternative or salvage pathway involves the regeneration of BH4 from dihydrobiopterin by pterin-4-alpha-carbinolamine dehydratase (PCBD1) and quinoid dihydropteridine reductase (QDPR) [24]. Although the major controlling point in BH4 biosynthesis is GCH1, defects in all enzymes, with the exception of SPR, can be a cause of HPA.
The proper folding and degradation of PAH is regulated by DNAJC12, a member of the subclass of the DNAJ/Hsp40 family of cochaperones, which modulate the activity of molecular chaperone Hsp70 [25]. In particular, DNAJC12 directly interacts with PAH and may play a role in the Hsp70-assisted folding of PAH and in the processing of misfolded ubiquitinated PAH [26]. The deficiency of DNAJC12 leads to decreased PAH protein levels and activity [27].
3. Genetics of Hyperphenylalaninemia
In Table 1, we list all the genes associated with HPA. About 98% of cases are caused by loss-of-function mutations in PAH that, as described before, encodes the enzyme performing the rate-limiting step in Phe catabolism. In a few cases, HPA is associated with mutations of DNAJC12, whose encoded protein controls proper folding and degradation of PAH [26,27]. In the remaining cases, HPA originates from defects in genes encoding enzymes involved in the biosynthesis (GCH1, PTS) or regeneration (PCBD1, QDPR) of BH4, the active cofactor of PAH [28]. Below is a description of each of these genes and their allelic variants.

Table 1.
Genetic causes of HPA.
HPA derived from mutations of PAH (chromosome 12q23.2, 13 exons) shows an autosomal recessive (AR) inheritance (Table 1). To date, more than 1000 mutations have been described and reported in the locus-specific PAH database (http://www.biopku.org, accessed on 23 December 2021), including single-nucleotide variants (SNVs), short insertions and deletions (InDels) and large structural variants (SVs) [29,30,31].
DNAJC12 (chromosome 10q21.3, 6 exons), also known as JDP1 or HPANBH4, encodes for a heat shock co-chaperone family member protein involved in proper folding of PAH [32]. The destabilization of this enzyme caused by AR mutations with subsequent loss of Phe, Tyr and neuronal tryptophan hydroxylases activity, leads to HPA and neurotransmitter deficiency [33,34] (Table 1). To date, different pathogenic or likely pathogenic variants have been associated with mild and non-BH4-deficient HPA, causing nonsense, frameshift, missense and splice-site mutations [35,36]. Recently, new heterozygous mutations in DNAJC12 were found by whole exome sequencing (WES), further supporting the importance of high-throughput screening methods for discovering and improving the neurodevelopmental outcome of HPA patients [32].
GCH1 (chromosome 14q22.2, 7 exons) encodes the first and rate-limiting enzyme of BH4 biosynthesis [18]. Its deficiency causes DOPA-responsive dystonia with or without HPA [37]. The most common dominant form, known as Segawa disease, responds well to dopamine replacement therapy, whereas the recessive form is more severe and is associated with malignant HPA [38,39,40]. In some patients with autosomal recessive GCH1 deficiency, the diagnosis can be late due to normal blood phenylalanine levels at NBS [41]. Different pathogenic GCH1 variants are known for producing a variety of molecular consequences (Table 1).
PTS (chromosome 11q23.1, 6 exons) encodes 6-pyruvoyl-tetrahydropterin synthase, an enzyme involved in the catalytic conversion of dihydroneopterin triphosphate to 6-pyruvoyl-tetrahydropterin and elimination of inorganic triphosphate from dihydroneopterin triphosphate, which is the second and irreversible step in the biosynthesis of BH4 [42,43]. Autosomal recessive genetic variations in PTS, which account for approximately 60% of all BH4 deficiencies, are associated with severe or mild forms of HPA [11,44]. Deletions, duplications, insertion and single nucleotide PTS variants (Table 1) can result in decreased or null enzyme activity, thus leading to little or no BH4 production and consequently to toxic levels of Phe in blood and other tissues [42].
PCBD1 (chromosome 10q22.1, 6 exons) encodes for Pterin-4 Alpha-Carbinolamine Dehydratase 1, an enzyme involved in the regeneration of BH4 whose defects are associated with a benign transient form of HPA [45]. Different PCBD1 variants (missense, frameshift, nonsense, and small deletions in exon 2 and 4) may reduce enzyme activity and result in pathogenic effects [39,46] (Table 1).
QDPR (chromosome 4p15.32, 7 exons) encodes the enzyme quinoid dihydropteridine reductase, catalyzing the regeneration of BH4 from quinonoid dihydropteridine (qBH2) [47]. Deficiency of QDPR causes an atypical PKU form due to the insufficient production of BH4 associated with severe neurological deterioration, microcephalia, psychomotor retardation, delayed development, tonal abnormalities, myoclonic epilepsy and dystonia [48]. Several pathogenic or likely pathogenic QDPR variants have been described [49] (Table 1).
4. Differential Diagnosis
Deficiencies in PAH or its cofactor BH4 can affect Phe homeostasis and lead to HPA. Most of the clinical manifestations associated with HPA are attributable to the increased levels of Phe and the depletion of monoamine neurotransmitters in the central nervous system [50]. The precise and early diagnosis of HPA represents the most important goal to avoid its harmful effects [51]. Indeed, the progressive neurologic manifestations, which include movement disorders, seizures, mental retardation, dyskinesias, microcephaly and hyperthermia, can be prevented or reduced with the choice of an early diagnosis and the right therapy [2,52]. The first step in the diagnostic strategy is the definition of the HPA subtypes. HPA with diverse severity degrees can be distinguished by different circulating blood Phe levels (a value up to 120 μmol/L is considered normal), response to diet and type of impaired enzymatic activity [53]. Specifically, patients with HPA can be classified as classic PKU (>1200 μM), moderate PKU (900–1200 μM), mild PKU (600–900 μM), mild HPA (<600 μM) or BH4 deficiency [54,55,56].
Tests used to diagnose and monitor patients with various degrees of severity of HPA include the quantification of Phe and Tyr concentrations by tandem mass spectrometry, the evaluation of pterin concentrations (neopterin, biopterin, primapterin, anapterin, and 6-oxo-primapterin) in urine or blood, the evaluation of PAH enzymatic activity in liver and kidney tissues, and the use of molecular genetic assays to screen for pathogenic variants in genes involved in HPA [57,58]. The latter is performed in infants with high levels of Phe and mainly involves genetic tests for PAH and/or other genes involved in the Phe metabolic pathway [59]. PAH mutations vary in their consequences for the residual level of PAH activity, from having little or no effect to abolishing PAH activity completely [60,61]. Once HPA is diagnosed at an early stage, the use of a specific diet can help to reduce the clinical outcomes of this disease. The use of BH4, alone or in addition to diet, can be used to further lower elevated blood Phe levels [3]. To this regard, sapropterin dihydrochloride (Kuvan, BioMarin Pharmaceutical Inc.) represents an orally active synthetic form of BH4 effective therapy that can be used in selected patients with HPA and mild-to-moderate PKU following a BH4 loading test [62,63].
Although biochemical NBS tests represent reliable diagnostic tools, they do not allow to identify the causes responsible for high Phe levels, which may be also transient and related to different factors, such as medical therapies, prematurity, liver metabolic immaturity, and parenteral nutrition [64]. Moreover, NBS tests are restricted to a limited number of metabolites associated with PHA, while the Phe metabolic pathway (Figure 1) is complex and involves additional key mediators with enzymatic, transporter or regulatory functions [14]. Since different metabolic phenotypes of HPA exist and depend upon variations in six different genes (Table 1), reaching a precise differential diagnosis and classification is not easy. To this end, genetic testing represents a suitable approach for a better genotype/phenotype correlation and, hopefully, improving the development of future innovative therapeutic interventions, such as gene therapy [65].
5. Current and Future Therapy for HPA
Without effective treatments, most people with HPA would develop neurological manifestations, among which intellectual disability is the most severe form [66]. To prevent neurological injuries, the mainstay of treatment for PAH deficiency consists of a carefully controlled Phe-restricted diet beginning the first days or weeks of life [67]. When diet therapy starts in early childhood, it helps to prevent the main manifestations of this metabolic disorder, although this treatment may not be as effective as the patient has to follow a complicated and unpleasant diet throughout the life. Adherence to dietary therapy in adolescents and adults is poor with up to 85–90% of patients exhibiting blood Phe concentrations above target levels [68,69]. Consequently, it is easy to assist to the development of a range of unsatisfactory outcomes, including neuropsychiatric symptoms [70]. The need to evaluate innovative therapies against HPA led researchers to investigate new ways to deal with this metabolic disorder, searching for new treatments that are not strictly dependent on dietary protein restriction. One of these is the gene correction strategy, which replaces defective genes with healthy ones and represents an attractive approach to the treatment of genetic diseases [71]. Thanks to the advent of new gene therapy technologies, such as the clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 system, which has revolutionized the field of molecular biology and medicine, the chance to cure genetic disorders such as HPA may not be far away [72,73,74,75,76,77,78,79,80,81,82]. In the perspective of gene therapy for HPA, its comprehensive genomic assessment will be necessary to group patients into diagnostic, prognostic or therapeutical clusters.
6. Genomic Testing to Improve the Management of HPA
In recent years, we have witnessed a new revolution in genetic testing, made possible by the fields of genomics and high-throughput technologies [83]. The field of genomics has evolved into a powerful approach to gain new biological insights, study the molecular pathways underlying health and disease, and the interaction between genes to find new approaches for the diagnosis, care delivery and development of therapies [84,85]. Based on these advances, we believe that genomic testing is not only useful in HPA, where the underlying causes are a number of genes and associated variants [86], but is nowadays feasible for clinical use [87,88]. Table 2 shows the advantages and challenges of various high-throughput methods. Among these, NGS represents the most powerful tool that may rapidly and effectively analyze HPA-associated genes, providing accurate results with a faster turnaround and a lower cost than traditional methods [89,90]. NGS-based targeted gene panels (TGPs) are particularly ideal for analyzing specific mutations or genes associated with HPA [11]. They offer greater coverage of selected regions of interest, faster turnaround time, and more clinically relevant data compared to broader genomic profiling, such as WES or whole genome sequencing (WGS) approaches [91] or CGH Microarray analysis [8,9,10,11]. The advantages of using TGPs are many: (i) they can be customized for different sample types and specific genomic regions of interest; (ii) the use of lower input amounts (1 ng compared to 100 ng required for WES); (iii) the possibility to identify rare variants; (iv) a workflow simpler and shorter than WES; (v) the possibility to process thousands of samples in a single sequencing run; and (vi) a minor cost than WGS, WES or CGH microarray analysis [92,93] (Table 2).
Table 2.
Advantages and challenges of various high-throughput methods.
In HPAs, the use of NGS-based TGP technology to search for new or rare variants may bring out a hitherto unexplored complexity and help to explain atypical phenotypes [94]. Back in 2014, Trujillano et al. showed that shifting from Sanger methods to high-throughput targeted resequencing improves differential diagnosis of HPA and produces a quicker establishment of specifically tailored treatments. The benefits also include a 60–80% cost savings per sample and a faster diagnostic process compared to traditional techniques [11]. In the same year, Y. Cao et al. used a customized NGS-based panel to detect mutations in HPA-related genes (PAH, PTS, QDPR, GCH1, and PCBD1), which provided a broader coverage, higher throughput, and a faster and more efficient solution compared with traditional molecular methods [95]. A 2017 study demonstrated the successful use of NGS to detect known and novel (one in PAH and two in PTS) causative mutations in PKU and BH4-deficiency cases, enabling accurate diagnosis and the appropriate effective treatment of patients [96,97].
Although different NGS platforms have been implemented, all NGS methods include steps performed on the laboratory bench (“wet bench”) and data analyses performed with bioinformatics pipelines (“dry bench”) [98,99]. Figure 2 shows a schematic representation of the NGS-based TGPs workflow performed with the Ion Torrent and the Illumina technologies.
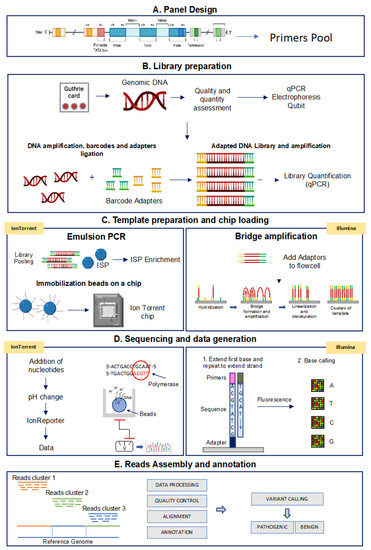
Figure 2.
Analytical workflow of targeted sequencing for the Ion Torrent and Illumina methods. (A) Panel design: the Designer software helps to create custom assays based on PCR target selection. (B) Library preparation: library construction is the preparation of the nucleic acid target into a form compatible with the sequencing system to be used. (C) Template preparation and chip loading: target enrichment is used in NGS workflows to capture only genomic DNA regions of interest. (D) Sequencing and data generation: IonTorrent platform: microwells of the chip is flooded by nucleotides that when binding to the complementary nucleotide on a template, release an ion. At each flow, the electrical signal at each well is measured, indicating that a reaction has occurred; Illumina platform: the fragments are clonally amplified on the slide utilizing fluorescently labeled reversible-terminator nucleotides; (E) Read assembly and annotation: starting from Binary Alignment Map (BAM) and Variant Call Format (VCF) files, variants are prioritized based on localization, functional effect, mode of inheritance, coverage and Minor Allelic Frequency (MAF) to obtain disease-correlated variants.
In the next sections, we represent the main steps of a TGP-based NGS analysis.
Panel design: a custom panel can be designed using the Ion AmpliSeq Designer Tool for Ion Torrent platform (Thermo Fisher Scientific) and the DesignStudio Sequencing Assay Designer for Illumina, and information contained in the NCBI (National Center for Biotechnology Information) ClinVar reference databases can be used to identify the clinical relevance of the identified variants. The two Designer tools allow the easy selection of genes ID or chromosomal coordinates across scientifically curated gene sets. The number of primers depends on the complexity and size of the genomic region to be analyzed. To estimate the number of samples that can be sequenced in multiplex assays, users need to consider different parameters, such as expected sequencing coverage and used chip type. In general, a 30× minimum coverage is recommended for germline detection mutations. Table 3 shows the key features of a custom panel created with both the AmpliSeq Designer Tool for the Ion Torrent platform and the DesignStudio Sequencing Assay Designer for the Illumina platform to analyze the genes associated with HPA. As indicated, less than 60 amplicons are needed to screen a total of 6 HPA related genes, with a 100% coverage per single amplicon.
Table 3.
NGS-targeted custom panels designed with AmpliSeqTM Designer Tool for Ion Torrent platform, and DesignStudio Sequencing Assay Designer for Illumina platform to analyze HPA related genes.
Library preparation: the first step of NGS-based TGPs workflow (Figure 2) involves library preparation. Genomic DNA is PCR amplified with the designed panel primers (see above) and then specific barcode adapters are incorporated to allow the later clonal amplification of libraries and the identification of each sample read after the pooling of libraries.
Template preparation and chip loading: following library quantification and normalization, the libraries can be pooled and used for template preparation. In this step, using the Ion Torrent platform, an emulsion-based PCR-amplification of each amplicon is performed around Ion Sphere Particles (ISPs) containing a primer complementary to one of the adapters added during the library preparation. When the concentration of the libraries is optimized, one sample amplicon is amplified around each ISP (clonal amplification). As a final step, the DNA strands are separated, and the single strands anchored to the ISP are ready to be loaded on the microwells of the semiconductor Ion Chip. In the Illumina platform, once the DNA is amplified and the adapters are added, the modified DNA is loaded onto a flow cell where the amplification and sequencing take place. The flow cell contains nanowells that space out fragments and help with overcrowding. Each nanowell contains oligonucleotides that provide an anchoring point for the adapters to attach. Once the fragments are attached, a phase called cluster generation begins. This step makes about a thousand copies of each fragment of DNA and it is performed by bridge amplification PCR.
Sequencing and data analysis: in this step, when using the Ion Torrent platform, microwells are flooded with a single species of deoxyribonucleotide triphosphate (dNTP). If the introduced dNTP is incorporated into the growing complementary strand, the release of a hydrogen ion triggers an ISFET ion sensor, which indicates that a reaction has occurred. The series of electrical pulses transmitted from each microwell of the chip to a computer is translated real time into a DNA sequence, which is then aligned to a genome and analyzed for the presence of variants. The Illumina platform, instead, adopts a sequencing-by-synthesis approach, utilizing fluorescently labeled reversible-terminator nucleotides, on clonally amplified DNA templates (bridge amplification) immobilized to an acrylamide coating on the surface of a glass flow cell.
Read assembly and annotation: the informatic pipeline includes different steps, such as signal processing, base calling, alignment of reads to a reference genome and variant calling. The entire process is performed using appropriate analysis software for variant annotation. The performance of the sequencing run can be evaluated by analyzing different metrics, such as uniformity of base coverage, base coverage and on-target reads.
An example of a sequencing run report of the Ion Torrent S5 sequencing output is shown in Figure 3. In the secondary analysis, variants can be filtered by different parameters, such as p-value (p < 0.001), phred quality score (p-read > 20), variant effect (missense, unknown, synonymous, InDels, SNVs), location (exon, intronic, splice-site, 5–3 UTR), Minor Allelic Frequency (MAF: 0.01–0.5) and allele frequency (40–60% for heterozygous; 90–100% for homozygous). To select disease-relevant pathogenic variants, effect prediction is performed using SIFT (Sorting Intolerant From Tolerant), PolyPhen (Polymorphism Phenotyping) or Fathmann score [100,101,102].
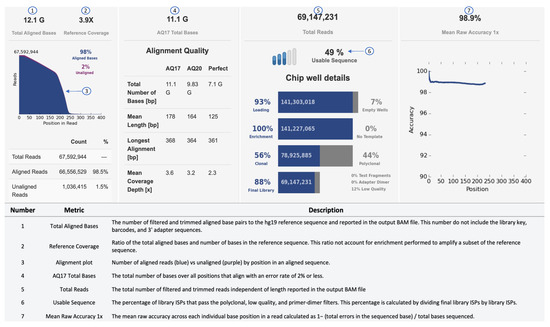
Figure 3.
Sequencing performance of Ion Torrent. Metrics, such as Raw Accuracy, Aligned Bases, and Total Reads, can be used to determine the performance of a run.
Considering the entire analytical NGS workflow (e.g., DNA isolation, library preparation and sequencing), the estimated cost for the analysis of HPA related genes is about EUR 120/sample (Table 3). This cost does not include equipment, labor or data analysis. The analysis of the HPA related genes using traditional methods would require higher costs and much longer times (Table 4).
Table 4.
Comparison between next-generation sequencing and Sanger sequencing.
7. Conclusions
HPA is the most commonly occurring amino acid metabolism genetic disorder characterized by serious clinical manifestations, including irreversible brain damage, intellectual deficiency and epilepsy. The precise and early diagnosis is remarkably successful in preventing these severe neurological features and ensuring healthy growth. Despite considerable progress having been made in the knowledge of this rare metabolic disorder, the diagnostic challenges are largely attributable to the marked clinical and genetic heterogeneity and the complexity of the Phe metabolic pathways involved, including additional unidentified key mediators with enzymatic, transporter, and regulatory functions. In this context, the advent of high-capacity and low-cost technologies and the use of ad hoc designed assays are producing a turning point for gene testing and clinical diagnosis of HPAs, improving our understanding of the basis of disease and the ability to better associate gene variants to specific phenotypes. The translation of fast, reliable and inexpensive genomic technologies into clinical practice will offer the opportunity for a better diagnosis of HPA in carrier patients, optimize clinical management, reduce the psychological burden and improve the development of early and effective therapeutic interventions.
Author Contributions
Conceptualization, E.A.T. and S.C.; Writing—Original draft, E.A.T.; Supervision, S.C.; Writing, review and editing, E.A.T., M.G., G.M. and S.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the PO FESR SICILIA 2014–2020 grant “PKU Smart-Sensor, realization and validation of a Point-of-Care system for home-testing monitoring of phenylalanine in patients suffering from hyperphenylalaninemia”, ID 08RG7211000341.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Blau, N.; Hennermann, J.B.; Langenbeck, U.; Lichter-Konecki, U. Diagnosis, classification, and genetics of phenylketonuria and tetrahydrobiopterin (BH4) deficiencies. Mol. Genet. Metab. 2011, 104, S2–S9. [Google Scholar] [CrossRef] [PubMed]
- Hillert, A.; Anikster, Y.; Belanger-Quintana, A.; Burlina, A.; Burton, B.K.; Carducci, C.; Chiesa, A.E.; Christodoulou, J.; Đorđević, M.; Desviat, L.R.; et al. The Genetic Landscape and Epidemiology of Phenylketonuria. Am. J. Hum. Genet. 2020, 107, 234–250. [Google Scholar] [CrossRef] [PubMed]
- Opladen, T.; Hoffmann, G.F.; Blau, N. An international survey of patients with tetrahydrobiopterin deficiencies presenting with hyperphenylalaninaemia. J. Inherit. Metab. Dis. 2012, 35, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Shintaku, H.; Ohura, T.; Takayanagi, M.; Kure, S.; Owada, M.; Matsubara, Y.; Yoshino, M.; Okano, Y.; Ito, T.; Okuyama, T.; et al. Guide for diagnosis and treatment of hyperphenylalaninemia. Pediatr. Int. 2021, 63, 8–12. [Google Scholar] [CrossRef]
- Levy, H.; Lamppu, D.; Anastosoaie, V.; Baker, J.L.; DiBona, K.; Hawthorne, S.; Lindenberger, J.; Kinch, D.; Seymour, A.; McIlduff, M.; et al. 5-year retrospective analysis of patients with phenylketonuria (PKU) and hyperphenylalaninemia treated at two specialized clinics. Mol. Genet. Metab. 2020, 129, 177–185. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Ma, D.; Zhang, Z.; Li, Y.; Yang, P.; Sun, Y.; Jiang, T. Neonatal screening and genotype-phenotype correlation of hyperphenylalaninemia in the Chinese population. Orphanet J. Rare Dis. 2021, 16, 214. [Google Scholar] [CrossRef]
- Muntau, A.C.; Du Moulin, M.; Feillet, F. Diagnostic and therapeutic recommendations for the treatment of hyperphenylalaninemia in patients 0–4 years of age. Orphanet J. Rare Dis. 2018, 13, 173. [Google Scholar] [CrossRef]
- de Baulny, H.O.; Abadie, V.; Feillet, F.; de Parscau, L. Management of phenylketonuria and hyperphenylalaninemia. J. Nutr. 2007, 137 (Suppl. 1), 1561S–1563S; discussion 1573S–1575S. [Google Scholar] [CrossRef]
- Waters, P.J. HowPAH gene mutations cause hyper-phenylalaninemia and why mechanism matters: Insights from in vitro expression. Hum. Mutat. 2003, 21, 357–369. [Google Scholar] [CrossRef]
- Calzone, K.A.; Jenkins, J.; Nicol, N.; Skirton, H.; Feero, W.G.; Green, E.D. Relevance of Genomics to Healthcare and Nursing Practice. J. Nurs. Sch. 2013, 45, 1–2. [Google Scholar] [CrossRef]
- Trujillano, D.; Pérez, B.; González, J.; Tornador, C.; Navarrete, R.; Escaramís, G.; Ossowski, S.; Armengol, L.; Cornejo, V.; Desviat, L.R.; et al. Accurate molecular diagnosis of phenylketonuria and tetrahydrobiopterin-deficient hyperphenylalaninemias using high-throughput targeted sequencing. Eur. J. Hum. Genet. 2013, 22, 528–534. [Google Scholar] [CrossRef] [PubMed]
- La Cognata, V.; Cavallaro, S. A Comprehensive, Targeted NGS Approach to Assessing Molecular Diagnosis of Lysosomal Storage Diseases. Genes 2021, 12, 1750. [Google Scholar] [CrossRef] [PubMed]
- La Cognata, V.; Guarnaccia, M.; Morello, G.; Ruggieri, M.; Polizzi, A.; Cavallaro, S. Design and Validation of a Custom NGS Panel Targeting a Set of Lysosomal Storage Diseases Candidate for NBS Applications. Int. J. Mol. Sci. 2021, 22, 10064. [Google Scholar] [CrossRef] [PubMed]
- Schuck, P.F.; Malgarin, F.; Cararo, J.H.; Cardoso, F.; Streck, E.L.; Ferreira, G.C. Phenylketonuria Pathophysiology: On the Role of Metabolic Alterations. Aging Dis. 2015, 6, 390–399. [Google Scholar]
- Kaufman, S. The Phenylalanine Hydroxylating System. Adv. Enzymol. Relat. Areas. Mol. Biol. 2006, 67, 77–264. [Google Scholar]
- Kaufman, S. Regulation of the activity of hepatic phenylalanine hydroxylase. Adv. Enzym. Regul. 1986, 25, 37–64. [Google Scholar] [CrossRef]
- Moller, N.; Meek, S.; Bigelow, M.; Andrews, J.; Nair, K.S. The kidney is an important site for in vivo phenylalanine-to-tyrosine conversion in adult humans: A metabolic role of the kidney. Proc. Natl. Acad. Sci. USA 2000, 97, 1242–1246. [Google Scholar] [CrossRef]
- Fanet, H.; Capuron, L.; Castanon, N.; Calon, F.; Vancassel, S. Tetrahydrobioterin (BH4) Pathway: From Metabolism to Neuropsychiatry. Curr. Neuropharmacol. 2021, 19, 591–609. [Google Scholar] [CrossRef]
- Mitchell, J.J.; Trakadis, Y.J.; Scriver, C.R. Phenylalanine hydroxylase deficiency. Genet. Med. 2011, 13, 697–707. [Google Scholar] [CrossRef]
- Kim, H.L.; Park, Y.S. Maintenance of cellular tetrahydrobiopterin homeostasis. BMB Rep. 2010, 43, 584–592. [Google Scholar] [CrossRef]
- Watschinger, K.; Keller, M.A.; Hermetter, A.; Golderer, G.; Werner-Felmayer, G.; Werner, E.R. Glyceryl ether monooxygenase resembles aromatic amino acid hydroxylases in metal ion and tetrahydrobiopterin dependence. Biol. Chem. 2008, 390, 3–10. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Werner, E.R.; Blau, N.; Thöny, B. Tetrahydrobiopterin: Biochemistry and pathophysiology. Biochem. J. 2011, 438, 397–414. [Google Scholar] [CrossRef] [PubMed]
- Bonafé, L.; Thöny, B.; Penzien, J.M.; Czarnecki, B.; Blau, N. Mutations in the Sepiapterin Reductase Gene Cause a Novel Tetrahydrobiopterin-Dependent Monoamine-Neurotransmitter Deficiency without Hyperphenylalaninemia. Am. J. Hum. Genet. 2001, 69, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Thony, B.; Auerbach, G.; Blau, N. Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochem. J. 2000, 347 Pt 1, 1–16. [Google Scholar] [CrossRef]
- Choi, J.; Djebbar, S.; Fournier, A.; Labrie, C. The co-chaperone DNAJC12 binds to Hsc70 and is upregulated by endoplasmic reticulum stress. Cell Stress Chaperones 2014, 19, 439–446. [Google Scholar] [CrossRef]
- Jung-Kc, K.; Himmelreich, N.; Prestegård, K.S.; Shi, T.S.; Scherer, T.; Ying, M.; Jorge-Finnigan, A.; Thöny, B.; Blau, N.; Martinez, A. Phenylalanine hydroxylase variants interact with the co-chaperone DNAJC12. Hum. Mutat. 2019, 40, 483–494. [Google Scholar] [CrossRef]
- Anikster, Y.; Haack, T.B.; Vilboux, T.; Pode-Shakked, B.; Thöny, B.; Shen, N.; Guarani, V.; Meissner, T.; Mayatepek, E.; Trefz, F.K.; et al. Biallelic Mutations in DNAJC12 Cause Hyperphenylalaninemia, Dystonia, and Intellectual Disability. Am. J. Hum. Genet. 2017, 100, 257–266. [Google Scholar] [CrossRef]
- Shintaku, H. Disorders of Tetrahydrobiopterin Metabolism and their Treatment. Curr. Drug Metab. 2002, 3, 123–131. [Google Scholar] [CrossRef]
- Neto, E.V.; Laranjeira, F.; Quelhas, D.; Ribeiro, I.; Seabra, A.; Mineiro, N.; Carvalho, L.D.M.; Lacerda, L.; Ribeiro, M.G. Mutation analysis of the PAH gene in phenylketonuria patients from Rio de Janeiro, Southeast Brazil. Mol. Genet. Genom. Med. 2018, 6, 575–591. [Google Scholar] [CrossRef]
- Wang, Z.-W.; Jiang, S.-W.; Zhou, B.-C. PAH mutation spectrum and correlation with PKU manifestation in north Jiangsu province population. Kaohsiung J. Med Sci. 2018, 34, 89–94. [Google Scholar] [CrossRef]
- Kuznetcova, I.; Gundorova, P.; Ryzhkova, O.; Polyakov, A. The study of the full spectrum of variants leading to hyperphenylalaninemia have revealed 10 new variants in the PAH gene. Metab. Brain Dis. 2019, 34, 1547–1555. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yang, Q.; Yi, S.; Qin, Z.; Luo, J.; Fan, X. Two novel mutations in DNAJC12 identified by whole-exome sequencing in a patient with mild hyperphenylalaninemia. Mol. Genet. Genom. Med. 2020, 8, e1303. [Google Scholar] [CrossRef] [PubMed]
- Veenma, D.; Cordeiro, D.; Sondheimer, N.; Mercimek-Andrews, S. DNAJC12-associated developmental delay, movement disorder, and mild hyperphenylalaninemia identified by whole-exome sequencing re-analysis. Eur. J. Hum. Genet. 2018, 26, 1867–1870. [Google Scholar] [CrossRef] [PubMed]
- van Spronsen, F.J.; Himmelreich, N.; Rüfenacht, V.; Shen, N.; van Vliet, D.; Al-Owain, M.; Ramzan, K.; Alkhalifi, S.M.; Lunsing, R.J.; Heiner-Fokkema, R.M.; et al. Heterogeneous clinical spectrum of DNAJC12-deficient hyperphenylalaninemia: From attention deficit to severe dystonia and intellectual disability. J. Med. Genet. 2018, 55, 249–253. [Google Scholar] [CrossRef]
- Gundorova, P.; Kuznetcova, I.A.; Baydakova, G.V.; Stepanova, A.A.; Itkis, Y.S.; Kakaulina, V.S.; Alferova, I.P.; Lyazina, L.V.; Andreeva, L.P.; Kanivets, I.; et al. BH4-deficient hyperphenylalaninemia in Russia. PLoS ONE 2021, 16, e0249608. [Google Scholar] [CrossRef] [PubMed]
- Odagiri, S.; Kabata, D.; Tomita, S.; Kudo, S.; Sakaguchi, T.; Nakano, N.; Yamamoto, K.; Shintaku, H.; Hamazaki, T. Clinical and Genetic Characteristics of Patients with Mild Hyperphenylalaninemia Identified by Newborn Screening Program in Japan. Int. J. Neonatal Screen. 2021, 7, 17. [Google Scholar] [CrossRef]
- Blau, N.; Bonafé, L.; Thöny, B. Tetrahydrobiopterin Deficiencies without Hyperphenylalaninemia: Diagnosis and Genetics of DOPA-Responsive Dystonia and Sepiapterin Reductase Deficiency. Mol. Genet. Metab. 2001, 74, 172–185. [Google Scholar] [CrossRef]
- Giri, S.; Naiya, T.; Roy, S.; Das, G.; Wali, G.M.; Das, S.K.; Ray, K.; Ray, J. A Compound Heterozygote for GCH1 Mutation Represents a Case of Atypical Dopa-Responsive Dystonia. J. Mol. Neurosci. 2019, 68, 214–220. [Google Scholar] [CrossRef]
- Thöny, B.; Blau, N. Mutations in the BH4-metabolizing genes GTP cyclohydrolase I, 6-pyruvoyl-tetrahydropterin synthase, sepiapterin reductase, carbinolamine-4a-dehydratase, and dihydropteridine reductase. Hum. Mutat. 2006, 27, 870–878. [Google Scholar] [CrossRef]
- Dayasiri, K.C.; Suraweera, N.; Nawarathne, D.; Senanayake, U.E.; Dayanath, B.K.T.P.; Jasinge, E.; Weerasekara, K. GTP-Cyclohydrolase I deficiency presenting as malignant hyperphenylalaninemia, recurrent hyperthermia and progressive neurological dysfunction in a South Asian child—A case report. BMC Pediatr. 2019, 19, 199. [Google Scholar] [CrossRef]
- Opladen, T.; Hoffmann, G.; Hörster, F.; Hinz, A.-B.; Neidhardt, K.; Klein, C.; Wolf, N. Clinical and biochemical characterization of patients with early infantile onset of autosomal recessive GTP cyclohydrolase I deficiency without hyperphenylalaninemia. Mov. Disord. 2011, 26, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Leuzzi, V.; Carducci, C.A.; Carducci, C.L.; Pozzessere, S.; Burlina, A.; Cerone, R.; Concolino, D.; Donati, M.A.; Fiori, L.; Meli, C.; et al. Phenotypic variability, neurological outcome and genetics background of 6-pyruvoyl-tetrahydropterin synthase deficiency. Clin. Genet. 2010, 77, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Hara, S.; Fukumura, S.; Ichinose, H. Reversible S-glutathionylation of human 6-pyruvoyl tetrahydropterin synthase protects its enzymatic activity. J. Biol. Chem. 2019, 294, 1420–1427. [Google Scholar] [CrossRef]
- Vatanavicharn, N.; Kuptanon, C.; Liammongkolkul, S.; Liu, T.-T.; Hsiao, K.-J.; Ratanarak, P.; Blau, N.; Wasant, P. Novel mutation affecting the pterin-binding site of PTS gene and review of PTS mutations in Thai patients with 6-pyruvoyltetrahydropterin synthase deficiency. J. Inherit. Metab. Dis. 2009, 32 (Suppl. 1), S279–S282. [Google Scholar] [CrossRef]
- MacKenzie, D.W.; Odds, F.C. Non-identity and authentication of two major reference strains of Candida albicans. J. Med. Vet. Mycol. 1991, 29, 255–261. [Google Scholar] [CrossRef]
- Thöny, B.; Neuheiser, F.; Kierat, L.; Blaskovics, M.; Arn, P.H.; Ferreira, P.; Rebrin, I.; Ayling, J.; Blau, N. Hyperphenylalaninemia with High Levels of 7-Biopterin is Associated with Mutations in the PCBD Gene Encoding the Bifunctional Protein Pterin-4a-Carbinolamine Dehydratase and Transcriptional Coactivator (DCoH). Am. J. Hum. Genet. 1998, 62, 1302–1311. [Google Scholar] [CrossRef]
- Longo, N. Disorders of biopterin metabolism. J. Inherit. Metab. Dis. 2009, 32, 333–342. [Google Scholar] [CrossRef]
- Romstad, A.; Kalkanoğlu, H.S.; Coşkun, T.; Demirkol, M.; Tokatli, A.; Dursun, A.; Baykal, T.; Özalp, I.; Guldberg, P.; Güttler, F. Molecular analysis of 16 Turkish families with DHPR deficiency using denaturing gradient gel electrophoresis (DGGE). Qual. Life Res. 2000, 107, 546–553. [Google Scholar] [CrossRef]
- Li, N.; Yu, P.; Rao, B.; Deng, Y.; Guo, Y.; Huang, Y.; Ding, L.; Zhu, J.; Yang, H.; Wang, J.; et al. Molecular genetics of tetrahydrobiopterin deficiency in Chinese patients. J. Pediatr. Endocrinol. Metab. 2018, 31, 911–916. [Google Scholar] [CrossRef]
- Winn, S.R.; Scherer, T.; Thöny, B.; Ying, M.; Martinez, A.; Weber, S.; Raber, J.; Harding, C.O. Blood phenylalanine reduction corrects CNS dopamine and serotonin deficiencies and partially improves behavioral performance in adult phenylketonuric mice. Mol. Genet. Metab. 2018, 123, 6–20. [Google Scholar] [CrossRef]
- Zurflüh, M.R.; Giovannini, M.; Fiori, L.; Fiege, B.; Gokdemir, Y.; Baykal, T.; Kierat, L.; Gärtner, K.H.; Thöny, B.; Blau, N. Screening for tetrahydrobiopterin deficiencies using dried blood spots on filter paper. Mol. Genet. Metab. 2005, 86, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Weglage, J.; Wiedermann, D.; Denecke, J.; Feldmann, R.; Koch, H.-G.; Ullrich, K.; Moller, H. Individual blood-brain barrier phenylalanine transport in siblings with classical phenylketonuria. J. Inherit. Metab. Dis. 2002, 25, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Cleary, M.; Trefz, F.; Muntau, A.C.; Feillet, F.; van Spronsen, F.J.; Burlina, A.; Bélanger-Quintana, A.; Giżewska, M.; Gasteyger, C.; Bettiol, E.; et al. Fluctuations in phenylalanine concentrations in phenylketonuria: A review of possible relationships with outcomes. Mol. Genet. Metab. 2013, 110, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Camp, K.M.; Parisi, M.A.; Acosta, P.B.; Berry, G.T.; Bilder, D.A.; Blau, N.; Bodamer, O.A.; Brosco, J.P.; Brown, C.S.; Burlina, A.B.; et al. Phenylketonuria Scientific Review Conference: State of the science and future research needs. Mol. Genet. Metab. 2014, 112, 87–122. [Google Scholar] [CrossRef]
- Vockley, J.; Andersson, H.C.; Antshel, K.M.; Braverman, N.E.; Burton, B.K.; Frazier, D.M.; Mitchell, J.; Smith, W.E.; Thompson, B.H.; Berry, S.A. Phenylalanine hydroxylase deficiency: Diagnosis and management guideline. Genet. Med. 2014, 16, 188–200. [Google Scholar] [CrossRef]
- Heintz, C.; Cotton, R.G.; Blau, N. Tetrahydrobiopterin, its Mode of Action on Phenylalanine Hydroxylase, and Importance of Genotypes for Pharmacological Therapy of Phenylketonuria. Hum. Mutat. 2013, 34, 927–936. [Google Scholar] [CrossRef]
- Groselj, U.; Murko, S.; Tansek, M.Z.; Kovač, J.; Bakija, A.T.; Lampret, B.R.; Battelino, T. Comparison of tandem mass spectrometry and amino acid analyzer for phenylalanine and tyrosine monitoring—Implications for clinical management of patients with hyperphenylalaninemia. Clin. Biochem. 2015, 48, 14–18. [Google Scholar] [CrossRef]
- McBride, K.L.; Pluciniczak, J.; Rhyand, T.; Bartholomew, D. Phenylalanine and tyrosine measurements across gestation by tandem mass spectrometer on dried blood spot cards from normal pregnant women. Genet. Med. 2019, 21, 1821–1826. [Google Scholar] [CrossRef]
- Tolve, M.; Artiola, C.; Pasquali, A.; Giovanniello, T.; D’Amici, S.; Angeloni, A.; Pizzuti, A.; Carducci, C.; Leuzzi, V.; Carducci, C. Molecular Analysis of PKU-Associated PAH Mutations: A Fast and Simple Genotyping Test. Methods Protoc. 2018, 1, 30. [Google Scholar] [CrossRef]
- Romano, V.; Lio, D.; Cali, F.; Scola, L.; Leggio, L.; D’Anna, C.; De Leo, G.; Salerno, A. A methodological strategy for PAH genotyping in populations with a marked molecular heterogeneity of hyperphenylalaninemia. Mol. Cell. Probes 2001, 15, 13–19. [Google Scholar] [CrossRef]
- Kim, S.-W.; Jung, J.; Oh, H.-J.; Kim, J.; Lee, K.-S.; Lee, D.-H.; Park, C.; Kimm, K.; Koo, S.K.; Jung, S.-C. Structural and functional analyses of mutations of the human phenylalanine hydroxylase gene. Clin. Chim. Acta 2006, 365, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Eshraghi, P.; Asl, S.N.; Bagheri, S.; Chalak, V. Response to sapropterin hydrochloride (Kuvan(R)) in children with phenylketonuria (PKU): A clinical trial. J. Pediatr. Endocrinol. Metab. 2019, 32, 885–888. [Google Scholar] [CrossRef] [PubMed]
- Trefz, F.K.; Muntau, A.C.; Lagler, F.B.; Moreau, F.; Alm, J.; Burlina, A.; Rutsch, F.; Bélanger-Quintana, A.; Feillet, F.; KAMPER Investigators. The Kuvan((R)) Adult Maternal Paediatric European Registry (KAMPER) Multinational Observational Study: Baseline and 1-Year Data in Phenylketonuria Patients Responsive to Sapropterin. JIMD Rep. 2015, 23, 35–43. [Google Scholar] [PubMed]
- Van Wegberg, A.M.J.; Macdonald, A.; Ahring, K.; BéLanger-Quintana, A.; Blau, N.; Bosch, A.M.; Burlina, A.; Campistol, J.; Feillet, F.; Giżewska, M.; et al. The complete European guidelines on phenylketonuria: Diagnosis and treatment. Orphanet J. Rare Dis. 2017, 12, 162. [Google Scholar] [CrossRef] [PubMed]
- Lowe, W.L.; Reddy, T.E. Genomic approaches for understanding the genetics of complex disease. Genome Res. 2015, 25, 1432–1441. [Google Scholar] [CrossRef]
- Anderson, P.; Wood, S.J.; Francis, D.E.; Coleman, L.; Anderson, V.; Boneh, A. Are Neuropsychological Impairments in Children with Early-Treated Phenylketonuria (PKU) Related to White Matter Abnormalities or Elevated Phenylalanine Levels? Dev. Neuropsychol. 2007, 32, 645–668. [Google Scholar] [CrossRef]
- MacLeod, E.L.; Ney, D.M. Nutritional Management of Phenylketonuria. Annales Nestlé (Engl. Ed.) 2010, 68, 58–69. [Google Scholar] [CrossRef]
- Walter, J.H.; White, F.J. Blood phenylalanine control in adolescents with phenylketonuria. Int. J. Adolesc. Med. Health 2004, 16, 41–46. [Google Scholar] [CrossRef]
- Jurecki, E.; Cederbaum, S.; Kopesky, J.; Perry, K.; Rohr, F.; Sanchez-Valle, A.; Viau, K.; Sheinin, M.; Cohen-Pfeffer, J. Adherence to clinic recommendations among patients with phenylketonuria in the United States. Mol. Genet. Metab. 2017, 120, 190–197. [Google Scholar] [CrossRef]
- Bilder, D.A.; Noel, J.K.; Baker, E.R.; Irish, W.; Chen, Y.; Merilainen, M.J.; Prasad, S.; Winslow, B.J. Systematic Review and Meta-Analysis of Neuropsychiatric Symptoms and Executive Functioning in Adults with Phenylketonuria. Dev. Neuropsychol. 2016, 41, 245–260. [Google Scholar] [CrossRef]
- Harding, C.O.; Gillingham, M.B.; Hamman, K.; Clark, H.; Goebel-Daghighi, E.; Bird, A.; Koeberl, D.D. Complete correction of hyperphenylalaninemia following liver-directed, recombinant AAV2/8 vector-mediated gene therapy in murine phenylketonuria. Gene Ther. 2006, 13, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Rees, H.A.; Liu, D.R. Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018, 19, 770–788. [Google Scholar] [CrossRef] [PubMed]
- Adikusuma, F.; Piltz, S.; Corbett, M.A.; Turvey, M.; McColl, S.R.; Helbig, K.; Beard, M.R.; Hughes, J.; Pomerantz, R.T.; Thomas, P.Q. Large deletions induced by Cas9 cleavage. Nature 2018, 560, E8–E9. [Google Scholar] [CrossRef]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef]
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. CRISPR–Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018, 24, 927–930. [Google Scholar] [CrossRef]
- Ihry, R.J.; Worringer, K.A.; Salick, M.R.; Frias, E.; Ho, D.; Theriault, K.; Kommineni, S.; Chen, J.; Sondey, M.; Ye, C.; et al. p53 inhibits CRISPR–Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018, 24, 939–946. [Google Scholar] [CrossRef]
- Zeballos, C.M.; Gaj, T. Next-Generation CRISPR Technologies and Their Applications in Gene and Cell Therapy. Trends Biotechnol. 2021, 39, 692–705. [Google Scholar] [CrossRef]
- Grisch-Chan, H.M.; Schwank, G.; Harding, C.O.; Thöny, B. State-of-the-Art 2019 on Gene Therapy for Phenylketonuria. Hum. Gene Ther. 2019, 30, 1274–1283. [Google Scholar] [CrossRef]
- Molla, K.A.; Yang, Y. CRISPR/Cas-Mediated Base Editing: Technical Considerations and Practical Applications. Trends Biotechnol. 2019, 37, 1121–1142. [Google Scholar] [CrossRef]
- Villiger, L.; Grisch-Chan, H.M.; Lindsay, H.; Ringnalda, F.; Pogliano, C.B.; Allegri, G.; Fingerhut, R.; Häberle, J.; Matos, J.; Robinson, M.D.; et al. Treatment of a metabolic liver disease by in vivo genome base editing in adult mice. Nat. Med. 2018, 24, 1519–1525. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, R.A.; Carlson, D.F.; Allen, K.L.; Webster, D.A.; VanLith, C.J.; Nicolas, C.T.; Hillin, L.G.; Yu, Y.; Kaiser, C.W.; Wahoff, W.R.; et al. Development of a porcine model of phenylketonuria with a humanized R408W mutation for gene editing. PLoS ONE 2021, 16, e0245831. [Google Scholar] [CrossRef] [PubMed]
- Lightbody, G.; Haberland, V.; Browne, F.; Taggart, L.; Zheng, H.; Parkes, E.; Blayney, J.K. Review of applications of high-throughput sequencing in personalized medicine: Barriers and facilitators of future progress in research and clinical application. Brief. Bioinform. 2019, 20, 1795–1811. [Google Scholar] [CrossRef] [PubMed]
- Green, E.D.; Gunter, C.; Biesecker, L.G.; Di Francesco, V.; Easter, C.L.; Feingold, E.A.; Felsenfeld, A.L.; Kaufman, D.J.; Ostrander, E.A.; Pavan, W.J.; et al. Strategic vision for improving human health at The Forefront of Genomics. Nature 2020, 586, 683–692. [Google Scholar] [CrossRef]
- Voy, B.H. Systems Genetics: A Powerful Approach for Gene-Environment Interactions. J. Nutr. 2011, 141, 515–519. [Google Scholar] [CrossRef]
- Rajabi, F.; Levy, H.L. Hyperphenylalaninemia and the genomic revolution. Mol. Genet. Metab. 2015, 114, 380–381. [Google Scholar] [CrossRef]
- Bilkey, G.A.; Burns, B.L.; Coles, E.P.; Bowman, F.L.; Beilby, J.P.; Pachter, N.S.; Baynam, G.; Dawkins, H.J.S.; Nowak, K.J.; Weeramanthri, T.S. Genomic Testing for Human Health and Disease Across the Life Cycle: Applications and Ethical, Legal, and Social Challenges. Front. Public Health 2019, 7, 40. [Google Scholar] [CrossRef]
- Dixon-Salazar, T.J.; Silhavy, J.L.; Udpa, N.; Schroth, J.; Bielas, S.; Schaffer, A.E.; Olvera, J.; Bafna, V.; Zaki, M.S.; Abdel-Salam, G.H.; et al. Exome Sequencing Can Improve Diagnosis and Alter Patient Management. Sci. Transl. Med. 2012, 4, 138ra78. [Google Scholar] [CrossRef]
- Di Resta, C.; Galbiati, S.; Carrera, P.; Ferrari, M. Next-generation sequencing approach for the diagnosis of human diseases: Open challenges and new opportunities. EJIFCC 2018, 29, 4–14. [Google Scholar]
- Fernandez-Marmiesse, A.; Gouveia, S.; Couce, M.L. NGS Technologies as a Turning Point in Rare Disease Research, Diagnosis and Treatment. Curr. Med. Chem. 2018, 25, 404–432. [Google Scholar] [CrossRef]
- Xue, Y.; Ankala, A.; Wilcox, W.R.; Hegde, M.R. Solving the molecular diagnostic testing conundrum for Mendelian disorders in the era of next-generation sequencing: Single-gene, gene panel, or exome/genome sequencing. Genet. Med. 2015, 17, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Brunelli, L.; Jenkins, S.M.; Gudgeon, J.M.; Bleyl, S.B.; Miller, C.E.; Tvrdik, T.; Dames, S.A.; Ostrander, B.; Daboub, J.A.F.; Zielinski, B.A.; et al. Targeted gene panel sequencing for the rapid diagnosis of acutely ill infants. Mol. Genet. Genom. Med. 2019, 7, e00796. [Google Scholar] [CrossRef] [PubMed]
- Reid, E.S.; Papandreou, A.; Drury, S.; Boustred, C.; Yue, W.W.; Wedatilake, Y.; Beesley, C.; Jacques, T.S.; Anderson, G.; Abulhoul, L.; et al. Advantages and pitfalls of an extended gene panel for investigating complex neurometabolic phenotypes. Brain 2016, 139, 2844–2854. [Google Scholar] [CrossRef] [PubMed]
- Russell, L.E.; Schwarz, U.I. Variant discovery using next-generation sequencing and its future role in pharmacogenetics. Pharmacogenomics 2020, 21, 471–486. [Google Scholar] [CrossRef]
- Cao, Y.-Y.; Qu, Y.-J.; Song, F.; Zhang, T.; Bai, J.-L.; Jin, Y.-W.; Wang, H. Fast clinical molecular diagnosis of hyperphenylalaninemia using next-generation sequencing-based on a custom AmpliSeq™ panel and Ion Torrent PGM sequencing. Mol. Genet. Metab. 2014, 113, 261–266. [Google Scholar] [CrossRef]
- Chaiyasap, P.; Ittiwut, C.; Srichomthong, C.; Sangsin, A.; Suphapeetiporn, K.; Shotelersuk, V. Massive parallel sequencing as a new diagnostic approach for phenylketonuria and tetrahydrobiopterin-deficiency in Thailand. BMC Med Genet. 2017, 18, 102. [Google Scholar] [CrossRef]
- Trier, C.; Fournous, G.; Strand, J.M.; Stray-Pedersen, A.; Pettersen, R.D.; Rowe, A.D. Next-generation sequencing of newborn screening genes: The accuracy of short-read mapping. NPJ Genom. Med. 2020, 5, 36. [Google Scholar] [CrossRef]
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of Next-Generation Sequencing Technologies. Curr. Protoc. Mol. Biol. 2018, 122, e59. [Google Scholar] [CrossRef]
- Kumar, K.R.; Cowley, M.J.; Davis, R.L. Next-Generation Sequencing and Emerging Technologies. Semin. Thromb. Hemost. 2019, 45, 661–673. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 7, 7.20.1–7.20.41. [Google Scholar] [CrossRef] [PubMed]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.A.; Edwards, K.J.; Day, I.N.M.; Gaunt, T.R. Predicting the Functional, Molecular, and Phenotypic Consequences of Amino Acid Substitutions using Hidden Markov Models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).