

Targeted Resequencing of Otosclerosis Patients from Different Populations Replicates Results from a Previous Genome-Wide Association Study
 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Selection of Cases and Controls
2.2. Gene Selection and Targeted Enrichment
2.3. Variant Calling and Statistical Testing
3. Results
3.1. Sample Collection and Gene Selection
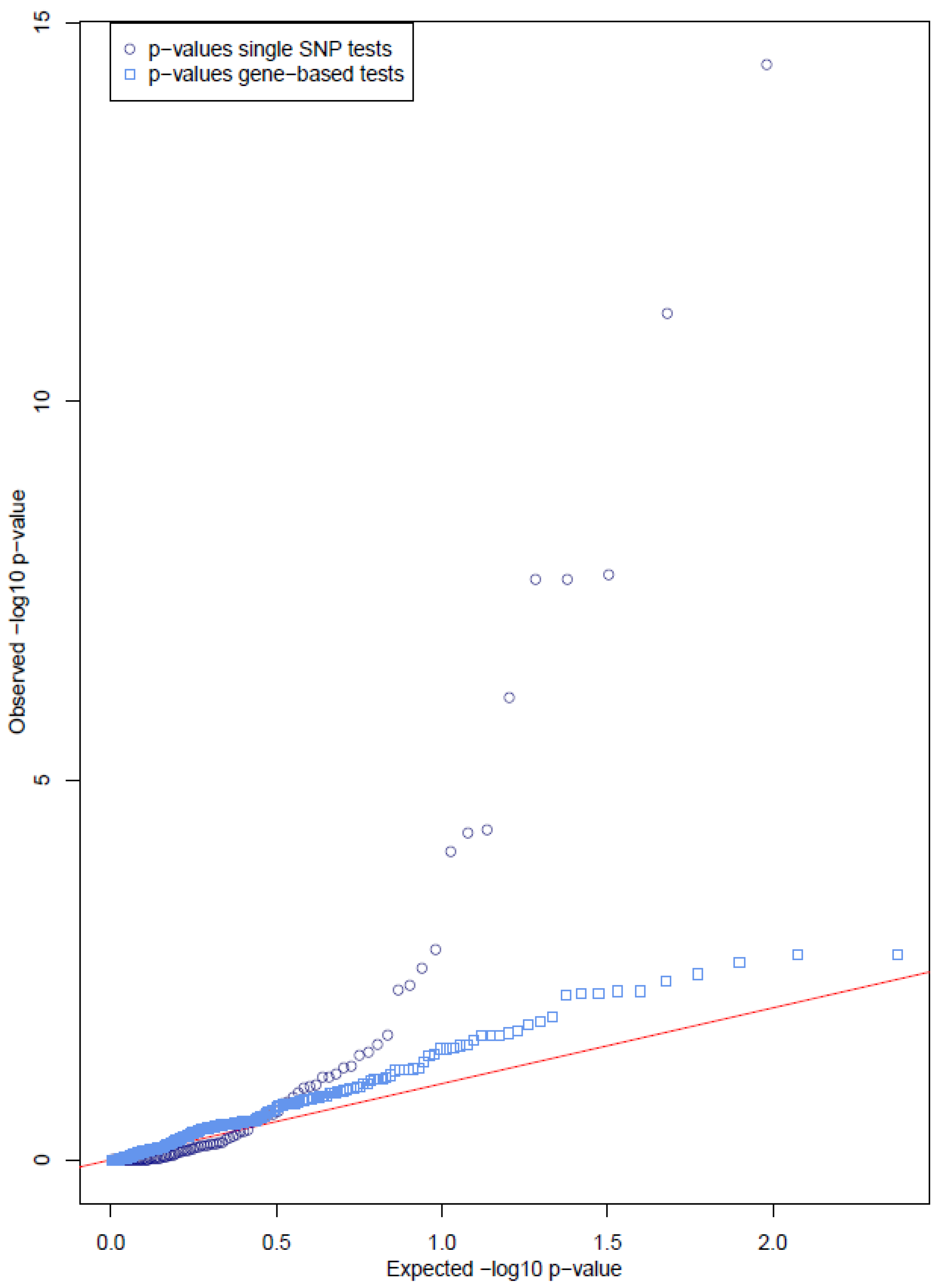
3.2. Single Variant Tests Show Significant Results in Several Genes
3.3. Gene-Based Tests Show No Cumulative Effect of Rare Variants
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Milroy, C.M.; Michaels, L. Pathology of the otic capsule. J. Laryngol. Otol. 1990, 104, 83–90. [Google Scholar] [CrossRef]
- Guild, S. Histologic otosclerosis. Ann. Otol. Rhinol. Laryngol. 1944, 53, 246–267. [Google Scholar]
- Balle, V.; Linthicum, F.H. Histologically Proven Cochlear Otosclerosis with Pure Sensorineural Hearing Loss. Ann. Otol. Rhinol. Laryngol. 1984, 93, 105–111. [Google Scholar]
- Browning, G.G.; Gatehouse, S. Sensorineural Hearing Loss in Stapedial Otosclerosis. Ann. Otol. Rhinol. Laryngol. 1984, 93, 13–16. [Google Scholar] [CrossRef]
- Declau, F.; Van Spaendonck, M.; Timmermans, J.-P.; Michaels, L.; Liang, J.; Qiu, J.P.; Van de Heyning, P. Prevalence of Otosclerosis in an Unselected Series of Temporal Bones. Otol. Neurotol. 2001, 22, 596–602. [Google Scholar] [CrossRef]
- Larsson, A. Otosclerosis. A genetic and clinical study. Acta Otolaryngol. Suppl. 1960, 154, 1–86. [Google Scholar] [PubMed]
- Morrison, A.W. Genetic factors in otosclerosis. Ann. R. Coll. Surg. Engl. 1967, 41, 202–237. [Google Scholar] [PubMed]
- Ali, I.B.H.; Thys, M.; Beltaief, N.; Schrauwen, I.; Hilgert, N.; Vanderstraeten, K.; Dieltjens, N.; Mnif, E.; Hachicha, S.; Besbes, G.; et al. A new locus for otosclerosis, OTSC8, maps to the pericentromeric region of chromosome 9. Qual. Life Res. 2008, 123, 267–272. [Google Scholar]
- Brownstein, Z.; Goldfarb, A.; Levi, H.; Frydman, M.; Avraham, K.B. Chromosomal Mapping and Phenotypic Characterization of Hereditary Otosclerosis Linked to the OTSC4 Locus. Arch. Otolaryngol.-Head Neck Surg. 2006, 132, 416–424. [Google Scholar] [CrossRef]
- Chen, W.; Campbell, C.A.; Green, G.E.; Bogaert, K.V.D.; Komodikis, C.; Manolidis, L.S.; Aconomou, E.; Kyamides, Y.; Christodoulou, K.; Faghel, C.; et al. Linkage of otosclerosis to a third locus (OTSC3) on human chromosome 6p21.3-22.3. J. Med. Genet. 2002, 39, 473–477. [Google Scholar] [CrossRef]
- Schrauwen, I.; Weegerink, N.; Fransen, E.; Claes, C.; Pennings, R.; Cremers, C.; Huygen, P.; Kunst, H.; Van Camp, G. A new locus for otosclerosis, OTSC10, maps to chromosome 1q41-44. Clin. Genet. 2011, 79, 495–497. [Google Scholar] [CrossRef]
- Thys, M.; Van Den Bogaert, K.; Iliadou, V.; Vanderstraeten, K.; Dieltjens, N.; Schrauwen, I.; Van Camp, G. A seventh locus for otosclerosis, OTSC7, maps to chromosome 6q13–16.1. Eur. J. Hum. Genet. 2007, 15, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Tomek, M.S.; Brown, M.R.; Mani, S.R.; Ramesh, A.; Srisailapathy, C.R.S.; Coucke, P.; Zbar, R.I.S.; Bell, A.M.; McGuirt, W.T.; Fukushima, K.; et al. Localization of a gene for otosclerosis to chromosome 15q25-q26. Hum. Mol. Genet. 1998, 7, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Bogaert, K.V.D.; De Leenheer, E.M.R.; Chen, W.; Lee, Y.; Nürnberg, P.; Pennings, R.J.E.; Vanderstraeten, K.; Thys, M.; Cremers, C.W.R.J.; Smith, R.; et al. A fifth locus for otosclerosis, OTSC5, maps to chromosome 3q22-24. J. Med. Genet. 2004, 41, 450–453. [Google Scholar] [CrossRef] [PubMed]
- Van Den Bogaert, K.; Govaerts, P.J.; Schatteman, I.; Brown, M.R.; Caethoven, G.; Offeciers, F.E.; Van Camp, G. A second gene for otosclerosis, OTSC2, maps to chromosome 7q34-36. Am. J. Hum. Genet. 2001, 68, 495–500. [Google Scholar] [CrossRef]
- Højland, A.T.; Tavernier, L.J.M.; Schrauwen, I.; Sommen, M.; Topsakal, V.; Schatteman, I.; Dhooge, I.; Huber, A.; Zanetti, D.; Kunst, H.P.M.; et al. A wide range of protective and predisposing variants in aggrecan influence the susceptibility for otosclerosis. Qual. Life Res. 2021, 141, 951–963. [Google Scholar] [CrossRef]
- Schrauwen, I.; Valgaeren, H.; Tomas-Roca, L.; Sommen, M.; Altunoglu, U.; Wesdorp, M.; Beyens, M.; Fransen, E.; Nasir, A.; Vandeweyer, G.; et al. Variants affecting diverse domains of MEPE are associated with two distinct bone disorders, a craniofacial bone defect and otosclerosis. Genet. Med. 2019, 21, 1199–1208. [Google Scholar] [CrossRef]
- Rämö, J.T.; Kiiskinen, T.; Karjalainen, J.; Krebs, K.; Kurki, M.; Havulinna, A.S.; Palotie, A. Genome-wide Screen of Otosclerosis in Population Biobanks: 18 Loci and Shared Heritability with Skeletal Structure. medRxiv 2020. [Google Scholar]
- Schrauwen, I.; Ealy, M.; Fransen, E.; Vanderstraeten, K.; Thys, M.; Meyer, N.C.; Cosgarea, M.; Huber, A.; Mazzoli, M.; Pfister, M.; et al. Genetic variants in the RELN gene are associated with otosclerosis in multiple European populations. Qual. Life Res. 2010, 127, 155–162. [Google Scholar] [CrossRef]
- Schrauwen, I.; Khalfallah, A.; Ealy, M.; Fransen, E.; Claes, C.; Huber, A.; Murillo, L.R.; Masmoudi, S.; Smith, R.; Van Camp, G. COL1A1 association and otosclerosis: A meta-analysis. Am. J. Med. Genet. Part A 2012, 158A, 1066–1070. [Google Scholar] [CrossRef]
- Schrauwen, I.; Thys, M.; Vanderstraeten, K.; Fransen, E.; Dieltjens, N.; Huyghe, J.R.; Van Camp, G. Association of bone morphogenetic proteins with otosclerosis. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2008, 23, 507–516. [Google Scholar] [CrossRef]
- Thys, M.; Schrauwen, I.; Vanderstraeten, K.; Dieltjens, N.; Fransen, E.; Ealy, M.; Cremers, C.W.R.J.; van de Heyning, P.; Vincent, R.; Offeciers, E.; et al. Detection of Rare Nonsynonymous Variants in TGFB1 in Otosclerosis Patients. Ann. Hum. Genet. 2009, 73, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Valgaeren, H.; Sommen, M.; Beyens, M.; Vandeweyer, G.; Schrauwen, I.; Schepers, A.; Schatteman, I.; Topsakal, V.; Dhooge, I.; Kunst, H.; et al. Insufficient evidence for a role of SERPINF1 in otosclerosis. Mol. Genet. Genomics. 2019, 294, 1001–1006. [Google Scholar] [CrossRef]
- Boyle, E.A.; O’Roak, B.; Martin, B.K.; Kumar, A.; Shendure, J. MIPgen: Optimized modeling and design of molecular inversion probes for targeted resequencing. Bioinformatics 2014, 30, 2670–2672. [Google Scholar] [CrossRef]
- Hiatt, J.B.; Pritchard, C.C.; Salipante, S.J.; O’Roak, B.J.; Shendure, J. Single molecule molecular inversion probes for targeted, high-accuracy detection of low-frequency variation. Genome Res. 2013, 23, 843–854. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Lucas, F.A.S.; Wang, G.; Scheet, P.; Peng, B. Integrated annotation and analysis of genetic variants from next-generation sequencing studies with variant tools. Bioinformatics 2011, 28, 421–422. [Google Scholar] [CrossRef] [PubMed]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef] [PubMed]
- The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Thys, M.; Schrauwen, I.; Vanderstraeten, K.; Janssens, K.; Dieltjens, N.; Van Den Bogaert, K.; Van Camp, G. The coding polymorphism T263I in TGF-beta1 is associated with otosclerosis in two independent populations. Hum. Mol. Genet. 2007, 16, 2021–2030. [Google Scholar] [CrossRef] [PubMed]
- Helmholz, H.; Adejube, B.; Luthringer-Feyerabend, B.; Willumeit-Römer, R. In Vitro Investigation on Degradable Mg-Based Biomaterial under the Impact of the Serum Glycoprotein Fetuin. Materials 2021, 14, 5005. [Google Scholar] [CrossRef]
- Herrmann, M.; Kinkeldey, A.; Jahnen-Dechent, W. Fetuin-A Function in Systemic Mineral Metabolism. Trends Cardiovasc. Med. 2012, 22, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Khalfallah, A.; Schrauwen, I.; Mnejja, M.; HadjKacem, H.; Dhouib, L.; Mosrati, M.A.; Hakim, B.; Lahmar, I.; Charfeddine, I.; Driss, N.; et al. Association of COL1A1 and TGFB1 Polymorphisms with Otosclerosis in a Tunisian Population. Ann. Hum. Genet. 2011, 75, 598–604. [Google Scholar] [CrossRef]
- Szweras, M.; Liu, D.; Partridge, E.A.; Pawling, J.; Sukhu, B.; Clokie, C.; Dennis, J.W. alpha 2-HS glycoprotein/fetuin, a transforming growth factor-beta/bone morphogenetic protein antagonist, regulates postnatal bone growth and remodeling. J. Biol. Chem. 2002, 277, 19991–19997. [Google Scholar] [CrossRef]
- Ransohoff, J.D.; Wei, Y.; Khavari, P.A. The functions and unique features of long intergenic non-coding RNA. Nat. Rev. Mol. Cell Biol. 2018, 19, 143–157. [Google Scholar] [CrossRef]
- Kim, S.K. Identification of 613 new loci associated with heel bone mineral density and a polygenic risk score for bone mineral density, osteoporosis and fracture. PLoS ONE 2018, 13, e0200785. [Google Scholar] [CrossRef]
- Calabrese, G.M.; Mesner, L.D.; Stains, J.P.; Tommasini, S.M.; Horowitz, M.C.; Rosen, C.J.; Farber, C.R. Integrating GWAS and Co-expression Network Data Identifies Bone Mineral Density Genes SPTBN1 and MARK3 and an Osteoblast Functional Module. Cell Syst. 2016, 4, 46–59.e4. [Google Scholar] [CrossRef]
- Estrada, K.; Styrkarsdottir, U.; Evangelou, E.; Hsu, Y.-H.; Duncan, E.L.; Ntzani, E.E.; Oei, L.; Albagha, O.M.E.; Amin, N.; Kemp, J.P.; et al. Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat. Genet. 2012, 44, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.-M.; Kung, A.W.C.; Sham, P.C.; Tan, K.C.B. Genetic Analysis of Recently Identified Osteoporosis Susceptibility Genes in Southern Chinese. J. Clin. Endocrinol. Metab. 2013, 98, E1827–E1834. [Google Scholar] [CrossRef]
- Schrauwen, I.; Ealy, M.; Huentelman, M.J.; Thys, M.; Homer, N.; Vanderstraeten, K.; Fransen, E.; Corneveaux, J.J.; Craig, D.W.; Claustres, M.; et al. A Genome-wide Analysis Identifies Genetic Variants in the RELN Gene Associated with Otosclerosis. Am. J. Hum. Genet. 2009, 84, 328–338. [Google Scholar] [CrossRef]
- Khalfallah, A.; Schrauwen, I.; Mnaja, M.; Fransen, E.; Lahmar, I.; Ealy, M.; Dhouib, L.; Ayadi, H.; Charfedine, I.; Driss, N.; et al. Genetic variants in RELN are associated with otosclerosis in a non-European population from Tunisia. Ann. Hum. Genet. 2010, 74, 399–405. [Google Scholar] [CrossRef]
- Priyadarshi, S.; Hansdah, K.; Singh, N.; Bouzid, A.; Ray, C.S.; Panda, K.C.; Biswal, N.C.; Desai, A.; Choudhury, J.C.; Tekari, A.; et al. The risks of RELN polymorphisms and its expression in the development of otosclerosis. PLoS ONE 2022, 17, e0269558. [Google Scholar] [CrossRef]
- Quattrocchi, C.C.; Wannenes, F.; Persico, A.M.; Ciafré, S.A.; D’Arcangelo, G.; Farace, M.G.; Keller, F. Reelin is a serine protease of the extracellular matrix. J. Biol. Chem. 2002, 277, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Rice, S.J.; Aubourg, G.; Sorial, A.; Almarza, D.; Tselepi, M.; Deehan, D.; Reynard, L.N.; Loughlin, J. Identification of a novel, methylation-dependent, RUNX2 regulatory region associated with osteoarthritis risk. Hum. Mol. Genet. 2018, 27, 3464–3474. [Google Scholar] [CrossRef]
- Janssens, K.; ten Dijke, P.; Janssens, S.; Van Hul, W. Transforming growth factor-beta1 to the bone. Endocr. Rev. 2005, 26, 743–774. [Google Scholar] [CrossRef]
- Castaño-Betancourt, M.C.; Evans, D.S.; Ramos, Y.F.M.; Boer, C.G.; Metrustry, S.; Liu, Y.; Hollander, W.D.; van Rooij, J.; Kraus, V.B.; Yau, M.S.; et al. Novel Genetic Variants for Cartilage Thickness and Hip Osteoarthritis. PLoS Genet. 2016, 12, e1006260. [Google Scholar] [CrossRef] [PubMed]
- Gordon, K.J.; Blobe, G.C. Role of transforming growth factor-β superfamily signaling pathways in human disease. Biochim. et Biophys. Acta (BBA)-Mol. Basis Dis. 2008, 1782, 197–228. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Variant (rsID) | Gene | Region | Effect Allele in Cases/ Total Alleles in Cases | Effect Allele in Controls/Total Alleles in Controls | cDNA Change | Amino Acid Change | MAF GnomAD | Uncorrected p-Value | q-Value after FDR | Allelic OR (95% CI) |
|---|---|---|---|---|---|---|---|---|---|---|
| rs2273699 | MARK3 | intronic | 1508/3210 | 1147/3086 | c.413-4A > G | p.(=) | 0.36 | 3.58 × 10−15 | 3.40 × 10−13 | 1.42 (1.28–1.57) |
| rs11868207 | LINC01482 | ncRNA_exonic | 1151/3254 | 853/3114 | n.2589T > C | / | 0.25 | 6.87 × 10−12 | 3.26 × 10−10 | 1.40 (1.26–1.55) |
| rs4917 | AHSG | exonic | 1965/3260 | 2096/3126 | c.743T > C | p.(Met249Thr) | 0.66 | 1.90 × 10−8 | 4.25 × 10−7 | 0.70 (0.63–0.77) |
| rs4918 | AHSG | exonic | 1915/3202 | 2055/3084 | c.767G > C | p.(Ser257Thr) | 0.66 | 2.16 × 10−8 | 4.25 × 10−7 | 0.69 (0.62–0.76) |
| rs1071592 | AHSG | exonic | 2283/3234 | 2382/3102 | c.810A > C | p.(=) | 0.75 | 2.23 × 10−8 | 4.25 × 10−7 | 0.66 (0.59–0.74) |
| rs3744501 | LINC01482 | ncRNA_exonic | 1360/3254 | 1116/3122 | n.95A > C | / | 0.33 | 7.96 × 10−7 | 1.26 × 10−5 | 1.23 (1.11–1.36) |
| rs13987 | MARK3 | UTR3 | 940/3220 | 1055/3104 | c.*345= | / | 0.35 | 4.35 × 10−5 | 0.00057 | 0.76 (0.68–0.85) |
| rs11541718 | MARK3 | UTR5 | 912/3126 | 1028/3022 | c.-203= | / | 0.35 | 4.84 × 10−5 | 0.00057 | 0.76 (0.68–0.84) |
| rs2278445 | LINC01482 | ncRNA_exonic | 2293/3256 | 2046/3108 | n.332T > G | / | 0.67 | 8.49 × 10−5 | 0.00090 | 1.15 (1.04–1.28) |
| rs34216978 | LINC01482 | ncRNA_exonic | 326/3260 | 391/3128 | n.166A > G | / | 0.14 | 0.0017 | 0.016 | 0.76 (0.65–0.88) |
| rs9369514 | SUPT3H | UTR3 | 1224/3256 | 1284/3112 | c.*282= | / | 0.42 | 0.0029 | 0.025 | 0.82 (0.74–0.91) |
| rs529125 | SUPT3H | UTR3 | 1691/3260 | 1728/3120 | c.*465= | / | 0.70 | 0.0049 | 0.039 | 0.82 (0.75–0.91) |
| rs2229862 | RELN | exonic | 204/3270 | 145/3114 | c.8508C > T | p.(=) | 0.052 | 0.0058 | 0.043 | 1.32 (1.06–1.64) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tavernier, L.J.M.; Vanpoucke, T.; Schrauwen, I.; Van Camp, G.; Fransen, E. Targeted Resequencing of Otosclerosis Patients from Different Populations Replicates Results from a Previous Genome-Wide Association Study. J. Clin. Med. 2022, 11, 6978. https://doi.org/10.3390/jcm11236978
Tavernier LJM, Vanpoucke T, Schrauwen I, Van Camp G, Fransen E. Targeted Resequencing of Otosclerosis Patients from Different Populations Replicates Results from a Previous Genome-Wide Association Study. Journal of Clinical Medicine. 2022; 11(23):6978. https://doi.org/10.3390/jcm11236978
Chicago/Turabian StyleTavernier, Lisse J. M., Thomas Vanpoucke, Isabelle Schrauwen, Guy Van Camp, and Erik Fransen. 2022. "Targeted Resequencing of Otosclerosis Patients from Different Populations Replicates Results from a Previous Genome-Wide Association Study" Journal of Clinical Medicine 11, no. 23: 6978. https://doi.org/10.3390/jcm11236978
APA StyleTavernier, L. J. M., Vanpoucke, T., Schrauwen, I., Van Camp, G., & Fransen, E. (2022). Targeted Resequencing of Otosclerosis Patients from Different Populations Replicates Results from a Previous Genome-Wide Association Study. Journal of Clinical Medicine, 11(23), 6978. https://doi.org/10.3390/jcm11236978