
Single and Multiple Dose PK–PD Characterization for Carisoprodol. Part I: Pharmacokinetics, Metabolites, and 2C19 Phenotype Influence. Double-Blind, Placebo-Controlled Clinical Trial in Healthy Volunteers

 ,
,
Abstract
:1. Introduction
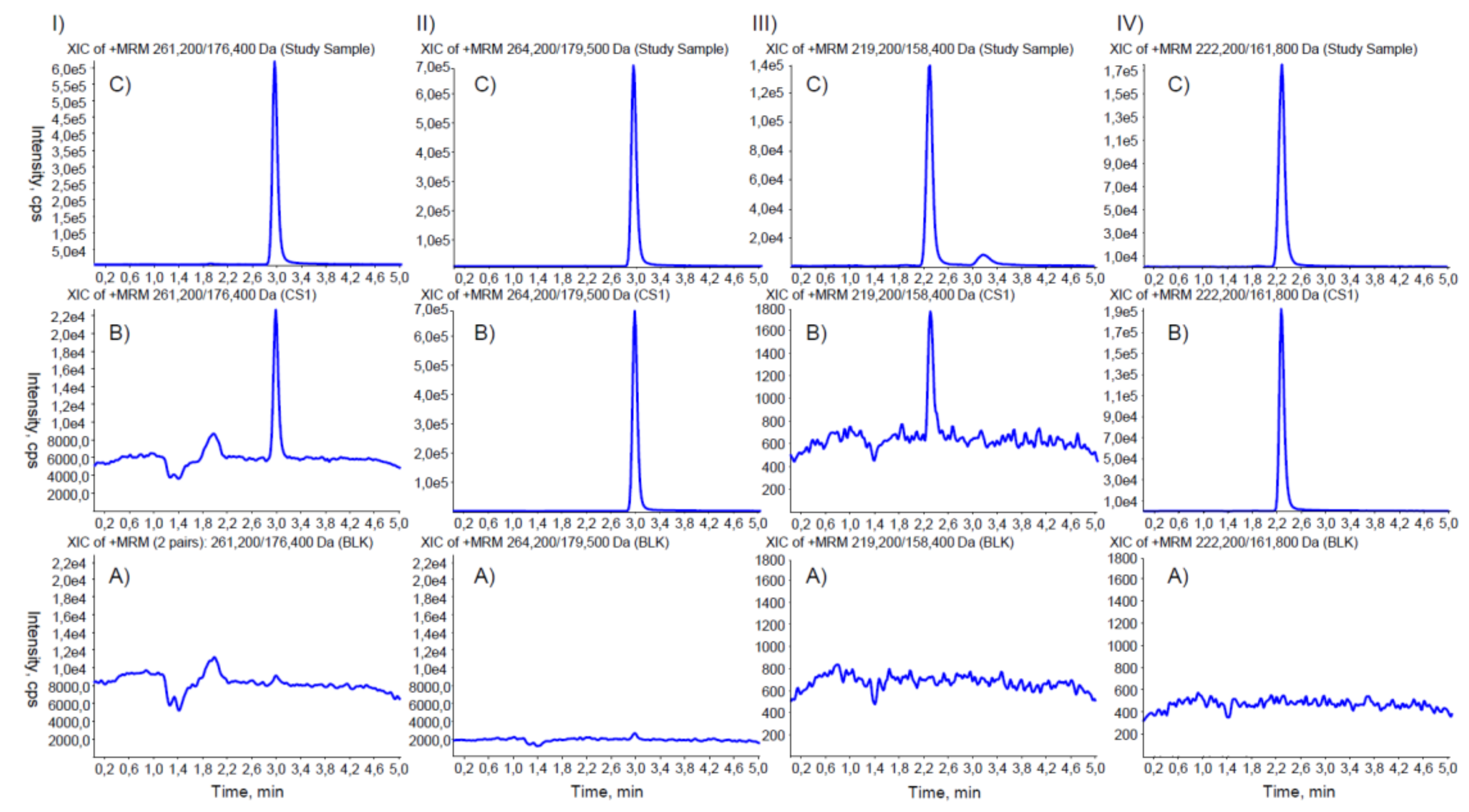
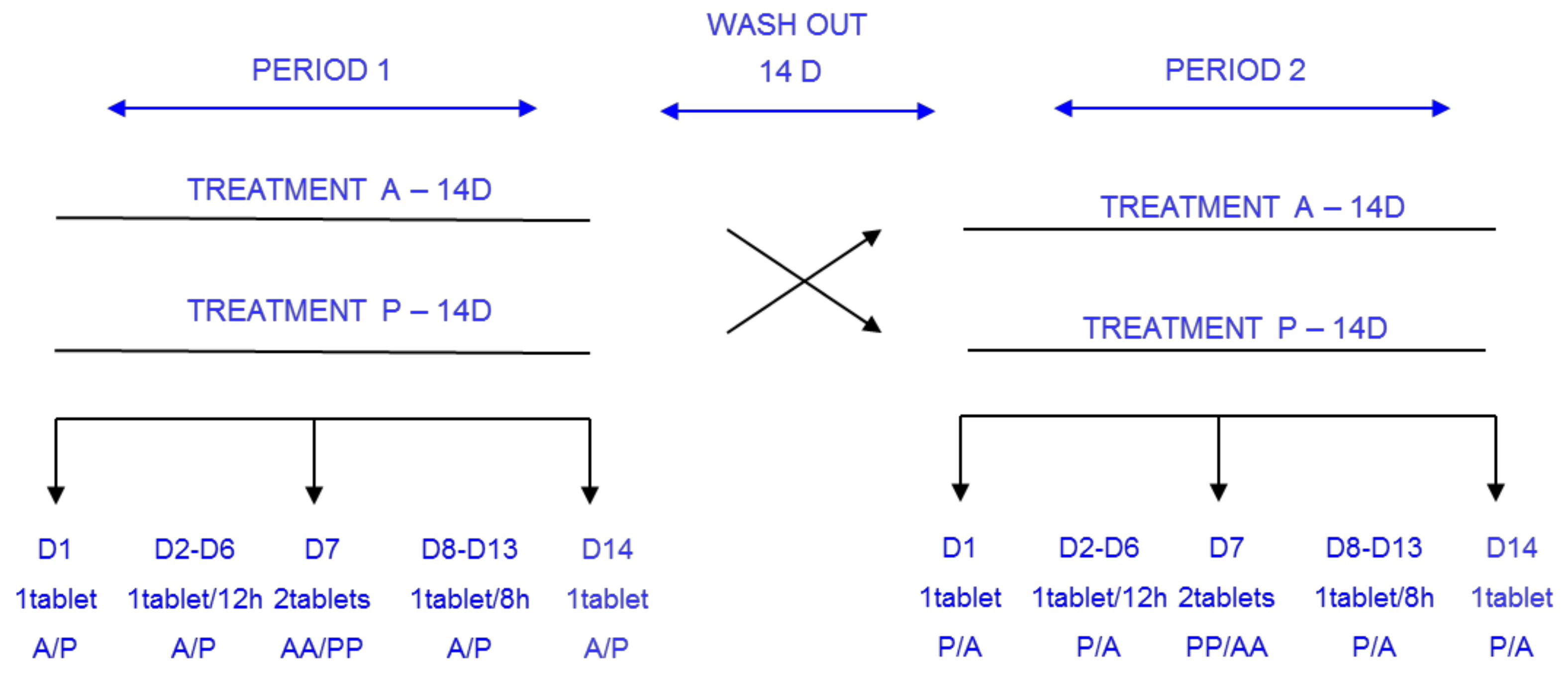
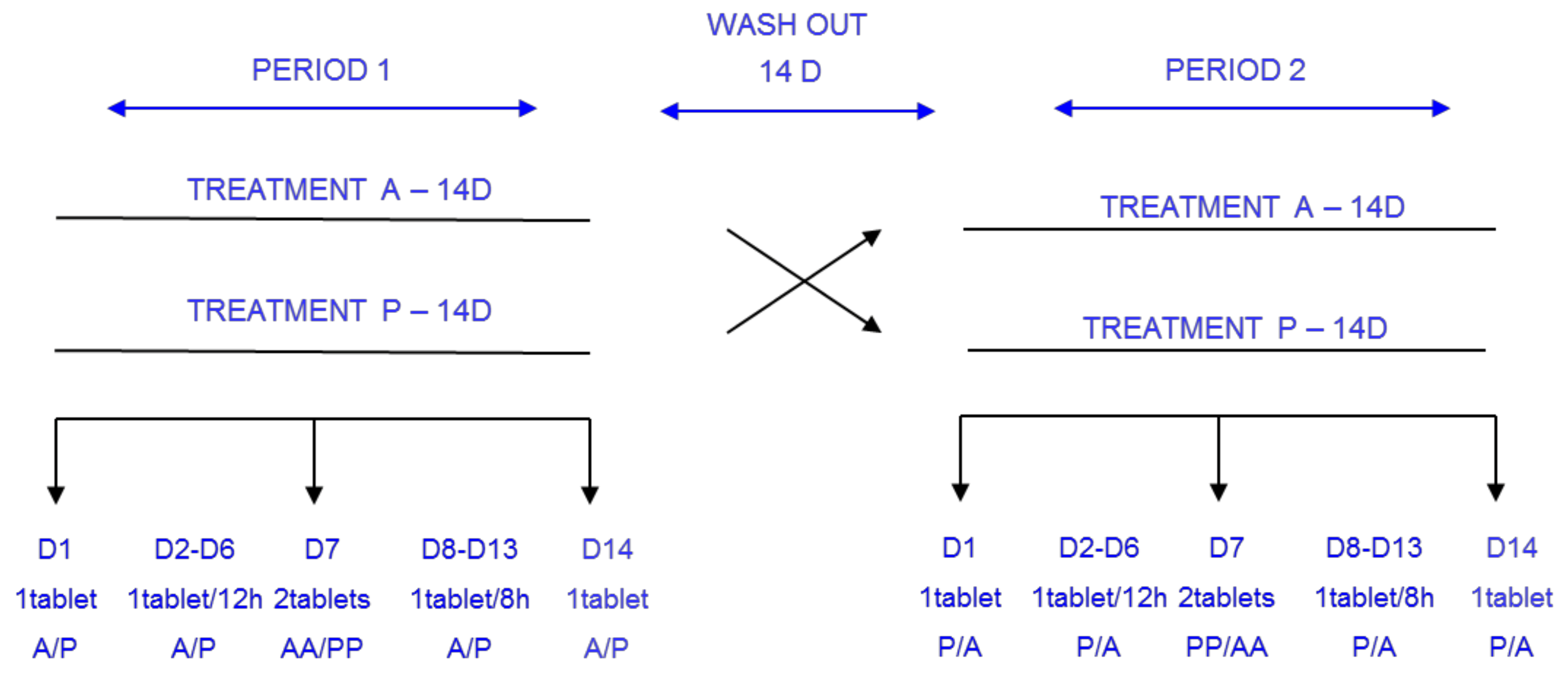
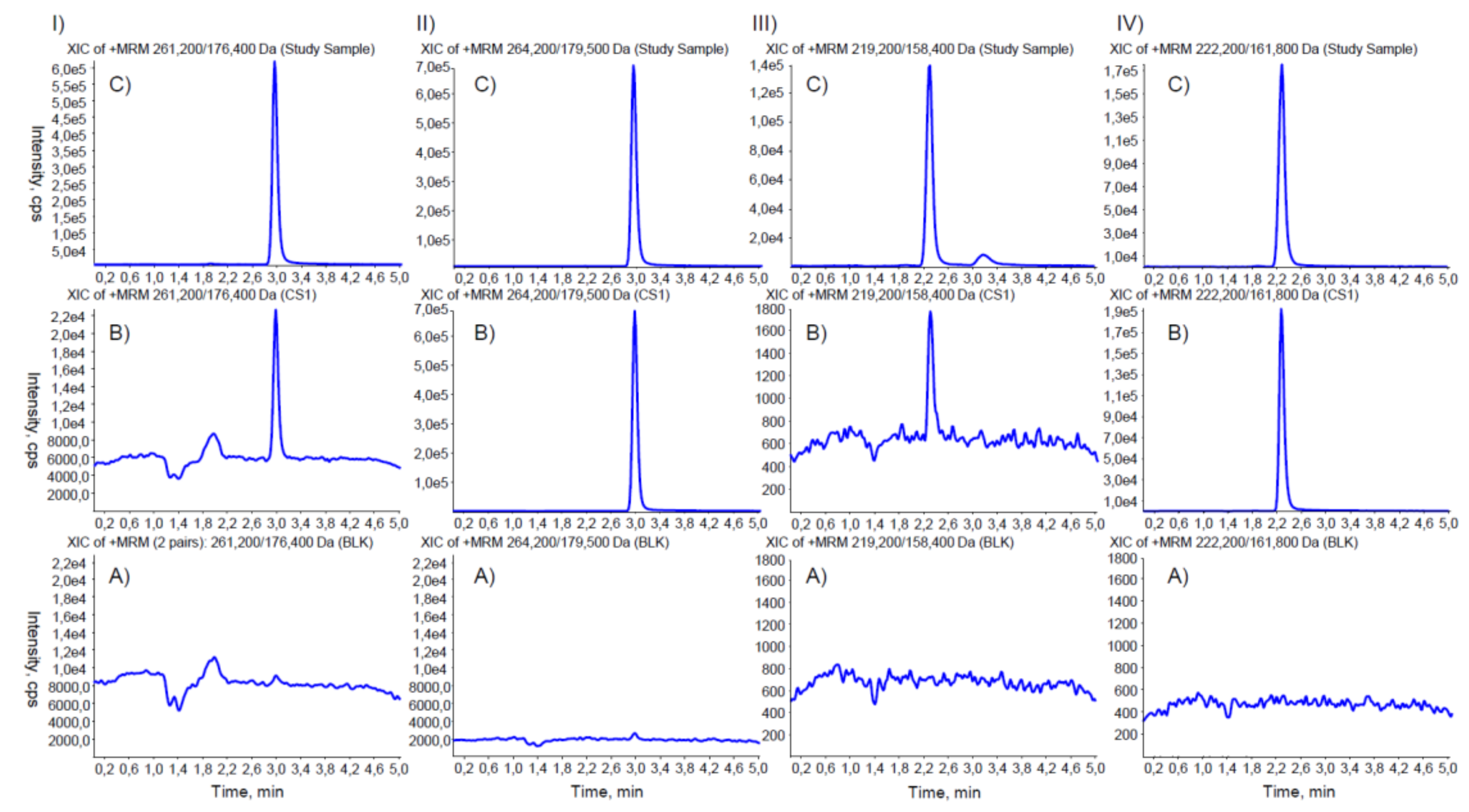
2. Materials and Methods
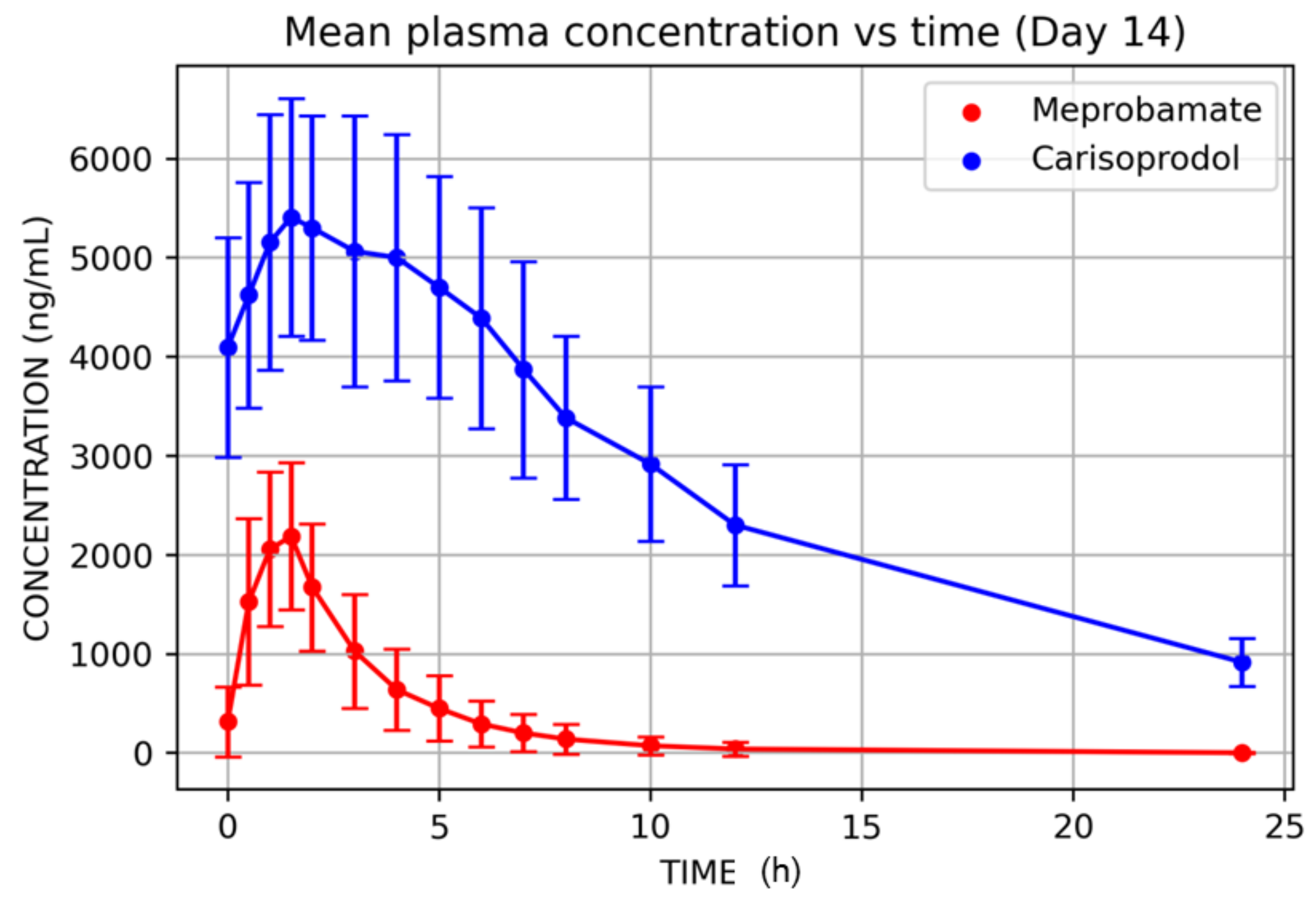
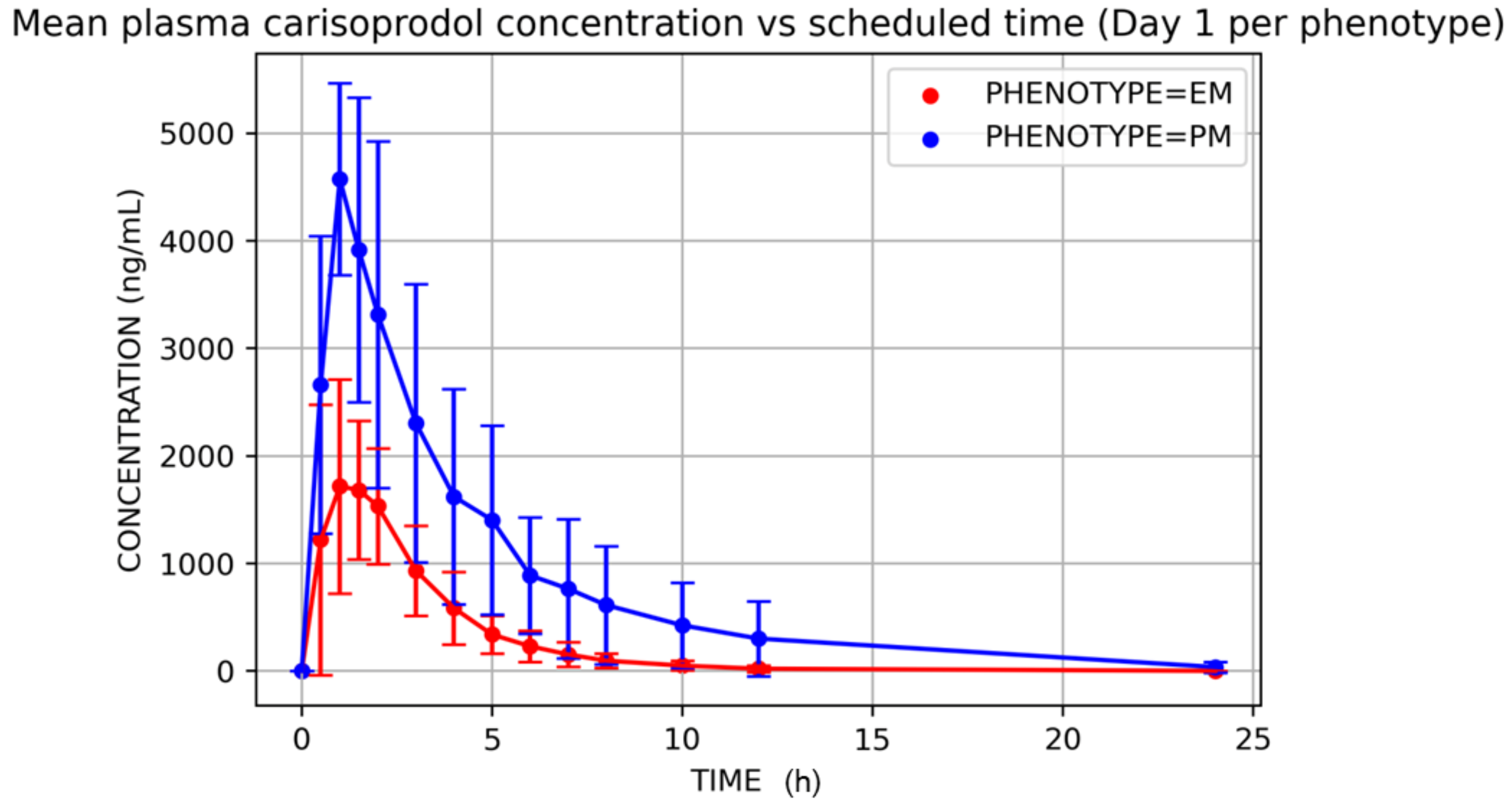
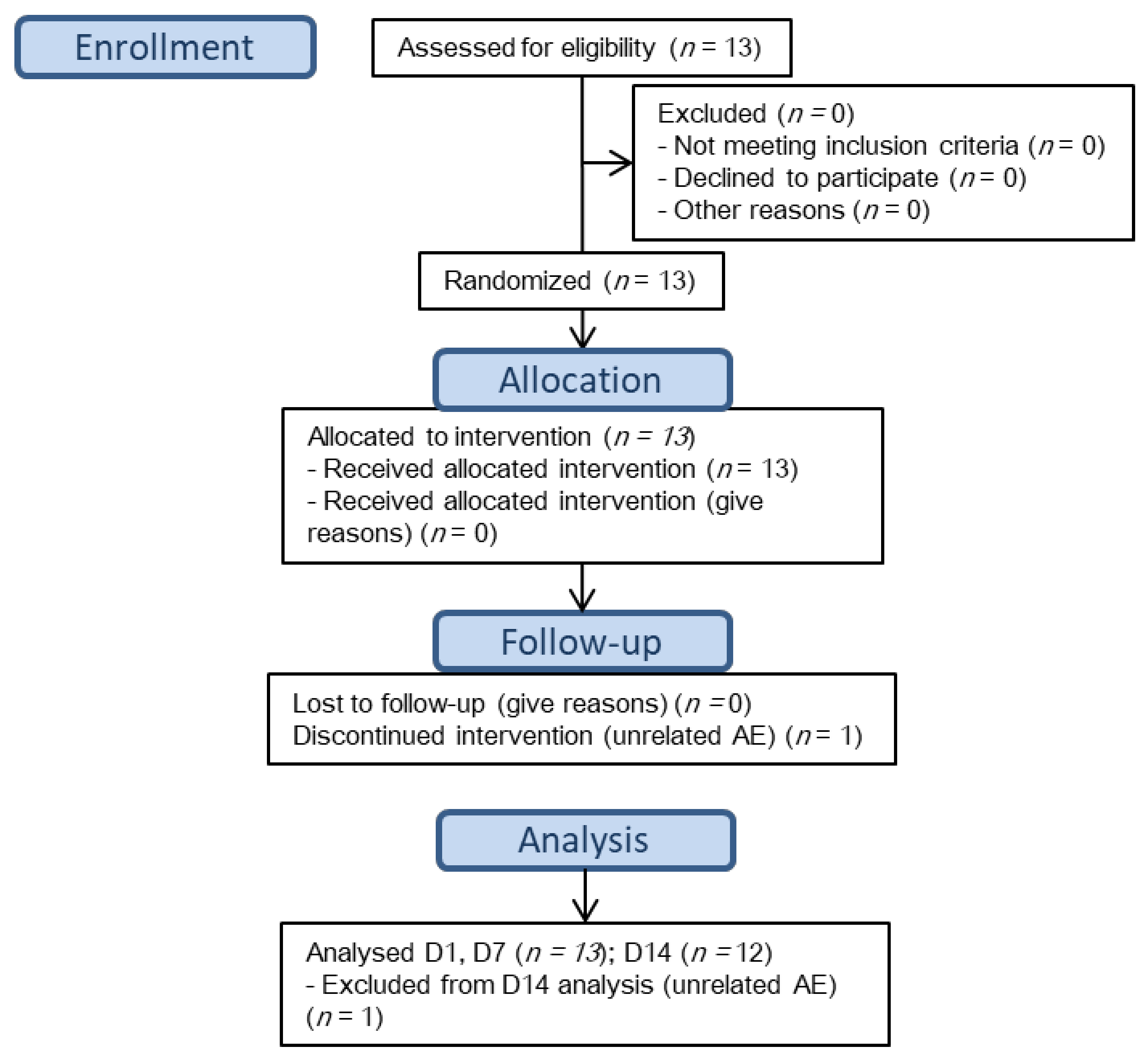
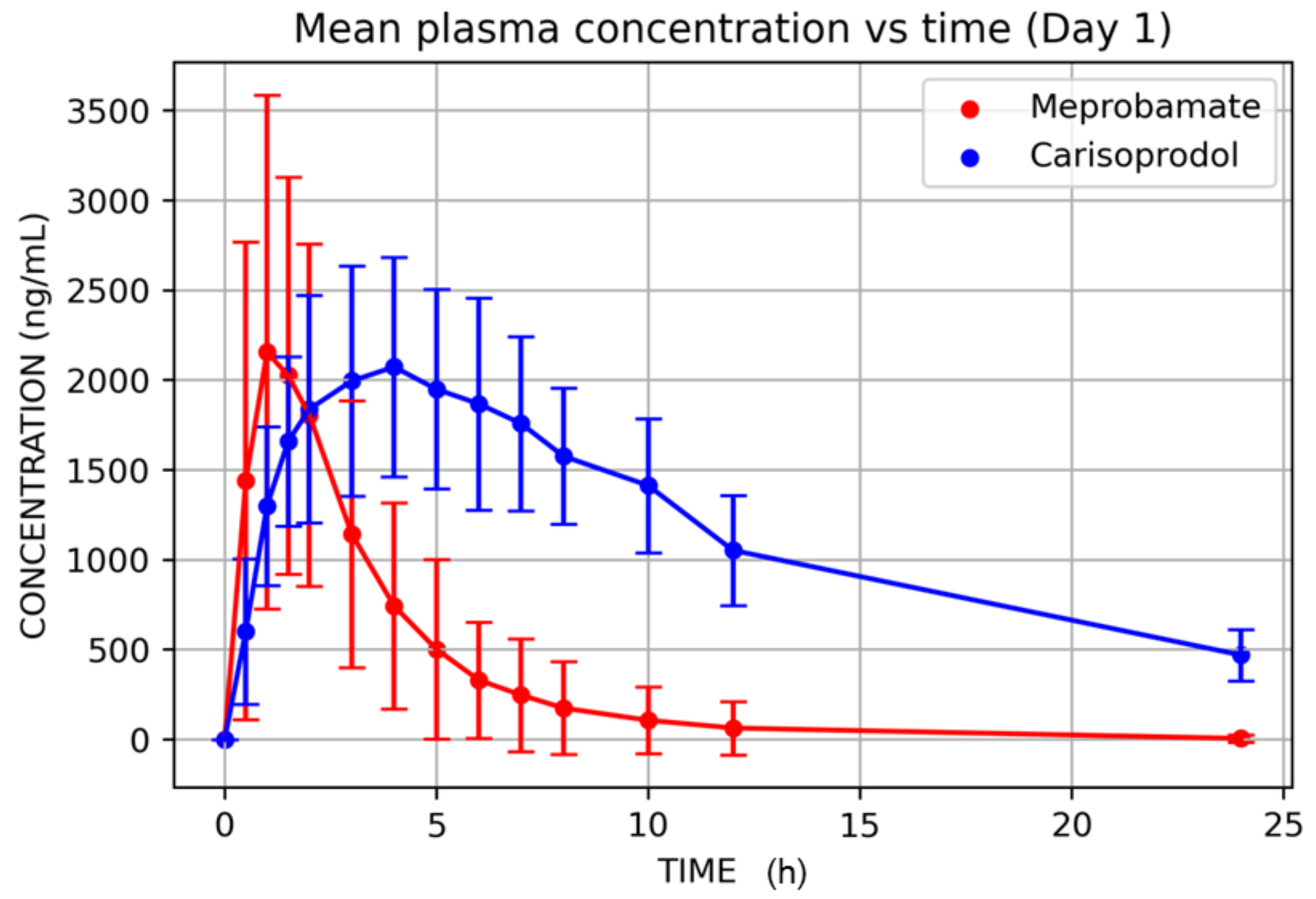
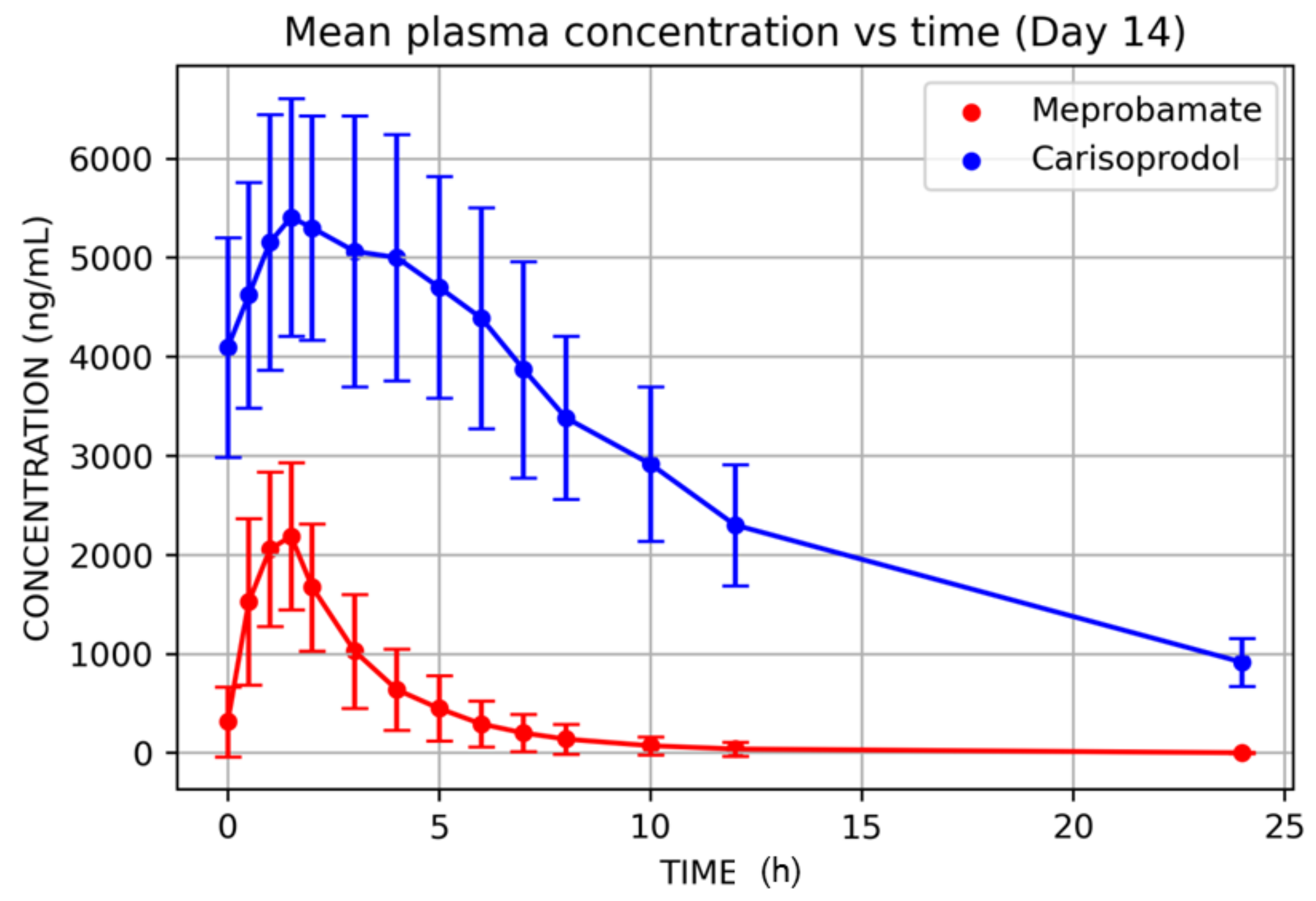
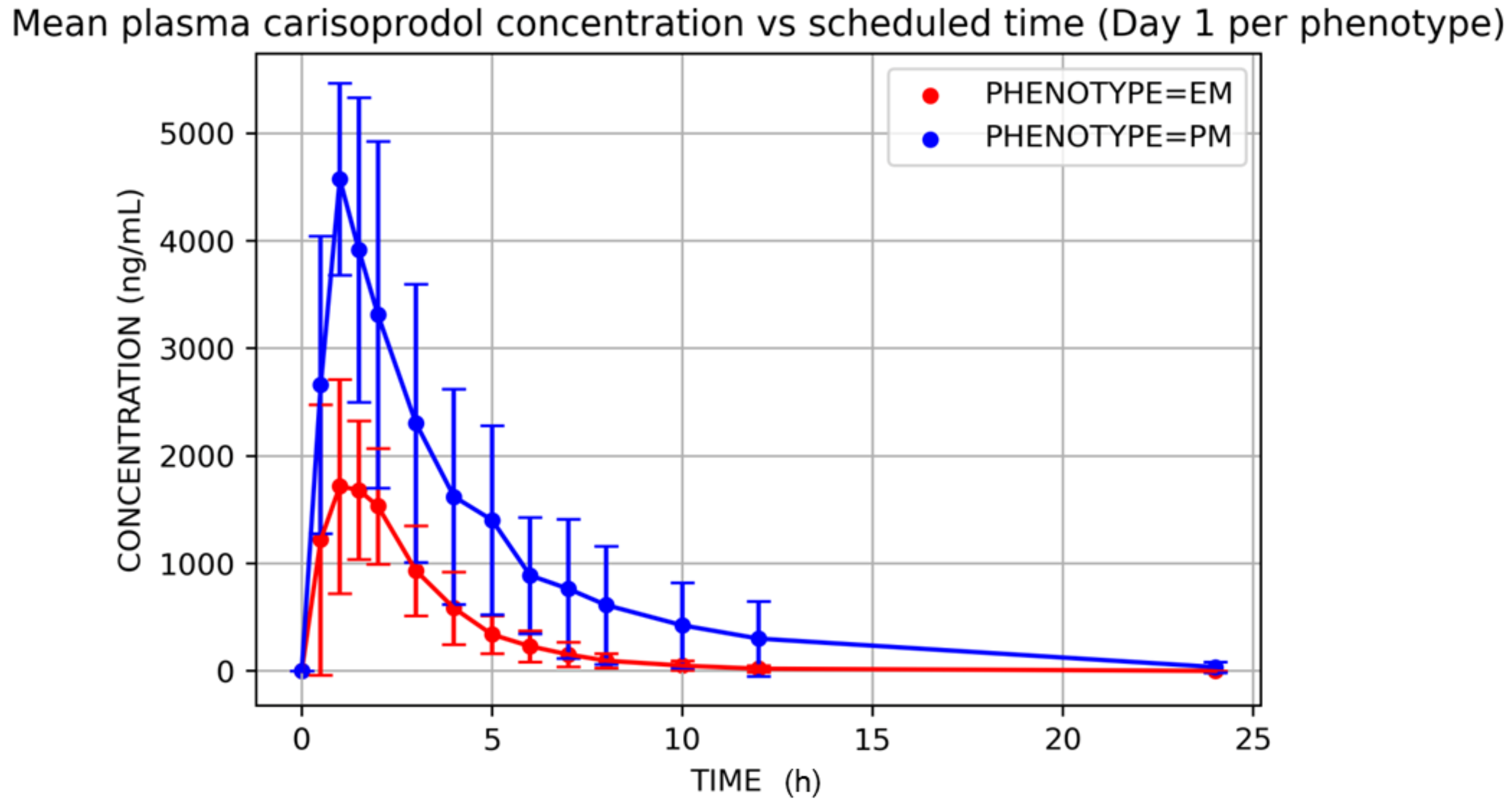
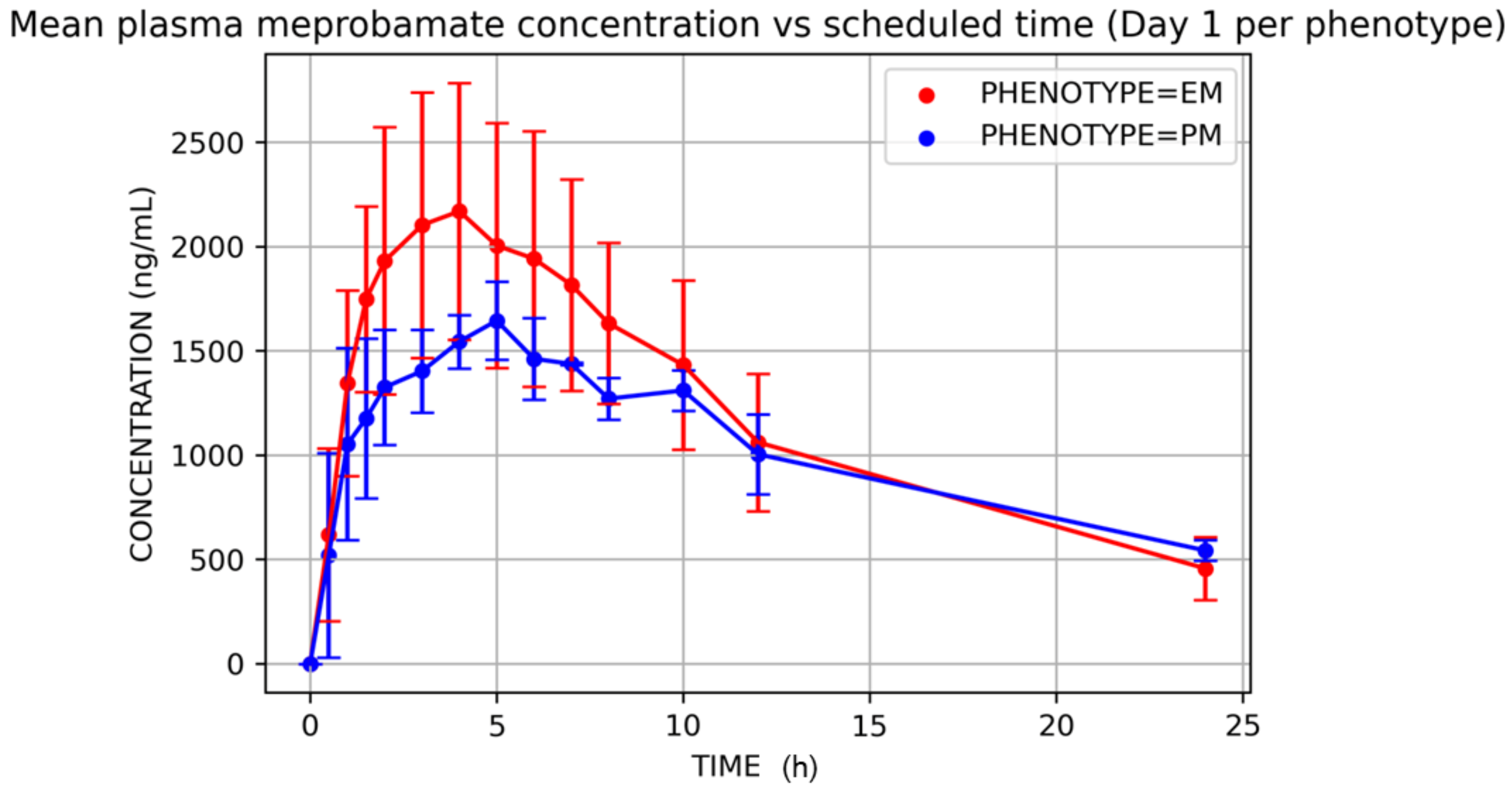
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Busto, U.; Sellers, E.M. Pharmacokinetic Determinants of Drug Abuse and Dependence: A Conceptual Perspective. Clin. Pharmacokinet. 2012, 11, 144–153. [Google Scholar] [CrossRef]
- Quinn, D.I.; Wodak, A.; Day, R.O. Pharmacokinetic and Pharmacodynamic Principles of Illicit Drug Use and Treatment of Illicit Drug Users. Clin. Pharmacokinet. 2012, 33, 344–400. [Google Scholar] [CrossRef]
- Wagner, J.G. History of pharmacokinetics. Pharmac. Ther. 1981, 12, 537–562. [Google Scholar] [CrossRef] [Green Version]
- Temin, P. The evolution of the Modern Pharmaceutical Industry; Library of the Massachusetts Institute of Technology: Cambridge, MA, USA, 1978; Volume 223, pp. 1–42. [Google Scholar]
- FDA. Opioid Analgesic Risk Evaluation and Mitigation Strategy (REMS). Available online: https://www.fda.gov/drugs/information-drug-class/opioid-analgesic-risk-evaluation-and-mitigation-strategy-rems (accessed on 1 October 2021).
- Seyler, T.; Giraudon, I.; Noor, A.; Mounteney, J.; Griffiths, P. Is Europe facing an opioid epidemic: What does European monitoring data tell us? Eur. J. Pain 2021, 5, 1072–1080. [Google Scholar] [CrossRef]
- Verhamme, K.M.C.; Bohnen, A. Are we facing an opioid crisis in Europe? Lancet Public Health 2019, 10, E483–E484. [Google Scholar] [CrossRef] [Green Version]
- European Monitoring Centre for Drugs and Drug Addiction. Health and Social Responses to Drug Problems: A European guide. 2021. Available online: https://www.emcdda.europa.eu/ (accessed on 1 October 2021).
- AEMPS. Fentanilo de Liberación Inmediata: Importancia de Respetar las Condiciones de Uso Autorizadas. 2018. Available online: https://www.aemps.gob.es/informa/notasinformativas/medicamentosusohumano-3/seguridad-1/2018/ni-muh_fv_5-2017-fentanilo/ (accessed on 1 October 2021).
- Drug Enforcement Administration Diversion Control Division Drug & Chemical Evaluation Section. Carisoprodol. 2019. Available online: https://www.deadiversion.usdoj.gov/drug_chem_info/carisoprodol/carisoprodol.pdf (accessed on 1 October 2021).
- EMA. Annex II, Scientific Conclusions and Grounds for Suspension of the Marketing Authorisation presented by the EMEA. 2007. Available online: https://www.ema.europa.eu/en/documents/referral/carisoprodol-article-107-procedures-annex-i_en.pdf (accessed on 1 October 2021).
- Drug Enforcement Administration. Schedules of Controlled Substances: Placement of Carisoprodol into Schedule IV. 2011. Available online: https://www.govinfo.gov/content/pkg/FR-2011-12-12/pdf/2011-31542.pdf (accessed on 1 October 2021).
- Li, Y.; Delcher, C.; Brown, J.D.; Wei, Y.J.; Reisfield, G.M.; Winterstein, A.G. Impact of Schedule IV controlled substance classification on carisoprodol utilization in the United States: An interrupted time series analysis. Drug Alcohol Depend. 2019, 202, 172–177. [Google Scholar] [CrossRef]
- ClinCalc. Carisoprodol. Drug Usage Statistics, United States, 2013–2019. Available online: https://clincalc.com/DrugStats/Drugs/Carisoprodol (accessed on 1 October 2021).
- Sikdar, S.; Basu, D.; Malhotra, A.K.; Varma, V.K.; Mattoo, S.K. Carisoprodol abuse: A report from India. Acta Psychiatr. Scand. 1993, 88, 302–303. [Google Scholar] [CrossRef]
- Littrell, R.; Hayes, L.; Stillner, V. Carisoprodol (Soma): A new and cautious perspective on an old agent. South Med. J. 1993, 86, 753–756. [Google Scholar] [CrossRef]
- Waldman, H. Centrally acting skeletal muscle relaxants and associated drugs. J. Pain Symptom Manag. 1994, 9, 434–441. [Google Scholar] [CrossRef]
- Logan, B.K.; Case, G.A.; Gordon, A.M. Carisoprodol, meprobamate, and driving impairment. J. Forensic Sci. 2000, 45, 619–623. [Google Scholar] [CrossRef]
- Bailey, D.N.; Briggs, J.R. Carisoprodol: An unrecognized drug of abuse. Am. J. Clin. Pathol. 2002, 117, 396–400. [Google Scholar] [CrossRef]
- Boothby, L.; Doering, P.; Hatton, R. Carisoprodol: A marginally effective skeletal muscle relaxant with serious abuse potential. Hosp. Pharm. 2003, 38, 337–345. [Google Scholar] [CrossRef]
- Reeves, R.R.; Beddingfield, J.J.; Mack, J.E. Carisoprodol withdrawal syndrome. Pharmacotherapy 2004, 24, 1804–1806. [Google Scholar] [CrossRef]
- Toth, P.P.; Urtis, J. Commonly used muscle relaxant therapies for acute low back pain: A review of Carisoprodol, cyclobenzaprine hydrochloride, and metaxalone. Clin Ther. 2004, 26, 1355–1367. [Google Scholar] [CrossRef]
- Owens, C.; Pugmire, B.; Salness, T.; Culbertson, V.; Force, R.; Cady, P.; Steiner, J. Abuse potential of Carisoprodol: A retrospective review of Idaho Medicaid pharmacy and medical claims data. Clin Ther. 2007, 29, 2222–2225. [Google Scholar] [CrossRef]
- Bramness, J.G.; Furu, K.; Engeland, A.; Skurtveit, S. Carisoprodol use and abuse in Norway: A pharmacoepidemiological study. Br. J. Clin. Pharmacol. 2007, 64, 210–218. [Google Scholar] [CrossRef] [Green Version]
- Olsen, H.; Koppang, E.; Alvan, G.; Morland, J. Carisoprodol elimination in humans. Ther Drug Monit. 1994, 16, 337–340. [Google Scholar] [CrossRef]
- Dalen, P.; Alvan, G.; Wakelkamp, M.; Olsen, H. Formation of meprobamate from Carisoprodol is catalysed by CYP2C19. Pharmacogenetics 1996, 6, 387–394. [Google Scholar] [CrossRef]
- FDA. Safety Labeling Changes Approved by FDA Center for Drug Evaluation and Research (CDER). 2009. Available online: http://www.fda.gov/Safety/MedWatch/SafetyInformation/ucm191961.htm (accessed on 1 March 2016).
- Simon, S.; D’Andrea, C.; Wheeler, W.J.; Sacks, H. Bioavailability of oral Carisoprodol 250 and 350 mg and metabolism to meprobamate: A single-dose crossover study. Curr. Ther. Res. Clin. Exp. 2010, 71, 50–59. [Google Scholar] [CrossRef] [Green Version]
- Bosó, V.; Herrero, M.J.; Buso, E.; Galán, J.; Almenar, L.; Sánchez-Lázaro, I. Genotype and allele frequencies of drug-metabolizing enzymes and drug transporter genes affecting immunosuppressants in the Spanish white population. Ther. Drug Monit. 2014, 36, 159–168. [Google Scholar] [CrossRef]
- Alonso-Navarro, H.; Jiménez-Jiménez, F.J.; García-Agúndez, J.A. The role of CYP2C19 polymorphism in the development of adverse effects to drugs and the risk for diseases. Med. Clin. 2006, 13, 697–706. [Google Scholar] [CrossRef]
- Tse, S.; Atayee, R.S.; Ma, J.D.; Best, B.M. Factors affecting Carisoprodol metabolism in pain patients using urinary excretion data. J. Anal. Toxicol. 2014, 38, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Høiseth, G.; Majid, U.; Mørland, J.; Bramness, J.G.; Molden, E. CYP2C19 genetics in fatal Carisoprodol intoxications. Eur. J. Clin. Pharmacol. 2012, 68, 1561–1565. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age | Weight (kg) | Height (cm) | Quetelet Index (kg/m2) | |
|---|---|---|---|---|
| Mean | 23.076 | 69.16 | 173 | 22.89 |
| SD | 1.07 | 12.85 | 8.13 | 2.57 |
| Pharmacokinetic Parameters of Carisoprodol after Single (350 mg) or Multiple Doses (up to 350 mg/8 h, 14 days) | ||||||||
| Day 1 | ||||||||
| Variable | N | Mean | SD | Min | Median | Max | CV% | Geometric Mean |
| Tmax (h) | 13 | 1.19 | 0.69 | 0.50 | 1.00 | 3.00 | 58.15 | 1.04 |
| Cmax (ng/mL) | 13 | 2579.82 | 1214.05 | 935.14 | 2294.50 | 5204.37 | 47.06 | 2316.47 |
| AUC0–8 (ng·h/mL) | 13 | 7219.99 | 4550.97 | 2283.54 | 6070.36 | 19,807.26 | 63.03 | 6165.38 |
| AUC0–12 (ng·h/mL) | 13 | 7671.52 | 5276.52 | 2334.17 | 6404.65 | 22,762.02 | 68.78 | 6427.87 |
| AUClast (ng·h/mL) | 13 | 7955.50 | 6184.89 | 2322.03 | 6404.65 | 26,473.74 | 77.74 | 6498.54 |
| AUC0–∞ (ng·h/mL) | 13 | 8071.70 | 6303.04 | 2348.37 | 6448.63 | 26,906.93 | 78.09 | 6578.31 |
| T1/2 (h) | 13 | 1.98 | 0.84 | 1.06 | 1.67 | 4.19 | 42.55 | 1.85 |
| Day 14 | ||||||||
| Variable | N | Mean | SD | Min | Median | Max | CV% | Geometric Mean |
| Tmax (h) | 12 | 1.08 | 0.42 | 0.50 | 1.00 | 1.50 | 38.53 | 1.00 |
| Cmax (ng/mL) | 12 | 2503.58 | 730.08 | 1408.42 | 2533.82 | 3556.65 | 29.16 | 2397.05 |
| AUC0–8 (ng·h/mL) | 12 | 6905.29 | 2793.14 | 3815.50 | 6471.21 | 14,151.96 | 40.45 | 6477.15 |
| AUC0–12 (ng·h/mL) | 12 | 7233.72 | 3174.64 | 3917.30 | 6776.70 | 15,668.25 | 43.89 | 6727.29 |
| AUClast (ng·h/mL) | 12 | 7362.86 | 3582.20 | 3917.30 | 6776.70 | 17,299.65 | 48.65 | 6774.03 |
| AUC0–∞ (ng·h/mL) | 12 | 7451.07 | 3614.80 | 3953.15 | 6918.11 | 17,425.04 | 48.51 | 6854.52 |
| T1/2 (h) | 12 | 1.98 | 0.73 | 1.27 | 1.84 | 3.62 | 36.75 | 1.87 |
| Pharmacokinetic Parameters of Meprobamate after Single (350 mg) or Multiple Doses of Carisoprodol (up to 350 mg/8 h, 14 Days) | ||||||||
| Day 1 | ||||||||
| Variable | N | Mean | SD | Min | Median | Max | CV% | Geometric Mean |
| Tmax (h) | 13 | 3.77 | 1.47 | 1.50 | 4.00 | 6.00 | 38.91 | 3.45 |
| Cmax (ng/mL) | 13 | 2181.38 | 605.49 | 1432.63 | 1944.80 | 3278.09 | 27.76 | 2108.30 |
| AUC0–8 (ng·h/mL) | 13 | 13,594.11 | 3590.73 | 9129.46 | 12,747.47 | 21,042.61 | 26.41 | 13,182.44 |
| AUC0–12 (ng·h/mL) | 13 | 19,050.75 | 4692.05 | 13,675.75 | 18,323.23 | 27,906.62 | 24.63 | 18,552.82 |
| AUClast (ng·h/mL) | 13 | 28,204.26 | 6115.74 | 21,040.18 | 25,706.36 | 39,340.37 | 21.68 | 27,628.98 |
| AUC0–∞ (ng·h/mL) | 13 | 34,529.06 | 7747.10 | 23,490.27 | 33,655.06 | 48,232.05 | 22.44 | 33,746.03 |
| T1/2 (h) | 13 | 9.01 | 1.86 | 6.42 | 8.91 | 12.11 | 20.65 | 8.83 |
| Day 14 | ||||||||
| Variable | N | Mean | SD | Min | Median | Max | CV% | Geometric Mean |
| Tmax (h) | 12 | 1.79 | 0.66 | 1.00 | 1.50 | 3.00 | 36.60 | 1.69 |
| Cmax (ng/mL) | 12 | 5758.05 | 1255.01 | 3999.98 | 5408.53 | 7875.76 | 21.80 | 5637.93 |
| AUC0–8 (ng·h/mL) | 12 | 37,323.09 | 8626.89 | 25,221.71 | 34,654.74 | 52,790.79 | 23.11 | 36,466.47 |
| AUC0–12 (ng·h/mL) | 12 | 48,845.72 | 11,387.45 | 33,030.74 | 45,743.04 | 69,808.33 | 23.31 | 47,701.24 |
| AUClast (ng·h/mL) | 12 | 68,036.28 | 16,013.98 | 47,744.16 | 64,970.83 | 97,947.49 | 23.54 | 66,401.03 |
| AUC0–∞ (ng·h/mL) | 12 | 79,699.89 | 17,978.55 | 57,653.89 | 78,557.16 | 112,876.64 | 22.56 | 77915.42 |
| T1/2 (h) | 12 | 8.68 | 1.39 | 6.66 | 8.09 | 10.97 | 15.97 | 8.58 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calvo, A.; Alonso, S.; Prieto, E.; Ascaso-del-Rio, A.; Ortuño, J.; Fernandez, N.; Portolés, A. Single and Multiple Dose PK–PD Characterization for Carisoprodol. Part I: Pharmacokinetics, Metabolites, and 2C19 Phenotype Influence. Double-Blind, Placebo-Controlled Clinical Trial in Healthy Volunteers. J. Clin. Med. 2022, 11, 858. https://doi.org/10.3390/jcm11030858
Calvo A, Alonso S, Prieto E, Ascaso-del-Rio A, Ortuño J, Fernandez N, Portolés A. Single and Multiple Dose PK–PD Characterization for Carisoprodol. Part I: Pharmacokinetics, Metabolites, and 2C19 Phenotype Influence. Double-Blind, Placebo-Controlled Clinical Trial in Healthy Volunteers. Journal of Clinical Medicine. 2022; 11(3):858. https://doi.org/10.3390/jcm11030858
Chicago/Turabian StyleCalvo, Aitana, Saioa Alonso, Esther Prieto, Ana Ascaso-del-Rio, Jordi Ortuño, Nieves Fernandez, and Antonio Portolés. 2022. "Single and Multiple Dose PK–PD Characterization for Carisoprodol. Part I: Pharmacokinetics, Metabolites, and 2C19 Phenotype Influence. Double-Blind, Placebo-Controlled Clinical Trial in Healthy Volunteers" Journal of Clinical Medicine 11, no. 3: 858. https://doi.org/10.3390/jcm11030858
APA StyleCalvo, A., Alonso, S., Prieto, E., Ascaso-del-Rio, A., Ortuño, J., Fernandez, N., & Portolés, A. (2022). Single and Multiple Dose PK–PD Characterization for Carisoprodol. Part I: Pharmacokinetics, Metabolites, and 2C19 Phenotype Influence. Double-Blind, Placebo-Controlled Clinical Trial in Healthy Volunteers. Journal of Clinical Medicine, 11(3), 858. https://doi.org/10.3390/jcm11030858