
Cystic Kidney Diseases That Require a Differential Diagnosis from Autosomal Dominant Polycystic Kidney Disease (ADPKD)
 ,
,  , , , , , , , ,
, , , , , , , ,  add
Show full author list
add
Show full author list
Abstract
1. Introduction
2. Cystic Kidney Diseases That Need to Be Excluded from an ADPKD Diagnosis
2.1. Multiple Simple Renal Cysts
2.2. Acquired Cystic Kidney Disease (ACKD)
2.3. Multilocular Renal Cyst (Also Referred to as Multilocular Cystic Nephroma or Polycystic Nephroma)
2.4. Multicystic Kidney/Multicystic Dysplastic Kidney (MCDK)
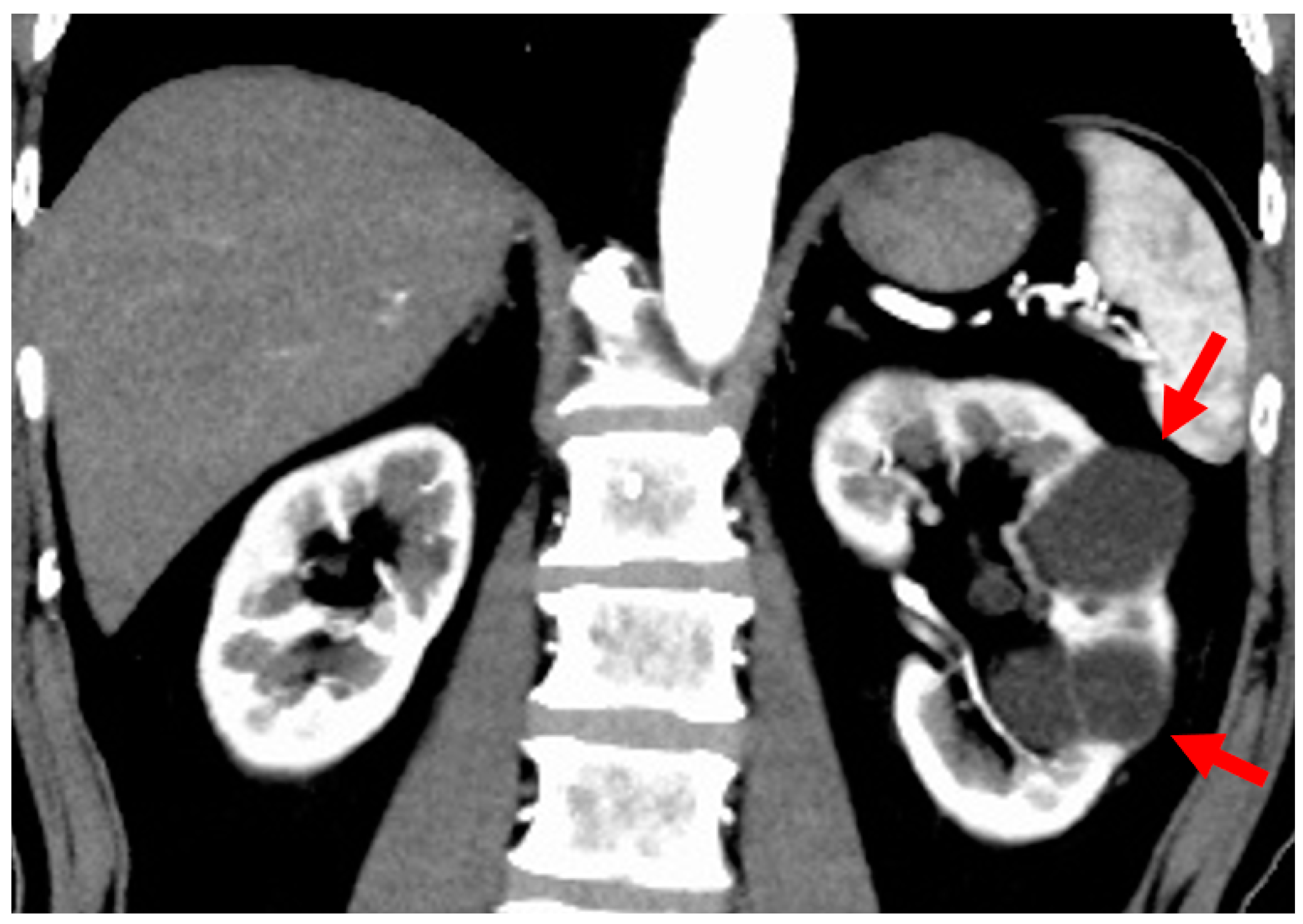
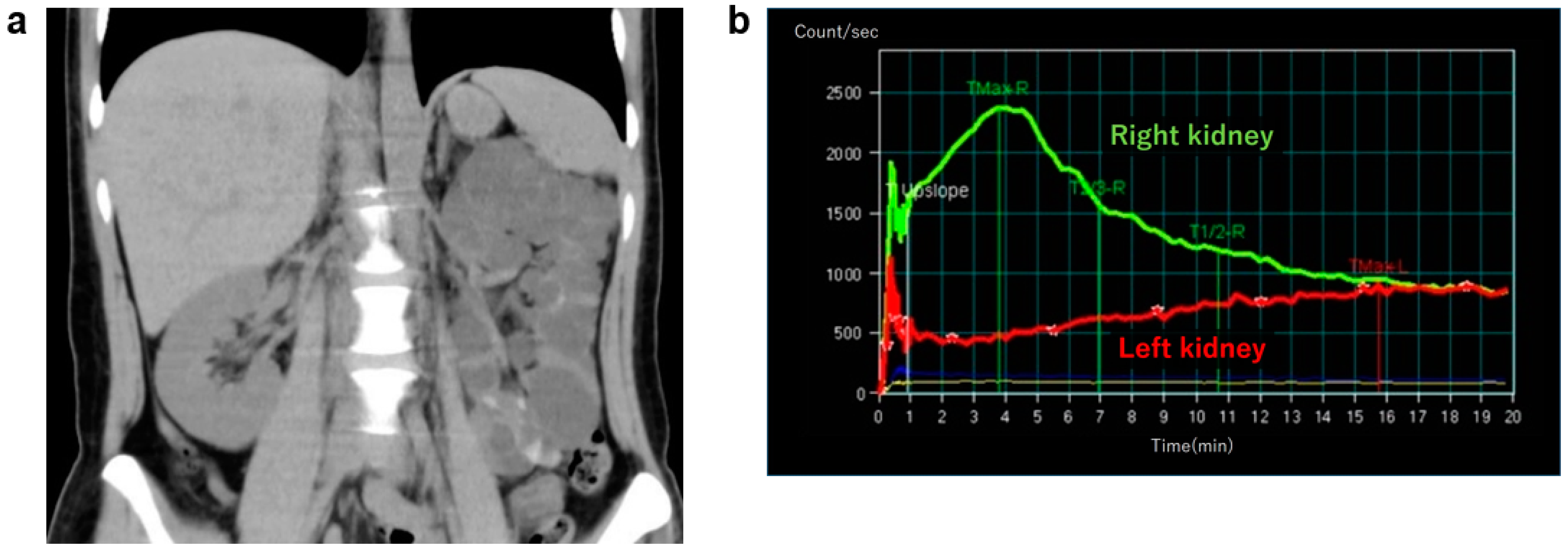
2.5. Unilateral Renal Cystic Disease (URCD)
3. Cystic Kidney Diseases That Require a Differential Diagnosis from ADPKD
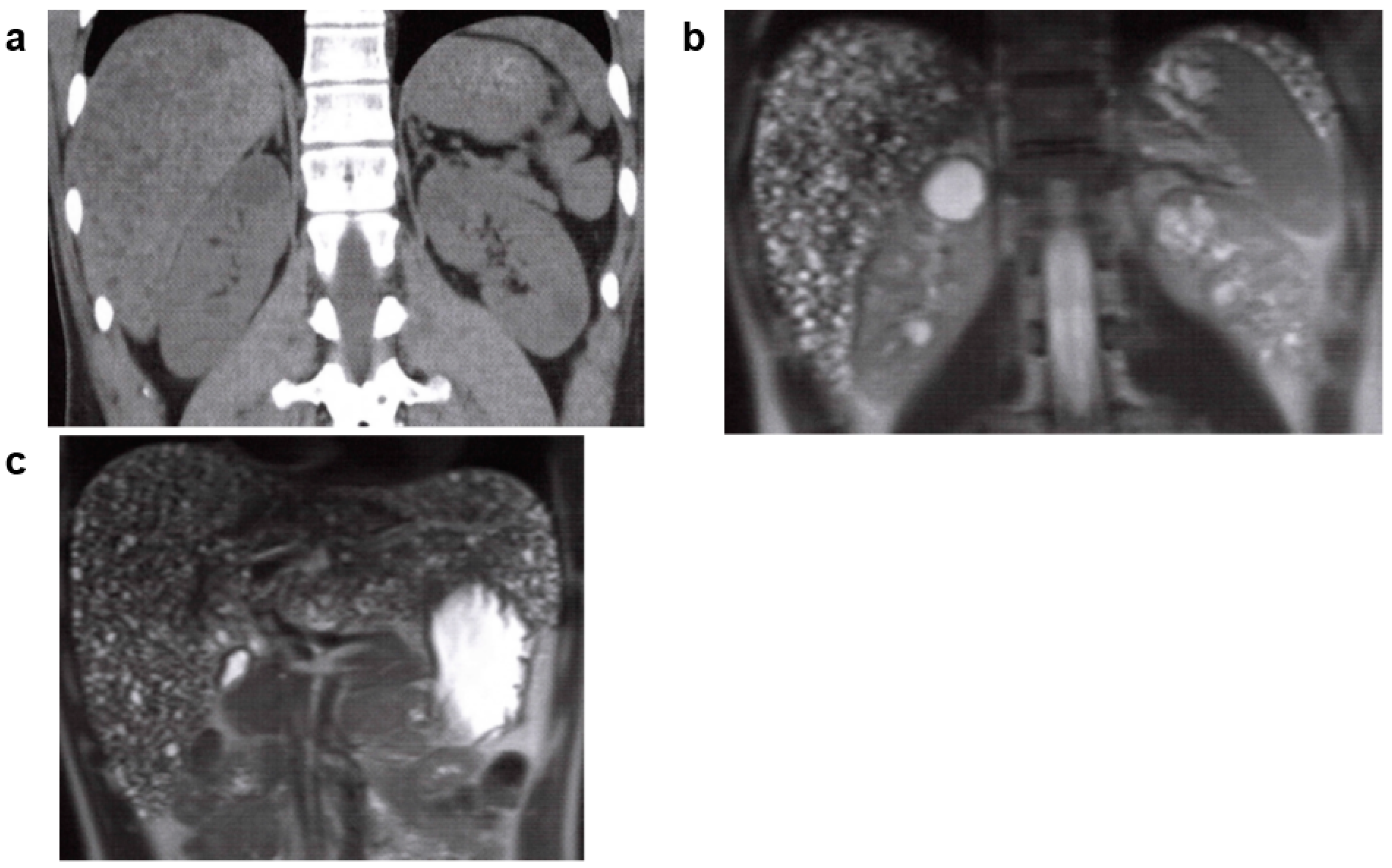
3.1. Autosomal Recessive Polycystic Kidney Disease (ARPKD)
3.2. Autosomal Dominant Tubulointerstitial Kidney Disease (ADTKD)
3.3. Nephronophthisis (NPH)
3.4. Multiple Abnormalities Accompanying Cystic Kidney Disease (OFD1, NPHP/SLS, JSRD, MKS, BBS, OFDS)
3.5. Oral-Facial-Digital Syndrome (OFD) Type 1
3.6. Neoplastic Cystic Kidney Diseases (Tuberous Sclerosis, Von Hippel-Lindau Syndrome)
3.6.1. Tuberous Sclerosis (TSC)
3.6.2. Von Hippel-Lindau (VHL) Syndrome
4. Summary
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cornec-Le Gall, E.; Alam, A.; Perrone, R.D. Autosomal dominant polycystic kidney disease. Lancet 2019, 393, 919–935. [Google Scholar] [CrossRef]
- Audrézet, M.P.; Cornec-Le Gall, E.; Chen, J.M.; Redon, S.; Quéré, I.; Creff, J.; Bénech, C.; Maestri, S.; Le Meur, Y.; Férec, C. Autosomal dominant polycystic kidney disease: Comprehensive mutation analysis of PKD1 and PKD2 in 700 unrelated patients. Hum. Mutat. 2012, 33, 1239–1250. [Google Scholar] [CrossRef]
- Rossetti, S.; Consugar, M.B.; Chapman, A.B.; Torres, V.E.; Guay-Woodford, L.M.; Grantham, J.J.; Bennett, W.M.; Meyers, C.M.; Walker, D.L.; Bae, K.; et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 2007, 18, 2143–2160. [Google Scholar] [CrossRef] [PubMed]
- Cornec-Le Gall, E.; Audrézet, M.P.; Chen, J.M.; Hourmant, M.; Morin, M.P.; Perrichot, R.; Charasse, C.; Whebe, B.; Renaudineau, E.; Jousset, P.; et al. Type of PKD1 mutation influences renal outcome in ADPKD. J. Am. Soc. Nephrol. 2013, 24, 1006–1013. [Google Scholar] [CrossRef]
- Hwang, Y.H.; Conklin, J.; Chan, W.; Roslin, N.M.; Liu, J.; He, N.; Wang, K.; Sundsbak, J.L.; Heyer, C.M.; Haider, M.; et al. Refining Genotype-Phenotype Correlation in Autosomal Dominant Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2016, 27, 1861–1868. [Google Scholar] [CrossRef] [PubMed]
- Iliuta, I.A.; Kalatharan, V.; Wang, K.; Cornec-Le Gall, E.; Conklin, J.; Pourafkari, M.; Ting, R.; Chen, C.; Borgo, A.C.; He, N.; et al. Polycystic Kidney Disease without an Apparent Family History. J. Am. Soc. Nephrol. 2017, 28, 2768–2776. [Google Scholar] [CrossRef] [PubMed]
- Fujimaru, T.; Mori, T.; Sekine, A.; Mandai, S.; Chiga, M.; Kikuchi, H.; Ando, F.; Mori, Y.; Nomura, N.; Iimori, S.; et al. Kidney enlargement and multiple liver cyst formation implicate mutations in PKD1/2 in adult sporadic polycystic kidney disease. Clin. Genet. 2018, 94, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.E.; Chapman, A.B.; Devuyst, O.; Gansevoort, R.T.; Grantham, J.J.; Higashihara, E.; Perrone, R.D.; Krasa, H.B.; Ouyang, J.; Czerwiec, F.S. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N. Engl. J. Med. 2012, 367, 2407–2418. [Google Scholar] [CrossRef]
- Torres, V.E.; Chapman, A.B.; Devuyst, O.; Gansevoort, R.T.; Perrone, R.D.; Dandurand, A.; Ouyang, J.; Czerwiec, F.S.; Blais, J.D. Multicenter, open-label, extension trial to evaluate the long-term efficacy and safety of early versus delayed treatment with tolvaptan in autosomal dominant polycystic kidney disease: The TEMPO 4:4 Trial. Nephrol. Dial. Transplant. 2018, 33, 477–489. [Google Scholar] [CrossRef]
- Torres, V.E.; Chapman, A.B.; Devuyst, O.; Gansevoort, R.T.; Perrone, R.D.; Koch, G.; Ouyang, J.; McQuade, R.D.; Blais, J.D.; Czerwiec, F.S.; et al. Tolvaptan in Later-Stage Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 2017, 377, 1930–1942. [Google Scholar] [CrossRef]
- Eknoyan, G. A clinical view of simple and complex renal cysts. J. Am. Soc. Nephrol. 2009, 20, 1874–1876. [Google Scholar] [CrossRef] [PubMed]
- Terada, N.; Ichioka, K.; Matsuta, Y.; Okubo, K.; Yoshimura, K.; Arai, Y. The natural history of simple renal cysts. J. Urol. 2002, 167, 21–23. [Google Scholar] [CrossRef]
- Rule, A.D.; Sasiwimonphan, K.; Lieske, J.C.; Keddis, M.T.; Torres, V.E.; Vrtiska, T.J. Characteristics of renal cystic and solid lesions based on contrast-enhanced computed tomography of potential kidney donors. Am. J. Kidney Dis. 2012, 59, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Simms, R.J.; Ong, A.C. How simple are ‘simple renal cysts’? Nephrol. Dial. Transpl. 2014, 29 (Suppl. S4), iv106–iv112. [Google Scholar] [CrossRef]
- Al-Said, J.; Brumback, M.A.; Moghazi, S.; Baumgarten, D.A.; O’Neill, W.C. Reduced renal function in patients with simple renal cysts. Kidney Int. 2004, 65, 2303–2308. [Google Scholar] [CrossRef]
- Helenon, O.; Crosnier, A.; Verkarre, V.; Merran, S.; Mejean, A.; Correas, J.M. Simple and complex renal cysts in adults: Classification system for renal cystic masses. Diagn. Interv. Imaging 2018, 99, 189–218. [Google Scholar] [CrossRef]
- Silverman, S.G.; Pedrosa, I.; Ellis, J.H.; Hindman, N.M.; Schieda, N.; Smith, A.D.; Remer, E.M.; Shinagare, A.B.; Curci, N.E.; Raman, S.S.; et al. Bosniak Classification of Cystic Renal Masses, Version 2019: An Update Proposal and Needs Assessment. Radiology 2019, 292, 475–488. [Google Scholar] [CrossRef]
- Truong, L.D.; Choi, Y.J.; Shen, S.S.; Ayala, G.; Amato, R.; Krishnan, B. Renal cystic neoplasms and renal neoplasms associated with cystic renal diseases: Pathogenetic and molecular links. Adv. Anat. Pathol. 2003, 10, 135–159. [Google Scholar] [CrossRef]
- Choyke, P.L. Acquired cystic kidney disease. Eur. Radiol. 2000, 10, 1716–1721. [Google Scholar] [CrossRef]
- Grantham, J.J. Acquired cystic kidney disease. Kidney Int. 1991, 40, 143–152. [Google Scholar] [CrossRef]
- Rahbari-Oskoui, F.; Mittal, A.; Mittal, P.; Chapman, A. Renal relevant radiology: Radiologic imaging in autosomal dominant polycystic kidney disease. Clin. J. Am. Soc. Nephrol. 2014, 9, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, I.; Morita, K.; Hayama, S.; Nakazawa, T.; Araki, I.; Higashi, K.; Miyazawa, K.; Suzuki, K.; Nojima, T. Imaging of acquired cystic disease-associated renal cell carcinoma by contrast-enhanced ultrasonography with perflubutane microbubbles and positron emission tomography-computed tomography. Clin. Exp. Nephrol. 2011, 15, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, K.; Yamamoto, S.; Kawanaka, Y.; Katsuura, T.; Fujita, M.; Nakanishi, Y.; Yamada, Y.; Hashimoto, T.; Suzuki, T.; Go, S.; et al. Imaging of renal cell carcinoma in patients with acquired cystic disease of the kidney: Comparison (11)C-choline and FDG PET/CT with dynamic contrast-enhanced CT. Jpn. J. Radiol. 2019, 37, 165–177. [Google Scholar] [CrossRef]
- Ishikawa, I.; Yuri, T.; Kitada, H.; Shinoda, A. Regression of acquired cystic disease of the kidney after successful renal transplantation. Am. J. Nephrol. 1983, 3, 310–314. [Google Scholar] [CrossRef]
- Leveridge, M.; Musquera, M.; Evans, A.; Cardella, C.; Pei, Y.; Jewett, M.; Robinette, M.; Finelli, A. Renal cell carcinoma in the native and allograft kidneys of renal transplant recipients. J. Urol. 2011, 186, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.H.; Vajdic, C.M.; van Leeuwen, M.T.; Amin, J.; Webster, A.C.; Chapman, J.R.; McDonald, S.P.; Grulich, A.E.; McCredie, M.R. The pattern of excess cancer in dialysis and transplantation. Nephrol. Dial. Transplant. 2009, 24, 3225–3231. [Google Scholar] [CrossRef]
- Schwarz, A.; Vatandaslar, S.; Merkel, S.; Haller, H. Renal cell carcinoma in transplant recipients with acquired cystic kidney disease. Clin. J. Am. Soc. Nephrol. 2007, 2, 750–756. [Google Scholar] [CrossRef]
- Jevremovic, D.; Lager, D.J.; Lewin, M. Cystic nephroma (multilocular cyst) and mixed epithelial and stromal tumor of the kidney: A spectrum of the same entity? Ann. Diagn. Pathol. 2006, 10, 77–82. [Google Scholar] [CrossRef]
- Wilkinson, C.; Palit, V.; Bardapure, M.; Thomas, J.; Browning, A.J.; Gill, K.; Biyani, C.S. Adult multilocular cystic nephroma: Report of six cases with clinical, radio-pathologic correlation and review of literature. Urol. Ann. 2013, 5, 13–17. [Google Scholar] [CrossRef]
- Granja, M.F.; O’Brien, A.T.; Trujillo, S.; Mancera, J.; Aguirre, D.A. Multilocular Cystic Nephroma: A Systematic Literature Review of the Radiologic and Clinical Findings. Am. J. Roentgenol. 2015, 205, 1188–1193. [Google Scholar] [CrossRef]
- Adsay, N.V.; Eble, J.N.; Srigley, J.R.; Jones, E.C.; Grignon, D.J. Mixed epithelial and stromal tumor of the kidney. Am. J. Surg. Pathol. 2000, 24, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Scala, C.; McDonnell, S.; Murphy, F.; Leone Roberti Maggiore, U.; Khalil, A.; Bhide, A.; Thilaganathan, B.; Papageorghiou, A.T. Diagnostic accuracy of midtrimester antenatal ultrasound for multicystic dysplastic kidneys. Ultrasound Obstet. Gynecol. 2017, 50, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Cardona-Grau, D.; Kogan, B.A. Update on Multicystic Dysplastic Kidney. Curr. Urol. Rep. 2015, 16, 67. [Google Scholar] [CrossRef] [PubMed]
- Wacksman, J.; Phipps, L. Report of the Multicystic Kidney Registry: Preliminary findings. J. Urol. 1993, 150, 1870–1872. [Google Scholar] [CrossRef]
- Rabêlo, E.A.; Oliveira, E.A.; Silva, J.M.; Bouzada, M.C.; Sousa, B.C.; Almeida, M.N.; Tatsuo, E.S. Conservative management of multicystic dysplastic kidney: Clinical course and ultrasound outcome. J. Pediatr. 2005, 81, 400–404. [Google Scholar] [CrossRef]
- Chetty, S. Multicystic dysplastic kidney. Am. J. Obstet. Gynecol. 2021, 225, B21–B22. [Google Scholar] [CrossRef]
- Gaither, T.W.; Patel, A.; Patel, C.; Chuang, K.W.; Cohen, R.A.; Baskin, L.S. Natural History of Contralateral Hypertrophy in Patients with Multicystic Dysplastic Kidneys. J. Urol. 2018, 199, 280–286. [Google Scholar] [CrossRef]
- Erlich, T.; Lipsky, A.M.; Braga, L.H. A meta-analysis of the incidence and fate of contralateral vesicoureteral reflux in unilateral multicystic dysplastic kidney. J. Pediatr. Urol. 2019, 15, 77.e71–77.e77. [Google Scholar] [CrossRef]
- Mansoor, O.; Chandar, J.; Rodriguez, M.M.; Abitbol, C.L.; Seeherunvong, W.; Freundlich, M.; Zilleruelo, G. Long-term risk of chronic kidney disease in unilateral multicystic dysplastic kidney. Pediatr. Nephrol. 2011, 26, 597–603. [Google Scholar] [CrossRef]
- Raaijmakers, A.; Corveleyn, A.; Devriendt, K.; van Tienoven, T.P.; Allegaert, K.; Van Dyck, M.; van den Heuvel, L.; Kuypers, D.; Claes, K.; Mekahli, D.; et al. Criteria for HNF1B analysis in patients with congenital abnormalities of kidney and urinary tract. Nephrol. Dial. Transplant. 2015, 30, 835–842. [Google Scholar] [CrossRef]
- Eckardt, K.U.; Alper, S.L.; Antignac, C.; Bleyer, A.J.; Chauveau, D.; Dahan, K.; Deltas, C.; Hosking, A.; Kmoch, S.; Rampoldi, L.; et al. Autosomal dominant tubulointerstitial kidney disease: Diagnosis, classification, and management—A KDIGO consensus report. Kidney Int. 2015, 88, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Nozu, K.; Goto, Y.; Kamei, K.; Ito, S.; Sato, H.; Emi, M.; Nakanishi, K.; Tsuchiya, S.; Iijima, K. HNF1B alterations associated with congenital anomalies of the kidney and urinary tract. Pediatr. Nephrol. 2010, 25, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Levine, E.; Huntrakoon, M. Unilateral renal cystic disease: CT findings. J. Comput. Assist. Tomogr. 1989, 13, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Middlebrook, P.F.; Nizalik, E.; Schillinger, J.F. Unilateral renal cystic disease: A case presentation. J. Urol. 1992, 148, 1221–1223. [Google Scholar] [CrossRef]
- Punia, R.P.; Mohan, H.; Bal, A.; Bansal, V.K. Unilateral and segmental cystic disease of the kidney. Int. J. Urol. 2005, 12, 308–310. [Google Scholar] [CrossRef]
- Hwang, D.Y.; Ahn, C.; Lee, J.G.; Kim, S.H.; Oh, H.Y.; Kim, Y.Y.; Lee, E.S.; Han, J.S.; Kim, S.; Lee, J.S. Unilateral renal cystic disease in adults. Nephrol. Dial. Transplant. 1999, 14, 1999–2003. [Google Scholar] [CrossRef] [PubMed]
- Poster, D.; Kistler, A.D.; Krauer, F.; Blumenfeld, J.D.; Rennert, H.; Weishaupt, D.; Wüthrich, R.P.; Serra, A.L. Kidney function and volume progression in unilateral autosomal dominant polycystic kidney disease with contralateral renal agenesis or hypoplasia: A case series. Am. J. Kidney Dis. 2009, 54, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Porch, P.; Noe, H.N.; Stapleton, F.B. Unilateral presentation of adult-type polycystic kidney disease in children. J. Urol. 1986, 135, 744–746. [Google Scholar] [CrossRef]
- Bergmann, C.; Guay-Woodford, L.M.; Harris, P.C.; Horie, S.; Peters, D.J.M.; Torres, V.E. Polycystic kidney disease. Nat. Rev. Dis. Prim. 2018, 4, 50. [Google Scholar] [CrossRef]
- Bergmann, C.; Senderek, J.; Windelen, E.; Küpper, F.; Middeldorf, I.; Schneider, F.; Dornia, C.; Rudnik-Schöneborn, S.; Konrad, M.; Schmitt, C.P.; et al. Clinical consequences of PKHD1 mutations in 164 patients with autosomal-recessive polycystic kidney disease (ARPKD). Kidney Int. 2005, 67, 829–848. [Google Scholar] [CrossRef]
- Guay-Woodford, L.M.; Bissler, J.J.; Braun, M.C.; Bockenhauer, D.; Cadnapaphornchai, M.A.; Dell, K.M.; Kerecuk, L.; Liebau, M.C.; Alonso-Peclet, M.H.; Shneider, B.; et al. Consensus expert recommendations for the diagnosis and management of autosomal recessive polycystic kidney disease: Report of an international conference. J. Pediatr. 2014, 165, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Burgmaier, K.; Kilian, S.; Bammens, B.; Benzing, T.; Billing, H.; Büscher, A.; Galiano, M.; Grundmann, F.; Klaus, G.; Mekahli, D.; et al. Clinical courses and complications of young adults with Autosomal Recessive Polycystic Kidney Disease (ARPKD). Sci. Rep. 2019, 9, 7919. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.J.; Hogan, M.C.; Rossetti, S.; Walker, D.; Sneddon, T.; Wang, X.; Kubly, V.; Cunningham, J.M.; Bacallao, R.; Ishibashi, M.; et al. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat. Genet. 2002, 30, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Onuchic, L.F.; Furu, L.; Nagasawa, Y.; Hou, X.; Eggermann, T.; Ren, Z.; Bergmann, C.; Senderek, J.; Esquivel, E.; Zeltner, R.; et al. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am. J. Hum. Genet. 2002, 70, 1305–1317. [Google Scholar] [CrossRef]
- Furu, L.; Onuchic, L.F.; Gharavi, A.; Hou, X.; Esquivel, E.L.; Nagasawa, Y.; Bergmann, C.; Senderek, J.; Avner, E.; Zerres, K.; et al. Milder presentation of recessive polycystic kidney disease requires presence of amino acid substitution mutations. J. Am. Soc. Nephrol. 2003, 14, 2004–2014. [Google Scholar] [CrossRef]
- Losekoot, M.; Haarloo, C.; Ruivenkamp, C.; White, S.J.; Breuning, M.H.; Peters, D.J. Analysis of missense variants in the PKHD1-gene in patients with autosomal recessive polycystic kidney disease (ARPKD). Hum. Genet. 2005, 118, 185–206. [Google Scholar] [CrossRef]
- Gunay-Aygun, M.; Tuchman, M.; Font-Montgomery, E.; Lukose, L.; Edwards, H.; Garcia, A.; Ausavarat, S.; Ziegler, S.G.; Piwnica-Worms, K.; Bryant, J.; et al. PKHD1 sequence variations in 78 children and adults with autosomal recessive polycystic kidney disease and congenital hepatic fibrosis. Mol. Genet Metab. 2010, 99, 160–173. [Google Scholar] [CrossRef]
- Gunay-Aygun, M.; Font-Montgomery, E.; Lukose, L.; Tuchman, M.; Graf, J.; Bryant, J.C.; Kleta, R.; Garcia, A.; Edwards, H.; Piwnica-Worms, K.; et al. Correlation of kidney function, volume and imaging findings, and PKHD1 mutations in 73 patients with autosomal recessive polycystic kidney disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 972–984. [Google Scholar] [CrossRef]
- Rossetti, S.; Harris, P.C. Genotype-phenotype correlations in autosomal dominant and autosomal recessive polycystic kidney disease. J. Am. Soc. Nephrol. 2007, 18, 1374–1380. [Google Scholar] [CrossRef]
- Erger, F.; Brüchle, N.O.; Gembruch, U.; Zerres, K. Prenatal ultrasound, genotype, and outcome in a large cohort of prenatally affected patients with autosomal-recessive polycystic kidney disease and other hereditary cystic kidney diseases. Arch. Gynecol. Obstet. 2017, 295, 897–906. [Google Scholar] [CrossRef]
- Burgmaier, K.; Brinker, L.; Erger, F.; Beck, B.B.; Benz, M.R.; Bergmann, C.; Boyer, O.; Collard, L.; Dafinger, C.; Fila, M.; et al. Refining genotype-phenotype correlations in 304 patients with autosomal recessive polycystic kidney disease and PKHD1 gene variants. Kidney Int. 2021, 100, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Galeano, M.C.R.; Ott, E.; Kaeslin, G.; Kausalya, P.J.; Kramer, C.; Ortiz-Brüchle, N.; Hilger, N.; Metzis, V.; Hiersche, M.; et al. Mutations in DZIP1L, which encodes a ciliary-transition-zone protein, cause autosomal recessive polycystic kidney disease. Nat. Genet. 2017, 49, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, C. Early and Severe Polycystic Kidney Disease and Related Ciliopathies: An Emerging Field of Interest. Nephron 2019, 141, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Dillman, J.R.; Trout, A.T.; Smith, E.A.; Towbin, A.J. Hereditary Renal Cystic Disorders: Imaging of the Kidneys and Beyond. Radiographics 2017, 37, 924–946. [Google Scholar] [CrossRef]
- Devuyst, O.; Olinger, E.; Weber, S.; Eckardt, K.U.; Kmoch, S.; Rampoldi, L.; Bleyer, A.J. Autosomal dominant tubulointerstitial kidney disease. Nat. Rev. Dis. Prim. 2019, 5, 60. [Google Scholar] [CrossRef]
- Bolar, N.A.; Golzio, C.; Zivna, M.; Hayot, G.; Van Hemelrijk, C.; Schepers, D.; Vandeweyer, G.; Hoischen, A.; Huyghe, J.R.; Raes, A.; et al. Heterozygous Loss-of-Function SEC61A1 Mutations Cause Autosomal-Dominant Tubulo-Interstitial and Glomerulocystic Kidney Disease with Anemia. Am. J. Hum. Genet. 2016, 99, 174–187. [Google Scholar] [CrossRef]
- Mabillard, H.; Sayer, J.A.; Olinger, E. Clinical and genetic spectra of autosomal dominant tubulointerstitial kidney disease. Nephrol. Dial. Transpl. 2021, gfab268. [Google Scholar] [CrossRef]
- Labriola, L.; Olinger, E.; Belge, H.; Pirson, Y.; Dahan, K.; Devuyst, O. Paradoxical response to furosemide in uromodulin-associated kidney disease. Nephrol. Dial. Transpl. 2015, 30, 330–335. [Google Scholar] [CrossRef][Green Version]
- Schaeffer, C.; Cattaneo, A.; Trudu, M.; Santambrogio, S.; Bernascone, I.; Giachino, D.; Caridi, G.; Campo, A.; Murtas, C.; Benoni, S.; et al. Urinary secretion and extracellular aggregation of mutant uromodulin isoforms. Kidney Int. 2012, 81, 769–778. [Google Scholar] [CrossRef]
- Bollee, G.; Dahan, K.; Flamant, M.; Moriniere, V.; Pawtowski, A.; Heidet, L.; Lacombe, D.; Devuyst, O.; Pirson, Y.; Antignac, C.; et al. Phenotype and outcome in hereditary tubulointerstitial nephritis secondary to UMOD mutations. Clin. J. Am. Soc. Nephrol. 2011, 6, 2429–2438. [Google Scholar] [CrossRef]
- Kirby, A.; Gnirke, A.; Jaffe, D.B.; Baresova, V.; Pochet, N.; Blumenstiel, B.; Ye, C.; Aird, D.; Stevens, C.; Robinson, J.T.; et al. Mutations causing medullary cystic kidney disease type 1 lie in a large VNTR in MUC1 missed by massively parallel sequencing. Nat. Genet. 2013, 45, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.M.; Bleyer, A.J.; Anis, K.; Herlitz, L.; Zivna, M.; Hulkova, H.; Markowitz, G.S.; Jim, B. Autosomal Dominant Tubulointerstitial Kidney Disease due to MUC1 Mutation. Am. J. Kidney Dis. 2018, 71, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Olinger, E.; Hofmann, P.; Kidd, K.; Dufour, I.; Belge, H.; Schaeffer, C.; Kipp, A.; Bonny, O.; Deltas, C.; Demoulin, N.; et al. Clinical and genetic spectra of autosomal dominant tubulointerstitial kidney disease due to mutations in UMOD and MUC1. Kidney Int. 2020, 98, 717–731. [Google Scholar] [CrossRef]
- Ayasreh, N.; Bullich, G.; Miquel, R.; Furlano, M.; Ruiz, P.; Lorente, L.; Valero, O.; Garcia-Gonzalez, M.A.; Arhda, N.; Garin, I.; et al. Autosomal Dominant Tubulointerstitial Kidney Disease: Clinical Presentation of Patients with ADTKD-UMOD and ADTKD-MUC1. Am. J. Kidney Dis. 2018, 72, 411–418. [Google Scholar] [CrossRef]
- Zivna, M.; Kidd, K.; Zaidan, M.; Vyletal, P.; Baresova, V.; Hodanova, K.; Sovova, J.; Hartmannova, H.; Votruba, M.; Treslova, H.; et al. An international cohort study of autosomal dominant tubulointerstitial kidney disease due to REN mutations identifies distinct clinical subtypes. Kidney Int. 2020, 98, 1589–1604. [Google Scholar] [CrossRef]
- Bleyer, A.J.; Zivná, M.; Hulková, H.; Hodanová, K.; Vyletal, P.; Sikora, J.; Zivný, J.; Sovová, J.; Hart, T.C.; Adams, J.N.; et al. Clinical and molecular characterization of a family with a dominant renin gene mutation and response to treatment with fludrocortisone. Clin. Nephrol. 2010, 74, 411–422. [Google Scholar] [CrossRef]
- Schaeffer, C.; Izzi, C.; Vettori, A.; Pasqualetto, E.; Cittaro, D.; Lazarevic, D.; Caridi, G.; Gnutti, B.; Mazza, C.; Jovine, L.; et al. Autosomal Dominant Tubulointerstitial Kidney Disease with Adult Onset due to a Novel Renin Mutation Mapping in the Mature Protein. Sci. Rep. 2019, 9, 11601. [Google Scholar] [CrossRef] [PubMed]
- Cornec-Le Gall, E.; Olson, R.J.; Besse, W.; Heyer, C.M.; Gainullin, V.G.; Smith, J.M.; Audrézet, M.P.; Hopp, K.; Porath, B.; Shi, B.; et al. Monoallelic Mutations to DNAJB11 Cause Atypical Autosomal-Dominant Polycystic Kidney Disease. Am. J. Hum. Genet. 2018, 102, 832–844. [Google Scholar] [CrossRef]
- Huynh, V.T.; Audrezet, M.P.; Sayer, J.A.; Ong, A.C.; Lefevre, S.; Le Brun, V.; Despres, A.; Senum, S.R.; Chebib, F.T.; Barroso-Gil, M.; et al. Clinical spectrum, prognosis and estimated prevalence of DNAJB11-kidney disease. Kidney Int. 2020, 98, 476–487. [Google Scholar] [CrossRef]
- Luo, F.; Tao, Y.H. Nephronophthisis: A review of genotype-phenotype correlation. Nephrology 2018, 23, 904–911. [Google Scholar] [CrossRef]
- Snoek, R.; van Setten, J.; Keating, B.J.; Israni, A.K.; Jacobson, P.A.; Oetting, W.S.; Matas, A.J.; Mannon, R.B.; Zhang, Z.; Zhang, W.; et al. NPHP1 (Nephrocystin-1) Gene Deletions Cause Adult-Onset ESRD. J. Am. Soc. Nephrol. 2018, 29, 1772–1779. [Google Scholar] [CrossRef]
- Hildebrandt, F.; Attanasio, M.; Otto, E. Nephronophthisis: Disease mechanisms of a ciliopathy. J. Am. Soc. Nephrol. 2009, 20, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.T. Nephronophthisis and related syndromes. Curr. Opin. Pediatr. 2015, 27, 201–211. [Google Scholar] [CrossRef]
- Georges, B.; Cosyns, J.P.; Dahan, K.; Snyers, B.; Carlier, B.; Loute, G.; Pirson, Y. Late-onset renal failure in Senior-Loken syndrome. Am. J. Kidney Dis. 2000, 36, 1271–1275. [Google Scholar] [CrossRef] [PubMed]
- Hoefele, J.; Nayir, A.; Chaki, M.; Imm, A.; Allen, S.J.; Otto, E.A.; Hildebrandt, F. Pseudodominant inheritance of nephronophthisis caused by a homozygous NPHP1 deletion. Pediatr. Nephrol. 2011, 26, 967–971. [Google Scholar] [CrossRef] [PubMed]
- König, J.; Kranz, B.; König, S.; Schlingmann, K.P.; Titieni, A.; Tönshoff, B.; Habbig, S.; Pape, L.; Häffner, K.; Hansen, M.; et al. Phenotypic Spectrum of Children with Nephronophthisis and Related Ciliopathies. Clin. J. Am. Soc. Nephrol. 2017, 12, 1974–1983. [Google Scholar] [CrossRef] [PubMed]
- Hudson, R.; Patel, C.; Hawley, C.M.; O’Shea, S.; Snelling, P.; Ho, G.; Holman, K.; Bennetts, B.; Crawford, J.; Francis, L.; et al. Adult-Diagnosed Nonsyndromic Nephronophthisis in Australian Families Caused by Biallelic NPHP4 Variants. Am. J. Kidney Dis. 2020, 76, 282–287. [Google Scholar] [CrossRef]
- Akira, M.; Suzuki, H.; Ikeda, A.; Iwasaki, M.; Honda, D.; Takahara, H.; Rinno, H.; Tomita, S.; Suzuki, Y. Atypical histological abnormalities in an adult patient with nephronophthisis harboring NPHP1 deletion: A case report. BMC Nephrol. 2021, 22, 261. [Google Scholar] [CrossRef]
- Macia, M.S.; Halbritter, J.; Delous, M.; Bredrup, C.; Gutter, A.; Filhol, E.; Mellgren, A.E.C.; Leh, S.; Bizet, A.; Braun, D.A.; et al. Mutations in MAPKBP1 Cause Juvenile or Late-Onset Cilia-Independent Nephronophthisis. Am. J. Hum. Genet. 2017, 100, 323–333. [Google Scholar] [CrossRef]
- Stokman, M.F.; van der Zwaag, B.; van de Kar, N.; van Haelst, M.M.; van Eerde, A.M.; van der Heijden, J.W.; Kroes, H.Y.; Ippel, E.; Schulp, A.J.A.; van Gassen, K.L.; et al. Clinical and genetic analyses of a Dutch cohort of 40 patients with a nephronophthisis-related ciliopathy. Pediatr. Nephrol. 2018, 33, 1701–1712. [Google Scholar] [CrossRef]
- Sugimoto, K.; Miyazawa, T.; Enya, T.; Nishi, H.; Miyazaki, K.; Okada, M.; Takemura, T. Clinical and genetic characteristics of Japanese nephronophthisis patients. Clin. Exp. Nephrol. 2016, 20, 637–649. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fujimaru, T.; Kawanishi, K.; Mori, T.; Mishima, E.; Sekine, A.; Chiga, M.; Mizui, M.; Sato, N.; Yanagita, M.; Ooki, Y.; et al. Genetic Background and Clinicopathologic Features of Adult-onset Nephronophthisis. Kidney Int. Rep. 2021, 6, 1346–1354. [Google Scholar] [CrossRef]
- Hamiwka, L.A.; Midgley, J.P.; Wade, A.W.; Martz, K.L.; Grisaru, S. Outcomes of kidney transplantation in children with nephronophthisis: An analysis of the North American Pediatric Renal Trials and Collaborative Studies (NAPRTCS) Registry. Pediatr. Transpl. 2008, 12, 878–882. [Google Scholar] [CrossRef] [PubMed]
- Harris, P.C.; Torres, V.E. Polycystic kidney disease. Annu. Rev. Med. 2009, 60, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Gurrieri, F.; Franco, B.; Toriello, H.; Neri, G. Oral-facial-digital syndromes: Review and diagnostic guidelines. Am. J. Med. Genet. A 2007, 143a, 3314–3323. [Google Scholar] [CrossRef] [PubMed]
- Romio, L.; Wright, V.; Price, K.; Winyard, P.J.; Donnai, D.; Porteous, M.E.; Franco, B.; Giorgio, G.; Malcolm, S.; Woolf, A.S.; et al. OFD1, the gene mutated in oral-facial-digital syndrome type 1, is expressed in the metanephros and in human embryonic renal mesenchymal cells. J. Am. Soc. Nephrol. 2003, 14, 680–689. [Google Scholar] [CrossRef]
- Feather, S.A.; Woolf, A.S.; Donnai, D.; Malcolm, S.; Winter, R.M. The oral-facial-digital syndrome type 1 (OFD1), a cause of polycystic kidney disease and associated malformations, maps to Xp22.2-Xp22.3. Hum. Mol. Genet. 1997, 6, 1163–1167. [Google Scholar] [CrossRef]
- Ferrante, M.I.; Giorgio, G.; Feather, S.A.; Bulfone, A.; Wright, V.; Ghiani, M.; Selicorni, A.; Gammaro, L.; Scolari, F.; Woolf, A.S.; et al. Identification of the gene for oral-facial-digital type I syndrome. Am. J. Hum. Genet. 2001, 68, 569–576. [Google Scholar] [CrossRef]
- Morleo, M.; Franco, B. OFD Type I syndrome: Lessons learned from a rare ciliopathy. Biochem. Soc. Trans. 2020, 48, 1929–1939. [Google Scholar] [CrossRef]
- Macca, M.; Franco, B. The molecular basis of oral-facial-digital syndrome, type 1. Am. J. Med. Genet. C Semin. Med. Genet. 2009, 151c, 318–325. [Google Scholar] [CrossRef]
- Feather, S.A.; Winyard, P.J.; Dodd, S.; Woolf, A.S. Oral-facial-digital syndrome type 1 is another dominant polycystic kidney disease: Clinical, radiological and histopathological features of a new kindred. Nephrol. Dial. Transplant. 1997, 12, 1354–1361. [Google Scholar] [CrossRef][Green Version]
- Curatolo, P.; Bombardieri, R.; Jozwiak, S. Tuberous sclerosis. Lancet 2008, 372, 657–668. [Google Scholar] [CrossRef]
- Dabora, S.L.; Jozwiak, S.; Franz, D.N.; Roberts, P.S.; Nieto, A.; Chung, J.; Choy, Y.S.; Reeve, M.P.; Thiele, E.; Egelhoff, J.C.; et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am. J. Hum. Genet. 2001, 68, 64–80. [Google Scholar] [CrossRef] [PubMed]
- Rakowski, S.K.; Winterkorn, E.B.; Paul, E.; Steele, D.J.; Halpern, E.F.; Thiele, E.A. Renal manifestations of tuberous sclerosis complex: Incidence, prognosis, and predictive factors. Kidney Int. 2006, 70, 1777–1782. [Google Scholar] [CrossRef]
- Northrup, H.; Krueger, D.A. Tuberous sclerosis complex diagnostic criteria update: Recommendations of the 2012 Iinternational Tuberous Sclerosis Complex Consensus Conference. Pediatr. Neurol. 2013, 49, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Wataya-Kaneda, M.; Tanaka, M.; Hamasaki, T.; Katayama, I. Trends in the prevalence of tuberous sclerosis complex manifestations: An epidemiological study of 166 Japanese patients. PLoS ONE 2013, 8, e63910. [Google Scholar] [CrossRef]
- Umeoka, S.; Koyama, T.; Miki, Y.; Akai, M.; Tsutsui, K.; Togashi, K. Pictorial review of tuberous sclerosis in various organs. Radiographics 2008, 28, e32. [Google Scholar] [CrossRef]
- Schillinger, F.; Montagnac, R. Chronic renal failure and its treatment in tuberous sclerosis. Nephrol. Dial. Transpl. 1996, 11, 481–485. [Google Scholar] [CrossRef]
- Barua, M.; Pei, Y. Diagnosis of autosomal-dominant polycystic kidney disease: An integrated approach. Semin. Nephrol. 2010, 30, 356–365. [Google Scholar] [CrossRef]
- Bissler, J.J.; Siroky, B.J.; Yin, H. Glomerulocystic kidney disease. Pediatr. Nephrol. 2010, 25, 2049–2056. [Google Scholar] [CrossRef]
- Siroky, B.J.; Towbin, A.J.; Trout, A.T.; Schäfer, H.; Thamann, A.R.; Agricola, K.D.; Tudor, C.; Capal, J.; Dixon, B.P.; Krueger, D.A.; et al. Improvement in Renal Cystic Disease of Tuberous Sclerosis Complex after Treatment with Mammalian Target of Rapamycin Inhibitor. J. Pediatr. 2017, 187, 318–322.e312. [Google Scholar] [CrossRef] [PubMed]
- Curatolo, P.; Maria, B.L. Tuberous sclerosis. Handb. Clin. Neurol. 2013, 111, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Martignoni, G.; Bonetti, F.; Pea, M.; Tardanico, R.; Brunelli, M.; Eble, J.N. Renal disease in adults with TSC2/PKD1 contiguous gene syndrome. Am. J. Surg. Pathol. 2002, 26, 198–205. [Google Scholar] [CrossRef]
- Sampson, J.R.; Maheshwar, M.M.; Aspinwall, R.; Thompson, P.; Cheadle, J.P.; Ravine, D.; Roy, S.; Haan, E.; Bernstein, J.; Harris, P.C. Renal cystic disease in tuberous sclerosis: Role of the polycystic kidney disease 1 gene. Am. J. Hum. Genet. 1997, 61, 843–851. [Google Scholar] [CrossRef]
- Pei, Y. Diagnostic approach in autosomal dominant polycystic kidney disease. Clin. J. Am. Soc. Nephrol. 2006, 1, 1108–1114. [Google Scholar] [CrossRef] [PubMed]
- Lonser, R.R.; Glenn, G.M.; Walther, M.; Chew, E.Y.; Libutti, S.K.; Linehan, W.M.; Oldfield, E.H. von Hippel-Lindau disease. Lancet 2003, 361, 2059–2067. [Google Scholar] [CrossRef]
- Maher, E.R.; Neumann, H.P.; Richard, S. von Hippel-Lindau disease: A clinical and scientific review. Eur. J. Hum. Genet. 2011, 19, 617–623. [Google Scholar] [CrossRef]
- Chittiboina, P.; Lonser, R.R. Von Hippel-Lindau disease. Handb. Clin. Neurol. 2015, 132, 139–156. [Google Scholar] [CrossRef]
- Ashouri, K.; Mohseni, S.; Tourtelot, J.; Sharma, P.; Spiess, P.E. Implications of Von Hippel-Lindau Syndrome and Renal Cell Carcinoma. J. Kidney Cancer VHL 2015, 2, 163–173. [Google Scholar] [CrossRef]
- Taylor, S.R.; Singh, J.; Sagoo, M.S.; Lightman, S.L. Clinical and molecular features associated with cystic visceral lesions in von hippel-lindau disease. Open Ophthalmol. J. 2012, 6, 83–85. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Gene | Inheritance | Prevalence | Clinical Features |
|---|---|---|---|---|
| ADPKD | PKD1, PKD2 | AD | 1/1000–1/2500 | Proliferative cysts in both kidneys increase in number and size as kidney function impairment advances, often introducing complications such as hypertension, multiple hepatic cysts, cerebral aneurysm, heart valve disease, and colonic diverticula. |
| Multiple simple renal cysts | Common | Usually a simple cyst of only unilateral, but with age the number of cysts increases in both kidneys. Normally asymptomatic. | ||
| ACKD | Common | In long-term dialysis patients, the renal parenchyma in both kidneys may show atrophy and proliferation of microcysts. Asymptomatic, but screening for hemorrhage and renal cancer development is necessary. | ||
| MCDK | 1/1000–1/4300 | Renal cysts of various sizes gather. Renal prognosis is favorable if no complications such as VUR in the contralateral kidney. | ||
| URCD | Unknown | Unilateral renal cysts resembling ADPKD in imaging. No family history of PKD, no progress to ESKD, and no complications such as hepatic cysts or cerebral aneurysm. | ||
| ARPKD | PKHD1, DZIP1L | AR | 1/8000–1/40,000 | Renal cystic lesions reflecting the expansion of the collecting duct with kidney function impairment, and congenital hepatic fibrosis characterized by bile duct dysplasia and intrahepatic periportal fibrogenesis. Thrombocytopenia, splenomegaly, and portal hypertension. |
| ADTKD | UMOD, MUC1, REN, HNF1B, SEC61A, DNAJB11 | AD | Unknown | Tubulointerstitial fibrogenesis and progressive kidney function impairment. Poor urinary findings, slow decline in kidney function. Small renal cysts without kidney enlargement. ADTKD-UMOD, -MUC1, -REN, -SEC61A; Hyperuricemia and Gout, ADTKD-HNF1B; diabetes mellitus and liver function impairment, ADTKD-SEC61A; congenital anemia, neutropenia, and hypogammaglobulinemia. ADTKD-DNAJB11; circulatory abnormalities such as intracranial aneurysm, thoracic aorta enlargement, and carotid artery dissociation. |
| NPH | NPHP1-20, NPHP1L, NPHP2L, TRAF31P1, AH11, CC2D2A | AR | 1/50,000–1/100,000 | Impaired urinary concentration, chronic tubulointerstitial nephritis, renal cystic lesions and accompanying kidney function impairment and progress to ESKD by the age of 30 years. Extrarenal lesions are reportedly found in 10–20% of patients, retinitis pigmentosa, cerebellar vermis hypoplasia, gaze palsy, hepatic fibrosis, and skeletal abnormalities. |
| OFD type1 | OFD1 | X-linked | 1/50,000–1/250,000 | Progress to kidney failure in adulthood with PKD with a variety of morphological abnormalities in the oral, facial, and finger and toe. Normal to large-sized kidneys. |
| TSC | TSC1, TSC2 | AD | 1/10,000 | Hamartoma in the skin, nervous system, kidney, lung, bone, and elsewhere. Renal lesions; angiomyolipoma (AML), renal cysts, renal cell carcinoma. |
| VHL syndrome | VHL | AD | 1/50,000 | Renal cell carcinoma, pancreatic cysts, central nervous system and retinal hemangioblastoma, and pheochromocytoma. Kidney function impairment is rare. |
| Class | |
|---|---|
| I | Hairline-thin wall; water attenuation; no septa, calcifications, or solid components; nonenhancing |
| II | Two types: 1. Few thin septa with or without perceived (not measurable) enhancement; fine calcification or a short segment of slightly thickened calcification in the wall or septa 2. Homogeneously high-attenuating masses ≤ 3 cm that are sharply marginated and do not enhance |
| IIF | Two types: 1. Minimally thickened or more than a few thin septa with or without perceived (not measurable) enhancement that may have thick or nodular calcification 2. Intrarenal nonenhancing hyperattenuating renal masses ≥ 3 cm |
| III | Thickened or irregular walls or septa with measurable enhancement |
| IV | Soft-tissue components (i.e., nodule[s]) with measurable enhancement |
| Major Features | Minor Features |
|---|---|
| 1. Hypomelanotic macules (≥3, at least 5-mm diameter) 2. Angiofibromas (≥3) or fibrous cephalic plaque 3. Ungual fibromas (≥2) 4. Shagreen patch 5. Multiple retinal hamartomas 6. Cortical dysplasias 7. Subependymal nodules 8. Subependymal giant cell astrocytoma 9. Cardiac rhabdomyoma 10. Lymphangioleiomyomatosis (LAM) 11. Angiomyolipomas (AML) (≥2) |
|
| Definitive diagnosis: 2 major features or 1 major feature with 2 minor features. Alternatively, the identification of either a TSC1 or TSC2 pathogenic mutation. | |
| Possible diagnosis: Either 1 major feature or ≥2 minor features | |
| (1) With a positive family history of VHL syndrome |
| Retinal angiomas, Spinal or cerebellar hemangioblastomas, Endolymphatic sac tumors, Renal cell carcinoma, Pheochromocytoma, Pancreatic lesions (Pancreatic cysts, Pancreatic neuroendocrine tumors), Epididymal cystadenomas, etc. |
| (2) With no clear family history of VHL syndrome |
| 1. Spinal or cerebellar hemangioblastomas or Retinal angiomas (≥2) 2. Spinal or cerebellar hemangioblastomas or Retinal angiomas (≥1) along with 1 of the following: a. Renal cell carcinoma, b. pheochromocytoma, c. Pancreatic cyst or Pancreatic neuroendocrine tumors, d. Epididymal cystadenomas, e. Endolymphatic sac tumors 3. 1 of the above clinical features with identification of a heterozygous germline pathogenic mutations in VHL on molecular genetic testing |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sekine, A.; Hidaka, S.; Moriyama, T.; Shikida, Y.; Shimazu, K.; Ishikawa, E.; Uchiyama, K.; Kataoka, H.; Kawano, H.; Kurashige, M.; et al. Cystic Kidney Diseases That Require a Differential Diagnosis from Autosomal Dominant Polycystic Kidney Disease (ADPKD). J. Clin. Med. 2022, 11, 6528. https://doi.org/10.3390/jcm11216528
Sekine A, Hidaka S, Moriyama T, Shikida Y, Shimazu K, Ishikawa E, Uchiyama K, Kataoka H, Kawano H, Kurashige M, et al. Cystic Kidney Diseases That Require a Differential Diagnosis from Autosomal Dominant Polycystic Kidney Disease (ADPKD). Journal of Clinical Medicine. 2022; 11(21):6528. https://doi.org/10.3390/jcm11216528
Chicago/Turabian StyleSekine, Akinari, Sumi Hidaka, Tomofumi Moriyama, Yasuto Shikida, Keiji Shimazu, Eiji Ishikawa, Kiyotaka Uchiyama, Hiroshi Kataoka, Haruna Kawano, Mahiro Kurashige, and et al. 2022. "Cystic Kidney Diseases That Require a Differential Diagnosis from Autosomal Dominant Polycystic Kidney Disease (ADPKD)" Journal of Clinical Medicine 11, no. 21: 6528. https://doi.org/10.3390/jcm11216528
APA StyleSekine, A., Hidaka, S., Moriyama, T., Shikida, Y., Shimazu, K., Ishikawa, E., Uchiyama, K., Kataoka, H., Kawano, H., Kurashige, M., Sato, M., Suwabe, T., Nakatani, S., Otsuka, T., Kai, H., Katayama, K., Makabe, S., Manabe, S., Shimabukuro, W., ... Muto, S. (2022). Cystic Kidney Diseases That Require a Differential Diagnosis from Autosomal Dominant Polycystic Kidney Disease (ADPKD). Journal of Clinical Medicine, 11(21), 6528. https://doi.org/10.3390/jcm11216528