Abstract
Multiple myeloma (MM) is the second most common hematological malignancy, and despite the introduction of innovative therapies, remains an incurable disease. Identifying early and minimally or non-invasive biomarkers for predicting clinical outcomes and therapeutic responses is an active field of investigation. Malignant plasma cells (PCs) reside in the bone marrow (BM) microenvironment (BMME) which comprises cells (e.g., tumour, immune, stromal cells), components of the extracellular matrix (ECM) and vesicular and non-vesicular (soluble) molecules, all factors that support PCs’ survival and proliferation. The interaction between PCs and BM stromal cells (BMSCs), a hallmark of MM progression, is based not only on intercellular interactions but also on autocrine and paracrine circuits mediated by soluble or vesicular components. In fact, PCs and BMSCs secrete various cytokines, including angiogenic cytokines, essential for the formation of specialized niches called “osteoblastic and vascular niches”, thus supporting neovascularization and bone disease, vital processes that modulate the pathophysiological PCs–BMME interactions, and ultimately promoting disease progression. Here, we aim to discuss the roles of cytokines and growth factors in pathogenetic pathways in MM and as prognostic and predictive biomarkers. We also discuss the potential of targeted drugs that simultaneously block PCs’ proliferation and survival, PCs–BMSCs interactions and BMSCs activity, which may represent the future goal of MM therapy.
1. Introduction
The balance between different cytokines is essential for tumor growth and progression. The proportion of inflammatory infiltrates and angiogenesis is determined by the vascular response generated by these pro-inflammatory factors [1]. This also occurs in multiple myeloma (MM), an incurable plasma cell (PC) dyscrasia that accounts for approximately 10% of hematological malignancies [2]. Typically, MM evolves from a pre-malignant stage termed a monoclonal gammopathy of undetermined significance (MGUS), which is observed in 1% of adults over 25 years of age. Every year, 1–2% of MGUSs progress to the malignant stage (active) of MM. A small subset of patients has an intermediate clinical phenotype between MGUS and MM, asymptomatic or smoldering multiple myeloma (SMM).
MM is a prototypic disease in which PCs subvert the local bone marrow microenvironment (BMME) to support their growth, elude immune surveillance and develop resistance to chemotherapy and/or immunotherapy. Hematopoietic stem cells (HSCs), mesenchymal stem cells (MSCs), bone marrow stromal cells (BMSCs), including fibroblasts (FBs), osteoblasts, osteoclasts, chondrocytes, endothelial cells (ECs) and endothelial progenitor cells (EPCs), coexist within the BMME with clonal malignant PCs, B and T lymphocytes, neutrophils, macrophages, mast cells, NK cells, erythrocytes, megakaryocytes, platelets and the extracellular matrix (ECM) [2]. The non-cellular compartment, on the other hand, includes soluble growth factors (i.e., interleukin (IL)-6, insulin-like growth factor (IGF)-1, vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF)-2, B-cell activating factor (BAFF), a proliferation-inducing ligand (APRIL), and stromal cell-derived factor (SDF-1)), adhesion molecules and extracellular vesicles [3,4].
Notably, the interaction between malignant PCs and BMSCs in the BMME is essential for the evolution of MM, for example through the production of IL-6 and the priming of the nuclear factor kappa-light-chain-enhancer of activated B cell (NF-κB) signaling [5].
The lack of an accurate prognostic biomarker panel in MM has led to recent research investigating angiogenic inflammatory cytokines and growth factors released in the BMME.
The identification of these “biomolecules” takes place in a unique site of the tumor BMME named the “vascular niche”, in which PCs survive undisturbed and proliferate [6]. The vascular niche originates from the de novo production of a vascular tree via the processes of neoangiogenesis, vasculogenesis and vascular mimicry [7]. The vascular niche allows PCs to leave the osteoblastic niche close to the endosteum and to enter the vascular system by migrating through the endothelium [5].
The deregulated conditions caused by hypoxic, proinflammatory and immuno-evasive conditions that occur in the “vascular niche” are currently under rigorous investigation to better understand the underlying mechanisms of cancer cells’ outgrowth and drug resistance in MM.
Cytokines play a critical role in PCs’ expansion, MM progression, cell-to-cell adhesion, angiogenesis and vasculogenesis. In general, the prevalence of proinflammatory cytokine secretion [IL-1, IL-6, IL-12, IL-15, IL-16, IL-17, IL-18, IL-22, IL-23, tumor necrosis factor (TNF)-α and interferon (IFN)-γ)] versus anti-inflammatory secretion [heat shock proteins (HSPs), IL-1 receptor (IL1R)α, IL-4, IL-10, IL-11, transforming growth factor (TGF)-β1, and lipoxin A4)] in MM patients is contingent on genetic alterations that originate from the cells within the microenvironment [8]. Consequently, MM ECs have an “activated” phenotype and genotype that determine the expression of adhesion molecules and receptors on these cells, stimulating their proliferation and new angiogenesis [7,9].
Here, we discuss the roles of cytokines and growth factors in pathogenetic pathways in MM and as prognostic and predictive biomarkers. We also elaborate on the major genetic and epigenetic aberrations that impact critical pathways responsible for the release of inflammatory and angiogenic cytokines [10]. Finally, we discuss the role of the BM microenvironment in drug resistance to anti-MM drugs [11].
2. Pro- and Anti-Inflammatory Cytokines Induce Pro- and Anti-Tumor Effects
Among the pro-inflammatory cytokines produced in the BMME, ILs certainly have the most significant influence on the malignant evolution of PCs. These cytokines make the PCs less differentiated and resistant to apoptosis and ultimately contribute to the malignant transformation from MGUS to MM [12].
The different stages of disease progression from MGUS to smoldering/symptomatic-MM are related to a progressive increase in cytokine levels (e.g., IL-1, IL-6, IL-12, IL-15, IL-17, IL-18, IL-22, IL-23, TNF-a and IFN-g) in the serum of MM patients. An eight-gene signature (IL-8, IL-10, IL-17, CCL3, CCL5, VEGFA, EBI3 and NOS2) involved in B lymphocyte inflammation has also been shown to distinguish the different phases of disease progression (MGUS/smoldering/symptomatic-MM) with an 84% accuracy [13].
The IL-1 family is a group of potent proinflammatory cytokines which includes IL-1α and IL-1β, regulated by different inhibitory receptors (e.g., IL-1Rα, IL-1RII) [14]. IL-1 family members have complex, divergent roles in the control of carcinogenesis and tumor progression [15]. IL-1 is produced by different types of cells in the BMME, such as FBs and B lymphocytes, and indirectly promotes the survival of PCs. In detail, low quantities of IL-1 stimulate the abundant production of IL-6, via a prostaglandin E2 (PGE2) loop [14,16]. According to several studies, the production of IL-1β is associated with the clonal evolution from MGUS to MM, revealing a minimal concentration in MGUS PCs [17,18,19,20] (Figure 1).
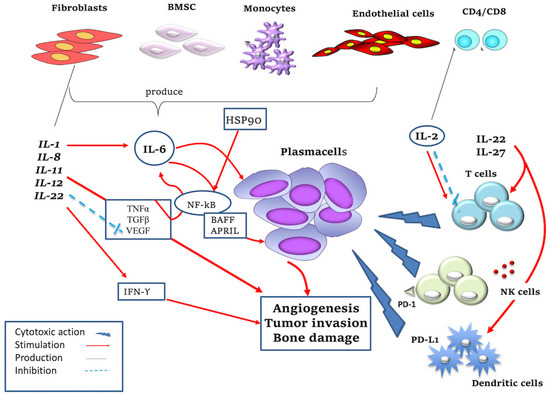
Figure 1.
Mechanism of action of pro- and anti-inflammatory cytokines. IL-1, produced by various cell types in the BM microenvironment (i.e., fibroblasts, B lymphocytes), promotes PCs’ survival by stimulating IL-6 production. IL-2 is produced by CD8+ and CD4+ T lymphocytes and promotes the growth of T and NK cells but at the same time induces apoptosis of T cells; it also stimulates the cytotoxic action of NK cells against malignant PCs. IL-6 promotes the growth of MM PCs and resistance to apoptosis; IL-6 is secreted by BMSCs, monocytes, endothelial cells, macrophages and fibroblasts thanks to the action of TGF-β, TNF-α, VEGF, IL-1. IL-1 and TNF are potent inducers of IL-6 synthesis. In detail, they regulate IL-6 gene expression at the level of transcription, which is mediated by upregulation of NF-kB activation. IL-6 stimulates the production of APRIL and BAFF, which activate NF-kB, phosphatidylinositol-3 (PI-3) kinase/AKT and MAPK pathways, promoting MM cells’ survival. IL-8 has a pro-tumor function promoting angiogenesis and tumor invasion. IL-11 promotes tumor invasion and in particular bone damage. IL-12 stimulates the production of IFN-γ with both pro-inflammatory and anti-angiogenic roles and this inhibits the production of VEGF. IL-22 is produced by T lymphocytes, activates the innate immune system and also regulates cell proliferation and resistance to drug-induced cell death in MM cells. IL-27 has antitumor activity, causing increased proliferation and differentiation of T cells. IL-27 stimulates the expression of immune checkpoint inhibitor programmed death (PD)-1 or PD-L1 in different cell types (i.e., lymphocytes and dendritic cells) and increases NK cells’ activity. HSP and HSP90 are produced by macrophages/monocytes and promote MM cell survival through TNF-mediated and NF-kB signaling pathways.
IL-2 is predominantly secreted by antigen-activated T cells and supports their proliferation and differentiation shortly after T cell receptor (TCR) stimulation [21], although its effects are controversial. In fact, IL-2 promotes the growth of T and NK cells but simultaneously induces the apoptosis of T cells [22] (Figure 1). IL-2 is used to treat immunogenic tumors such as melanoma, as it interferes with T cell-mediated responses. NK cells in MM disease express PD-L1 independently of IL-2, while in healthy subjects this receptor is expressed only in the presence of IL-2 [23]. Furthermore, hypoxia abrogates the cytotoxic activity of NK cells against multiple myeloma cells, but IL-2 can restore this ability. NK cell-based immunotherapy has been uncovered based on the above-mentioned findings [24].
IL-6, a pleiotropic cytokine with a variety of functions, is involved in immune responses, inflammation, hematopoiesis, bone metabolism and embryonic development, but above all it is an inducer of immunoglobulin production. IL-6 is a prototype member of the IL-6 family of cytokines, which is made up of 10 members, including IL-6, IL-11, IL-27, oncostatin M (OSM), leukemia inhibitory factor (LIF), ciliary neurotrophic factor (CNTF), cardiotrophin 1 (CT-1), factor 1 of cardiotropin-like cytokines (CLCF1), IL-35 and IL-39 [25]. IL-6 promotes MM PCs’ growth through several mechanisms involving signaling pathways with both pro- and anti-inflammatory effects [26]. The autocrine secretion of IL-6 is mainly associated with aggressive phenotypes. Frassanito et al. showed that autocrine levels of IL-6 produced by T cells are higher in patients with active disease than in patients in the plateau phase, remission or MGUS due to anticancer T helper 1 (Th1) cell dysfunction [27]. The paracrine mechanism occurs in the BM environment: IL-6 is secreted by BMSCs, monocytes, ECs, macrophages and FBs stimulated by TGF-β, TNF-α, VEGF, and IL-1. TNF-α induces IL-6 secretion in BMSCs activating NF-kB; on the contrary, the inhibition of NF-kB activity by the specific inhibitor of the IkB kinase (IKK) downregulates the secretion of IL-6 by BMSCs, promoting cell growth [26]. After binding to its receptor, IL-6 activates the signal janus kinase/signal transducer pathway (JAK/STAT) and the mitogen-activated GTPase/protein kinase pathway (RAS/MAPKs), inducing MM cell growth, the inhibition of apoptosis and proliferation. Due to the crucial role of Il-6 in MM progression, the blocking of its signaling pathways has been studied as a possible anti-MM target. This aspect was further investigated when the paracrine production of IL-6 was discovered in myeloid precursors [28]. BAFF and APRIL, new members of the TNF family, have been identified in recent years as potent inducers of MM cells. Both activate NF-kB, phosphatidylinositol-3 (PI-3) kinase/AKT and MAPK pathways and induce a strong up-regulation of the anti-apoptotic proteins Mcl-1 and Bcl-2 (Figure 1). The production of APRIL and its ligand BAFF is mediated by IL-6 production and has been recently considered as a new drug target, such as chimeric anti-IL-6 antibody Siltuximab [29,30].
IL-8, a member of the CXC family of chemokines, is produced by lymphocytes, monocytes, ECs, FBs, hepatocytes and keratinocytes. Substantially, it has a pro-tumor function by altering features of the tumor cells through the transition to a mesenchymal phenotype, to a migratory cell state, or by promoting proliferation [31]. Herrero et al. [32] investigated a possible role for IL-8 in osteoclastogenesis in a breast cancer model, in which IL-8 released by breast cancer cells increases osteoclast formation, probably contributing to bone metastasis. In this regard, it has been shown that in MM patients treated with melphalan and bortezomib, there is an increase in the production of IL-8 by BMSC and osteoclasts which favor bone damage [32]. Hence, the inhibition of IL-8 may represent a therapeutic target to counteract the aggressiveness of MM disease.
IL-11 belongs to the IL-6 family cytokines, along with inhibitory factor of leukemia (LIF), oncostatin M (OSM), ciliary neurotrophic factor, cardiotropin-1 and cytokines similar to cardiotrophin. These cytokines regulate immune responses in the acute phase of infections and also during processes such as hematopoiesis, the regeneration of the liver and neurons, embryonic development and fertility [33]. Accumulating evidence supports the notion that these cytokines and their related receptors play an important role in cancer biology and represent potential biomarkers for disease progression [34,35,36]. Giuliani et al. [37] demonstrated that IL-11 is involved in osteocytes and osteoclasts’ formation, inducing MM bone lesions. According to this study, the number of viable osteocytes decreases proportionally to bone invasion due to increased osteoclasts. On a bio-humoral level, this process corresponds to an increase in the production of osteoclastogenic cytokines, such as IL-11, found to be increased in patients with bone damage [37] (Figure 1).
IL-12 concentrations increase in MM patients compared to MGUS [11]. IL-12, mainly produced by mononuclear phagocytes, dendritic cells and neutrophils [32], is highly associated with pro-inflammatory, immunomodulatory and anti-tumor effects, stimulating the production of IFN-γ. The latter has an anti-angiogenic role by inhibiting the production of VEGF and FGF-2, and by stimulating the release of antiangiogenic chemokines [38] (Figure 1). Thalidomide, the progenitor of immunomodulatory drugs, exerts a robust suppression of the IL-12 production by peripheral blood mononuclear cells (PBMCs) [38]. Wang et al. showed that the synergistic effect of the proteasome inhibition with bortezomib and the immune treatment with IL-12 yields a more significant tumour growth inhibition [39]. In view of these broad-spectrum roles in the regulation of immune responses, IL-12 family cytokines are recognized as possible candidates for the modulation of antitumor immunity [40].
IL-27 is a cytokine that belongs to the IL-12 superfamily, which comprises IL-12, IL-23 and IL-35, which are produced by antigen-presenting cells in response to microbial or host immune stimuli and are involved in the regulation of immune responses against infections and tumor development [41]. IL-27 is composed of the EBI3 and p35 subunits and activates both STAT1 and STAT3 through a single IL-27 receptor, consisting of the WSX-1 receptor subunit coupled with the gp130 chain. The activation of STAT1 and STAT3 in T lymphocytes causes an increase in the proliferation of differentiated of CD4+ T helper and early Th1 cells and the suppression of the differentiation of Th2 and Th17 cells. Furthermore, IL-27 is involved in the production of regulatory T cells [42,43]. Several studies demonstrate that IL-27 activity may vary based on the type of target cell but in general appears to be increased in MM patients with an active disease. IL-27 primarily stimulates the expression of programmed death-ligand 1 (PD-L1) in different cell types, including CD4+ and CD8+ T cells, monocytes, dendritic cells, ECs and cancer cells [41]. In particular, as shown by Dondero et al., IL-27 maintains or increases the functions of NK cells, induced by suboptimal IL-15 concentrations, against ECs isolated from BM aspirates of MM patients. NK cells were shown to kill MM ECs and produce IFN-γ. Finally, IL-27 showed an extraordinary ability to increase the regulation of PD-L2 and HLA-I expression on the tumor endothelium, while it did not modify that of PD-L1 and HLA-II [44]. This therefore also reveals the antiangiogenic properties of IL-27 (Figure 1).
IL-22 belongs to the IL-10 superfamily cytokines which regulate the acute phase response, activating the innate immune system, cell migration and differentiation, and gene expression. Different cell types produce IL-22, including activated Th1 lymphocytes and endothelial cells. Through its receptor (IL22R), IL-22 activates JAK1, tyrosine-protein kinase (Tyk)2 and MAPK signaling pathways and therefore promotes cell proliferation and drug resistance in MM via the phosphorylation of STAT3 (Figure 1). Furthermore, the production of IL-22 in patients with active MM correlates with the release of IL-1β depicting the inflammatory component of the disease [45]. In general, studies conducted on this cytokine show that high secretion levels correlate with poor prognoses.
3. Angiogenic Cytokines and PCs Clonal Expansion
PCs function as primary inducers of angiogenesis and are crucial for the activation of BMSCs [46].
PCs express receptors such as αvβ3 integrin, which are important for the interaction with SCs, and secrete cytokines, such as VEGF-A, FGF-2, HGF-SF, angiotensin-1 (Ang-1), IGF-1, C-X-C motif chemokine (CXCL)12/SDF-1α and TNF-α.
These cytokines mediate cell growth (IL-6, IGF-1, SDF-1α, VEGF), survival (IL-6, IGF-1), drug resistance (IL-6, IGF-1, VEGF), migration (IGF-1, VEGF, MMP, SDF-1α) and angiogenesis (VEGF) in the BM.
3.1. Vascular Endothelial Growth Factor A (VEGF-A)
Vascular endothelial growth factor A (VEGF-A) belongs to a family of six proteins similar in structure and function: VEGF-A, -B, -C, -D, -E (viral factor), PDGF. These proteins exert their angiogenic (VEGF-A, -E/VEGFR-2-neuropilin-1,-2) activity or lymphangiogenic (VEGF-C, -D/VEGFR-2, -3) activity by binding to their respective receptors [7]. During the angiogenic switch, PCs acquire an angiogenic phenotype. This phenotype is the result of the expression of some oncogenes (c-myc, c-fos, c-Jun, ETS-1) that encode angiogenic factors and a shift from CD45-positive to CD45-negative PCs that are producers of VEGF [47]. Solimando et al. [48] demonstrated that a high expression of intercellular adhesion molecules, such as junctional adhesion molecule A (JAM-A), can essentially stimulate MM-associated angiogenesis.
In addition, genetic studies have introduced gene expression profiling 70 (GEP 70) [49], a microarray-based 70-gene classifier that identifies patients with a high risk for short progression-free survival and overall survival. GEP70 includes markers of angiogenesis, such as FABP5, BIRC5, AURKA, ALDOA, YWHAZ and ENO-1, strong mediators of neo-vasculogenesis [10].
VEGF-A is the most pro-angiogenic cytokine in MM: it is a regulator of the cell growth, survival and migration of ECs through VEGFR-2; in contrast, when acting through VEGFR-1, it regulates the growth of BMSCs.
Tumour-associated hypoxia promotes the production of VEGF-A via hypoxia inducible factor 1α (Hif-1α), an important transcription factor that regulates angiogenesis, mainly through the induction of VEGF transcription [50].
NF-κB, a transcription factor that plays a key role in the survival and proliferation of MM PCs, induces the overexpression of VEGF-A [5,51]. NF-κB can be activated by the phosphoinositide 3-kinase (PI3K)/Akt signaling pathway, implying that PI3K/Akt activation may play a role in angiogenesis and tumour progression in hematological malignancies [52]. VEGF-A directly stimulates the migration, proliferation, and survival of PCs through the autocrine and paracrine loops VEGF-A/VEGFR-2. Specifically, VEGF-A mediates the resistance to apoptosis via HSP90, which binds to Bcl-2 and Apaf-1, suppressing their apoptotic functions [53].
A recent clinical study involving MM patients (GIMEMA-MM0305 NCT01063179) has shown that high levels of VEGF and FGF-2 were associated with an unfavorable prognosis, supporting the notion that VEGF plays an important role in MM progression and suggesting that angiogenic factors may be used as non-invasive prognostic biomarkers in MM [54].
3.2. Tumour Necrosis Factor α (TNFα)
Tumour necrosis factor α (TNFα) is a proinflammatory cytokine produced by monocytes, macrophages, lymphocytes, and NK cells, which exerts its pro-tumoral action mainly on MM cells and BMSCs [55].
Bladè et al. reported that high serum concentrations of TNF-α in MGUS patients correlate with a higher probability of malignant progression when compared to patients with low serum concentrations [56].
By binding to its receptor, TNFR1, TNF induces the expression of pro-survival genes via the trimeric IκB kinase (IKK) complex/NKkB axis [57,58].
3.3. Insulin-like Growth Factor-1 (IGF-1)
Insulin-like growth factor-1 (IGF-1), secreted by the BMSC and osteoblasts, induces the growth, survival, and migration of cells by binding to its receptor on MM cells’ IGF-1R, with the subsequent activation of MAPK and PI3K/Akt signaling pathways. The Akt cascade leads to the activation of anti-apoptotic proteins Bcl-XL and Bcl-2 which promote PCs’ survival [59]. Furthermore, IGF-1 stimulates MM PCs to secrete VEGF. Finally, it promotes the proliferation, growth and chemotaxis of MMECs and BMSCs [7,59].
3.4. Matrix Metalloproteinases (MMP-2, MMP-9, uPA)
Matrix metalloproteinases (MMP-2, MMP-9, uPA) and their inhibitors (TIMP-1 and TIMP-2) are constantly produced by MM PCs (MMP-2 and -9) and BMSC (MMP-1 and -2). MMPs degrade collagen and fibronectin, allowing MM PCs to invade the stroma and the subendothelial basement membrane [60].
3.5. Fibroblast Growth Factor-2 (FGF-2)
Fibroblast growth factor-2 (FGF-2) promotes the proliferation, growth, and chemotaxis of MM ECs and BMSCs. It promotes the secretion of IL-6 and VEGF by BM SCs and is found to be upregulated in active MM. Its expression is related to the microvascular density of the BM [7].
FGF-2, together with VEGF and IGF, all secreted by MM plasma cells and inflammatory cells, promotes the recruitment of bone marrow stem cells and progenitor cells into the tumor microenvironment. Thus, MM endothelial cells are activated, participating in the formation of new vessel walls [61,62].
High levels of growth factors and angiogenic cytokines, such as VEGF-A, FGF-2, TNF-α, urokinase and MMPs, are secreted by the precursors of “cancer associated fibro-blasts” (CAFs). CAFs derive from cells that undergo the endothelial–mesenchymal phase or the mesenchymal transition and their precursors are resident FBs and progenitor cells that promote the formation of neovessels [6].
VEGF, FGF-2 and HGF can recruit and activate tumour-associated macrophages. Rajkumar et al. demonstrated that a higher proportion of CD68+ macrophages is present in patients with active MM compared to patients with an asymptomatic disease or MGUS. When exposed to VEGF and FGF-2, BM macrophages are capable of vasculogenic mimicry in vitro, making these cells endothelial-like cells. The latter can generate capillary networks in vitro. Indeed, tumor-associated macrophages express markers of both macrophages and endothelial cells with confocal laser microscopy [63].
3.6. Angiopoietin-1 and -2 (Ang-1 and -2)
Angiopoietin-1 and -2 (Ang-1 and -2) expression in MM patient serum and BM samples correlates with the BM microvascular density [64,65,66,67]. Several studies have shown that Ang-1 and Ang-2 are overexpressed in MM cell lines and primary PCs obtained from MM patients [65,68], and that the Tie-2 angiopoietin receptor is upregulated in the BM ECs in the presence of MM cells [69]. High Ang-1 and -2 expression levels were detected in patients with MM versus controls [64] and have been described as independent prognostic factors in these patients.
3.7. Platelet-Derived Growth Factor-BB (PDGF-BB)
Platelet-derived growth factor-BB (PDGF-BB) is strongly associated with the VEGF pathway and its altered secretion as well as other proangiogenic molecules, such as VEGF, FGF, EGF, TGFβ, angiopoietin in the tumor microenvironment, contributes to the mobilization and differentiation of EPCs and ECs’ proliferation [70].
Platelet-derived growth factor receptors-PDGFR AB and its α and β receptors in the serum of MM patients show a strong positive correlation with angiogenesis and bone marrow microvessel density. This also results in a prognostic value in terms of overall survival, as a higher PDGFR is associated with significantly higher microvessel density-MVD, which correlates with a lower survival rate [71].
PDGF and the PDGF-BB/PDGF-Rβ kinase axis expressed in the PCs of MM patients promotes tumor progression by activating ERK-1/2 and AKT [71,72]. In addition, PDGF secreted by MM PC and other SCs intervenes in the recruitment and differentiation of monocytes into active macrophages by activating the VEGF-A/VEGFR-1 and FGF-2/FGFR-1,-2,-3 pathways [73,74,75].
3.8. Hepatocyte Growth Factor (HGF)
Hepatocyte growth factor (HGF) is a potent angiogenic cytokine that induces the proliferation and migration of ECs by activating their specific tyrosine kinase receptor mesenchymal–epithelial transition factor cMET [74].
The HGF/cMET pathway triggers several signaling pathways involved in tumor growth, angiogenesis and metastatic spread. The HGF/cMET pathway acts on the pathogenesis of MM, enhancing the expression of VEGF/VEGFR-2 in MM ECs. When VEGF and HGF bind to their respective receptors, the dimerization and autophosphorylation of VEGFR and cMet induce the recruitment of signaling proteins to the binding site. Consequently, downstream pathways such as PI3K/AKT and Ras/ERK are activated, generating biological responses of cell survival, angiogenesis and tumor progression [7,74].
4. Cytokines and Bone Resorption
Osteolytic bone destruction is the most important effect of the dramatic changes in the BM microenvironment caused by the cross-talk between MM cells and the BM niche [75].
These lesions (“punched”) result from the increase in the number and activity of osteoclasts and the reduction in the number of osteoblasts and, therefore, the poor osteogenesis in the vicinity of MM cells [76].
The balance between the receptor activator of NF-kB-RANK and osteoprotegerin-OPG, which has a critical role in the regulation of osteoclastogenesis and bone remodeling activities, is maintained by the activity of different cytokines and pathways (IL-1, IL-6, TNF alpha, TNF/TRAF, PI3K, c-Src, Akt/PKB and mTOR) [77].
Several cytokines and growth factors are involved in the downregulation of osteoblastic activities. Exceptionally fundamental in bone formation and remodeling are the Wnt/dickkopf homolog 1 (DKK1) pathway, IL-3, IL-7, secreted frizzled-related protein-2 (sFRP-2), runt-related transcription factor 2 (Runx2), HGF, and TGF-β [78]. The activation of Runx2 and the Wnt pathways induce the differentiation of resident macrophages in osteoclasts and the transdifferentiation of PCs to functional osteoclasts [78,79].
DKK1 is overexpressed in patients with MM lytic bone lesions. Moreover, it also upregulates the production of Wnt-regulated OPG and RANKL by osteoblasts. If DKK1 is inhibited and Wnt signaling is activated, bone disease in MM is inhibited and the tumour mass is reduced [79]. The number of new, activated osteoclasts increases in areas near PCs, suggesting that bone lesions derive from the local production of osteoclast activation factors (OAF) secreted by MM PCs or SCs [79]. Many osteoclast-activating factors (IL-6, IL-1β, HGF and TNF-α) are released by the mutual interactions of tumour and bone marrow cells with a major involvement of RANKL, the decoy receptor OPG, and MIP-1α a.
RANKL is a member of the TNF family that binds to RANK on osteoclast precursors and promotes the production and differentiation of osteoclasts. OPG is a decoy receptor for RANKL. In MM, RANKL is increased, and OPG is decreased [75,80]. The binding between PCs and BMSCs through vascular cell adhesion molecule 1 (VCAM1) induces the overexpression of RANKL in both cell types, and inhibits the production of OPG by SCs [61,81,82]. MIP-1α, produced by MM PCs, promotes the proliferation and differentiation of osteoclasts’ maturation [83] (Figure 2).
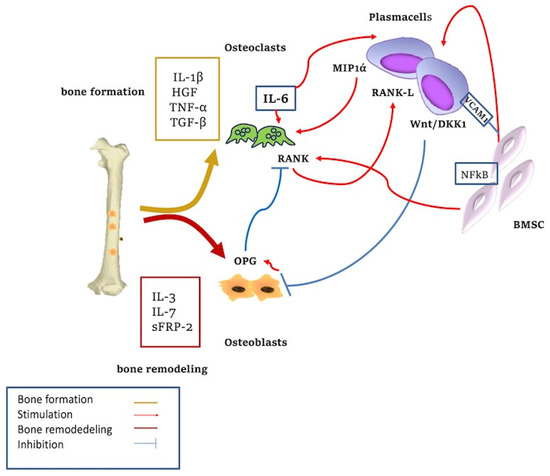
Figure 2.
Cytokine-mediated bone remodeling. The adhesion of plasma cells to stromal cells increases the regulation of IL6 with the activation of osteoclasts. The Wnt/DKK1 pathway is overexpressed in MM-associated lytic lesions. IL-3, IL-7, sFRP-2, and Runx2 are involved in bone formation; HGF and TGF-β act in bone remodeling. Wnt signaling stimulates osteoblasts’ (OB) differentiation. Blockade of this pathway through DKK1 inhibits OB formation. Wnt signaling upregulates RANKL expression from OB precursors, resulting in increased osteoclastic activity and bone resorption. Activation of Wnt pathway increases OPG production from OB which in turn downregulates RANKL-driven osteoclastogenesis. MIP-1α, produced by MM PC, promotes the proliferation and differentiation of osteoclast maturation. MM cells inhibit the osteoblastic activity through inhibitory factors and reduced production of cytokines.
In MM bone disease, osteoclasts can originate from resident monocyte-macrophages stimulated by RANKL overexpressed by the stromal cells [84,85].
It has been shown that CD38 is expressed by effectors and inhibitory cells and by osteoblasts. CD38 also appears to be involved in the formation and differentiation of osteoclasts and may play a role in bone remodeling [86,87]. Hence, there is a rationale for using the anti-CD38 monoclonal antibody (Daratumumab) against MM bone disease and the protection of MM PCs by BMSCs [86,87,88]. Large extracellular vesicles (i.e., microvesicles [4]) released after treatment with Daratumumab have been shown to be enriched in CD38. These microvesicles can be transported through the bloodstream and exert their action at distant sites where they contribute to ADO production [89], immunosuppression and bone remodeling within the BMME, via the modulation of pro- and anti-inflammatory cytokine release. Thus, microvesicles may be considered factors that determine the drug resistance to Daratumumab abrogating anti-MM immune responses [87,88,89].
5. Roles of Cytokines in Drug Resistance in Multiple Myeloma
Although the number of anti-MM treatments has dramatically increased in recent years, resulting in higher overall survival (OS) rates (up to 6–10 years), the emergence of drug resistance remains a major challenge for the management of MM [90,91,92]. Accumulating evidence suggests that targeting both PCs and the tumour microenvironment as well as the interactions between (cellular and non-cellular) BMME components and MM PCs may represent the ideal approach for treating MM patients [93]. A summary of anti-MM drugs is provided in Table 1 in which the mechanisms of action are indicated as well as possible implications of cytokines in drug actions or drug resistance.

Table 1.
Drugs, mechanisms of action, involved cytokines and related clinical studies.
Resistance to chemotherapy can be acquired or de novo [100]. The latter is innate and therefore occurs prior to drug exposure. Acquired resistance develops following genetic and epigenetic changes that make the tumor cell phenotype more and more complex and resistant to drugs [82]. The acquisition of drug resistance develops when malignant cells become genetically unstable and synthesize excessive quantities of abnormal proteins. Such genetic abnormalities, particularly epigenetic aberrations, influence DNA methylation patterns and histone modifications of genes, especially of tumor suppressor genes [101]. Furthermore, according to recent studies, factors such as microRNAs are involved in these mechanisms, modulating cellular signaling pathways that interfere with cell growth, proliferation, metastasis and drug resistance [102].
BMSCs have been shown to contribute to drug resistance in MM [103,104,105,106,107,108,109]. Different groups have shown that close interactions between MM and BMSC cells, hematopoietic niche components, endothelial cells and the extracellular matrix lead to so-called cell-mediated drug resistance (CAM-DR) [103,104]. CAM-DR is a mechanism in which MM cells escape the cytotoxic effects of anticancer therapy through adhesive interactions with BMSCs and/or ECM components using integrin family adhesion molecules (i.e., CD138/syndecan-1, Vascular Cell Adhesion Molecule-1/VCAM-1, Very Late Activation Antigen/VLA4/α4β1, VLA-5/α5β1, αvβ3 and β7 integrins) [105]. BMSCs are able to control the expression of anti-apoptotic proteins of the bcl-2 family and the ABC drug transporter proteins in myeloma cells, both involved in CAM-DR. Furthermore, BMSCs release IL-6 and IGF-1, which activate the signal transduction pathways that mediate drug resistance and increase the activation of HIF-1, affecting tumor metabolism and drug resistance [95,106,107,108,109,110,111,112,113,114].
Role of proteasome inhibitors. MM cells have been shown to be resistant to several drugs, such as doxorubicin, melphalan, vincristine, dexamethasone and mitoxantrone via the above-mentioned CAM-DR mechanisms [95,110,111,112,113,114]. Instead, the proteasome inhibitor bortezomib outperforms CAM-DR to vincristine and dexamethasone by inhibiting the adhesion of MM cells to fibronectin and BMSCs. Conversely, other integrins involved in CAM-DR, such as VLA-5 and β7, increase cell adhesion, migration and MM cell homing to the BM, and reduce bortezomib- and melphalan-induced apoptosis [115,116]. Proteasome inhibitors (i.e., bortezomib, carfilzomib, ixazomib) target the complex action of the proteasome, and by modulating NF-kB, they interfere with critical cellular processes, such as growth arrest, apoptosis, cell cycle progression, inflammation and immune surveillance [117]. The blockade of NF-kB inhibits the adhesion of MM cells to BMSCs, reduces the secretion of VEGF-2 and FGF-2 and angiogenic cytokines (e.g., VEGF, IL-6, IGF-I, Ang-1 and Ang-2), and consequently stops the growth of MM cells and creates new, activated ECs [118].
In addition to cell adhesion, numerous other mechanisms underlie the onset of primary and secondary resistance. The overexpression of the proteasome β5 subunit was the most frequent variation observed in bortezomib-resistant cell lines. The increase in the expression of the transcription factor of the oncogene c-Maf also affects the resistance to therapy with bortezomib and carfilzomib [108]. Finally, Zheng et al. [94] observed that the interaction between cytokine–cytokine receptors, autophagy, the ErbB signaling pathway and microRNAs can play a role in the resistance to carfilzomib in MM.
Role of Immunomodulatory drugs (IMIDs). IMIDs (thalidomide, lenalidomide, pomalidomide) selectively enhance the degradation of the transcription factors Ikaros (IKZF1) and Aiolos (IKZF3) vital for B-cell growth by binding to cereblon. In addition to their tumoricidal activity, IMIDs have critical antiangiogenic properties by reducing the expression of angiogenic cytokines [119,120]. They modulate the TNF-α signaling via direct and indirect effects on the tumor microenvironment [121], and they reduce the secretion of FGF-2 [112], VEGF and IL-6 by both MM cells and BMSCs [59].
Cell adhesion promotes the release of cytokines, chemokines and other factors that promote angiogenesis and immune suppression [10,59]. After extravasating into new tissue microenvironments, accommodating to survival signals, and colonizing extramedullary ecosystems, few plasma cells survive in the circulation [122,123].
Mechanisms of resistance to IMIDs studied to date include the downregulation of cerebron expression due to mutations in its gene. A recent study by Kortum et al. [96] identified three cerebron mutations developed by clonal selection after prolonged IMiD therapy.
New generations of more powerful cereblon E3 ligase modulators (CELMoDs®)—iberdomide (IBER) and CC-92480, are currently under investigation [99]. Their mechanism of action involves enhanced interactions with cereblon or substrates due to more extensive structures containing additional phenyl and morpholino fractions [124].
Role of immunotherapy. Increasing insights into the biology of MM, including the associated immune dysregulation, and the development of several immune-based therapies have renewed scientific interest in the clinical application of immunotherapy in MM. Monoclonal antibodies (mAbs) exert their cytotoxic function through different mechanisms: antibody-dependent cellular cytotoxicity (ADCC) through the engagement of immune effector cells, complement activation, antibody-dependent phagocytosis, and direct effects on target cells acting through different signaling pathways. Several mAbs are currently approved for the treatment of MM: elotuzumab, an IgG1κ mAb with a specificity against SLAMF7, daratumumab and isatuximab, humanizeds IgG1 mAb that targets CD38 [125,126]. Resistance to daratumumab is probably associated with an alteration in the CDC mechanism mediated by the overexpression of the complement inhibitory proteins CD55 and CD59 [97,126].
Role of B cell maturation antigen (BCMA) and CAR-T cells. Recently, BCMA has become an essential target for developing novel immunotherapeutics in MM. Targeted therapies against BCMA under evaluation include antibody-drug conjugates (ADCs), bispecific T cell engineers (BITE), and antigen receptor-modified T cell (CAR) chimeric therapies [127,128].
CAR T cells are genetically modified T cells with chimeric antigen receptors expressing a CAR against specific tumor-associated antigens. Binding to the specific antigen activates T lymphocytes, leading to cell lysis and death [129]. The immunosuppressive landscape of tumours is a critical obstacle to the effector activity of CAR-T cells. The tumor microenvironment, including its non-cellular components, various immune cells, abnormal tumor vasculature, immunosuppressive molecules and tumor metabolites, prevents CAR-T cells from exerting a high cytotoxicity. For example, fibroblasts and mesenchymal cells form physical barriers against the entry of T cells. Furthermore, the migration of T cells towards tumor lesions is increasingly challenged by the dysregulation of adhesion molecules, the mismatching of tumor-derived chemokines, and chemokine receptors expressed by immune cells.
Some immunosuppressive cytokine and chemokine as IL-2 recruit Treg cells, MDSCs and tumour-associated macrophages (TAMs) to specifically suppress cytotoxic T cell function. MDSCs have been shown to have a detrimental effect on CAR T cells because of their potent immunosuppressive capabilities directly targeting effector T cells [98].
Several studies have endeavored to integrate chemokines and CAR-T cells to fight cancer, with ongoing investigations focused on CXCR2-modified CAR-T cells that have been shown to induce complete tumour regression and immunological memory in aggressive tumors [130].
Furthermore, as demonstrated by several clinical studies as KarMMa and CARTITUDE-1, CAR- T can specifically recognize BCMA and kill BCMA-expressing MM cells [131].
Recent investigations have also shown that BCMA may represent a biomarker for the diagnosis of, monitoring of and therapeutic response to MM treatments, in both MM patients with secretory features (i.e., presence of tumour-related factors in peripheral blood) and in patients with non-secretory MM, in which it is more difficult to track the disease using conventional markers. Therefore, circulating BCMA may be helpful in detecting MM and also drug resistance [132].
As most of the therapies mentioned in this paragraph target the BMME and restore/activate an anti-MM immune response, BMME is believed to play a key role in therapy resistance for these drugs. As discussed by Neumeister et al. [133], BMME targeting drug resistance may be caused by the cross-talk of BMME and MM cells via soluble factors (e.g., IL-6, APRIL, growth factors), and via the integrin-mediated cell adhesion and Notch signaling, resulting in the inhibition of apoptosis [134,135,136], on which the major cytotoxic machinery of immune cells relies [137,138,139,140]. Drug resistance may also arise following the recruitment of immunosuppressive cells, including MDSCs, Tregs, Bregs and TAM, into the BMME of MM. These immunosuppressive cells might secrete nitric oxide (NO), arginase, reactive oxygen species (ROS), prostaglandin E2 (PGE2), or indoleamine 2,3-dioxygenase (IDO) [141,142,143] as well as immunosuppressive cytokines (e.g., IL-10 and TGF-β), inhibiting the proliferation and expansion of Th1 cells, CTLs and NK cells [144,145]. Furthermore, it has been shown that MSCs exert immunosuppressive actions mediated by the secretion of factors, such as IL-6, TGF-β, IL-10, PGE2, and by the upregulation of surface proteins, such as VCAM-1, ICAM-1, and CD40 [134,142,146,147,148,149]. For therapeutic approaches employing CAR T cells as well as bispecific antibodies, it was observed that MSCs could protect MM cells from highly lytic BCMA-CAR T cells [150], whereas the lytic function of BCMA/CD3 bispecific antibodies was not that much influenced by MSCs [151].
6. Conclusions
Cytokines play an important role in the pathogenesis and progression of MM. Some cytokines are produced directly by neoplastic cells as an effect of genetic mutations due to clonal evolution [152], and proinflammatory cytokines appear to be reduced in patients with MGUS compared to those with MM while anti-inflammatory cytokines can be both increased and reduced in MGUS. As described in this review, the role of cytokines in cancer and inflammation can be ambivalent. For this reason, the most critical proinflammatory cytokines involved in tumor progression, such as IL-6 and IL-1, cannot represent therapeutic targets without compromising the patients’ antitumor immunocompetence [153]. Anakinra, the recombinant form of IL-1Ra used for rheumatoid arthritis and periodic syndromes associated with crypirin [154], may be used in specific studies to evaluate IL-1 inhibition in MM.
The inhibition of proangiogenic cytokines has also been widely used in MM therapy. In addition to proteasome inhibitors and immunomodulators that have “indirect” antiangiogenic activity, bisphosphonates are other compounds that, although initially used to reduce bone loss in MM due to their anti-osteoclastic activity, have also been shown to have antiangiogenic activity [155,156]. Zoledronic acid has direct cytotoxic activity on cancer cells, suppresses angiogenesis, inhibits the FGF-2- and VEGF-dependent proliferation of endothelial cells, and inhibits VEGFR-2 in an autocrine cycle [157].
Future studies should aim at better understanding the delicate balance between the various cytokines in MM. The use of nanotechnology has been proposed to solve some of the limitations of conventional drug delivery systems, such as non-specific biodistribution and off-target effects, and to use cytokines for therapeutic purposes [158].
These strategies should aim at targeting both the malignant compartment (PCs) and the tumour microenvironment to overcome the onset of drug resistance and to possibly cure patients with MM.
Author Contributions
Conceptualization, A.M. and R.R.; writing of original draft, A.M., A.R., A.V. and R.R.; critical revision of the manuscript, A.R., I.S., V.D., S.C., A.L., M.A.F. and A.V.; English editing, A.R. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Programma Regionale—“Refine-Research for Innovation” POR Puglia FESR-FSE 2014–2020 (42F98C22) to A.M. and A.V. The sponsors of this study are public or non-profit organizations that support science in general.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| IMIDS | immunomodulatory agents |
| Anti-BCMA | anti-B cell maturation antigen |
| CRS | cytokine release syndrome |
| CRBN/DDB1 | complex-cereblon/DNA damage-binding protein1 |
| Treg | regulatory T cells |
References
- Akthar, S.; Tayyiba, A.; Faiyaz, A.; Khan, O.S.; Shadab Raza, S.; Kulinski, M.; El Omri, H.; Bhat, A.A.; Uddin, S. Cytokine-Mediated Dysregulation of Signaling Pathways in the Pathogenesis of Multiple Myeloma. Int. J. Mol. Sci. 2020, 21, 5002. [Google Scholar]
- Ria, R.; Reale, A.; Mangialardi, G.; Dammacco, F.; Ribatti, D.; Vacca, A. The Bone Marrow Microenvironment in Multiple Myeloma: Cellular and Molecular Basis of Disease Progression. In Cell Respiration and Cell Survival: Processes, Types and Effects; Nova Science Publishers: Hauppauge, NY, USA, 2009; Chapter 3; ISBN 978-1-60876-462-4. [Google Scholar]
- Ribatti, D.; Moschetta, M.; Vacca, A. Microenvironment and multiple myeloma spread. Thromb. Res. 2014, 133 (Suppl. S2), S102–S106. [Google Scholar] [CrossRef]
- Reale, A.; Carmichael, I.; Xu, R.; Mithraprabhu, S.; Khong, T.; Chen, M.; Fang, H.; Savvidou, I.; Ramachandran, M.; Bingham, N.; et al. Human myeloma cell- and plasma-derived extracellular vesicles contribute to functional regulation of stromal cells. Proteomics 2021, 21, e200011. [Google Scholar] [CrossRef]
- Dehghanifard, A.; Kaviani, S.; Abroun, S.; Mehdizadeh, M.; Saiedi, S.; Maali, A.; Ghaffari, S.; Azad, M. Various Signaling Pathways in Multiple Myeloma Cells and Effects of Treatment on These Pathways. Clin. Lymphoma Myeloma Leuk. 2018, 18, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Vacca, A. New Insights in Anti-Angiogenesis in Multiple Myeloma. Int. J. Mol. Sci. 2018, 19, 2031. [Google Scholar] [CrossRef] [PubMed]
- Ria, R.; Melaccio, A.; Racanelli, V.; Vacca, A. Anti-VEGF drugs in the treatment of multiple myeloma patients. J. Clin. Med. 2020, 9, 1765. [Google Scholar] [CrossRef]
- Musolino, C.; Allegra, A.; Innao, V.; Allegra, A.G.; Pioggia, G.; Gangemi, S. Inflammatory and Anti-Inflammatory Equilibrium, Proliferative and Antiproliferative Balance: The Role of Cytokines in Multiple Myeloma. Mediat. Inflamm. 2017, 24, 1852517. [Google Scholar] [CrossRef]
- Ria, R.; Todoerti, K.; Berardi, S.; Coluccia, A.M.L.; De Luisi, A.; Mattioli, M.; Ronchetti, D.; Morabito, F.; Guarini, A.; Petrucci, M.T.; et al. Gene expression profiling of bone marrow endothelial cells in patients with multiple myeloma. Clin. Cancer Res. 2009, 15, 5369–5378. [Google Scholar] [CrossRef]
- Solimando, A.G.; Da Vià, M.C.; Cicco, S.; Leone, P.; Di Lernia, G.; Giannico, D.; DeSantis, V.; Frassanito, M.A.; Morizio, A.; Delgado Tascon, J.; et al. High-Risk Multiple Myeloma: Integrated Clinical and Omics Approach Dissects the Neoplastic Clone and the Tumor Microenvironment. J. Clin. Med. 2019, 8, 977. [Google Scholar] [CrossRef]
- De Smedt, E.; Lui, H.; Maes, K.; De Veirman, K.; Menu, E.; Vanderkerken, K.; De Bruyne, E. The epigenomic in Multiple Myeloma: Impact in tumor cell plasticity and drug response. Front. Oncol. 2018, 11, 566. [Google Scholar] [CrossRef]
- Allegra, A.; Innao, V.; Allegra, A.G.; Pugliese, M.; Di Salvo, E.; Ventura-Spagnolo, E.; Musolino, C.; Gangemi, S. Lymphocyte Subsets and Inflammatory Cytokines of Monoclonal Gammopathy of Undetermined Significance and Multiple Myeloma. Int. J. Mol. Sci. 2019, 20, 2822. [Google Scholar] [CrossRef]
- Leone, P.; Solimando, A.G.; Malerba, E.; Fasano, R.; Buonavoglia, A.; Pappagallo, F.; De Re, V.; Argentiero, A.; Silvestris, N.; Vacca, A.; et al. Actors on the Scene: Immune Cells in the Myeloma Niche. Front. Oncol. 2020, 10, 599098. [Google Scholar] [CrossRef]
- Gabay, C.; Lamacchia, C.; Palmer, G. IL-1 pathways in inflammation and human diseases. Nat. Rev. Rheumatol. 2010, 6, 232–241. [Google Scholar] [CrossRef]
- Mantovani, A.; Dinarello, C.A.; Molgora, M.; Garlanda, C. Interleukin-1 and Related Cytokines in the Regulation of Inflammation and Immunity. Immunity 2019, 50, 778–795. [Google Scholar] [CrossRef]
- Lust, J.A.; QLacy, M.; Zeldenrust, S.R.; Dispenzieri, A.; Gertz, M.A.; Witzig, T.E.; Kumar, S.; Hayman, S.R.; Russell, S.J.; Buadi, F.K.; et al. Induction of a chronic disease state in patients with smoldering or indolent multiple myeloma by targeting interleukin 1β-induced Mediators of in flammation interleukin 6 production and the myeloma proliferative component. Mayo Clin. Proc. 2009, 84, 114–122. [Google Scholar] [CrossRef]
- Lichtenstein, A.; Berenson, J.; Norman, D.; Chang, M.P.; Carlile, A. Production of cytokines by bone marrow cells obtained from patients with multiple myeloma. Blood 1989, 74, 1266–1273. [Google Scholar] [CrossRef]
- Klein, B.; Lu, Z.Y.; Gaillard, J.P.; Harousseau, J.L.; Bataille, R. Inhibiting IL-6 in human multiple myeloma. Curr. Top. Microbiol. Immunol. 1992, 182, 237–244. [Google Scholar]
- Cozzolino, F.; Torcia, M.; Aldinucci, D.; Rubartelli, A.; Miliani, A.; Shaw, A.R.; Lansdorp, P.M.; Di Guglielmo, R. Production of interleukin-1 by bone marrow myeloma cells. Blood 1989, 74, 380–387. [Google Scholar] [CrossRef]
- Donovan, K.; Lacy, M.; Kline, M.; Ahmann, G.; Heimbach, J.; Kyle, R.; Lust, J. Contrast in cytokine expression between patients with monoclonal gammopathy of undetermined significance or multiple myeloma. Leukemia 1998, 12, 593–600. [Google Scholar] [CrossRef][Green Version]
- MacDonald, A.; Wu, T.-C.; Hung, C.-F. Interleukin 2-Based Fusion Proteins for the Treatment of Cancer. J. Immunol. Res. 2021, 2021, 11. [Google Scholar] [CrossRef]
- De la Rosa, M.; Rutz, S.; Dorninger, H.; Scheffold, A. Interleukin-2 is essential for CD4+ CD25+ regulatory T cell function. Eur. J. Immunol. 2004, 34, 2480–2488. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.M., Jr.; Bakan, C.E.; Mishra, A.; Hofmeister, C.C.; Efebera, Y.; Becknell, B.; Baiocchi, R.A.; Zhang, J.; Yu, J.; Smith, M.; et al. The PD-1/ PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: A therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody. Blood 2010, 116, 2286–2294. [Google Scholar] [CrossRef]
- Sarkar, S.; Germeraad, W.T.V.; Rouschop, K.M.A.; Steeghs, E.M.P.; Van Gelder, M.; Bos, G.M.J.; Wieten, L. Hypoxia-induced impairment of NK cell cytotoxicity against multiple myeloma can be overcome by IL-2 activation of the NK cells. PLoS ONE 2013, 8, e64835. [Google Scholar] [CrossRef] [PubMed]
- Toshio, H. IL-6 in inflammation, autoimmunity and cancer. Int. Immunol. 2020, 33, 127–148. [Google Scholar]
- Hideshima, T.; Podar, K.; Chauhan, D.; Anderson, K.C. Cytokines and signal transduction. Best Pract. Res. Clin. Haematol. 2005, 18, 509–524. [Google Scholar] [CrossRef]
- Frassanito, M.A.; Cusmai, A.; Dammacco, F. Deregulated cytokine network and defective Th1 immune response in multiple myeloma. Clin. Exp. Immunol. 2001, 125, 190–197. [Google Scholar] [CrossRef]
- Matthes, T.; Manfroi, B.; Huard, B. Revisiting IL-6 Antagonism in Multiple Myeloma. Crit. Rev. Oncol. Hematol. 2016, 105, 1–4. [Google Scholar] [CrossRef]
- Jing, P.; Sun, Y.; Zhang, N.; Li, J.; Ta, F.; Wei, W.; Yu, S.; Ai, L. Characteristics of BAFF and APRIL factor expression in multiple myeloma and clinical significance. Oncol. Lett. 2017, 14, 2657–2662. [Google Scholar]
- Markham, A.; Patel, T. Siltuximab: First global approval. Drugs 2014, 74, 1147–1152. [Google Scholar] [CrossRef]
- Fousek, F.; Horn, L.A.; Palena, C. Interleukin-8: A Chemokine at the Intersection of Cancer Plasticity, Angiogenesis, and Immune Suppression. Pharmacol. Ther. 2021, 219, 107692. [Google Scholar] [CrossRef]
- Herrero, A.B.; García-Gómez, A.; Garayoa, M.; Corchete, L.A.; Hernández, J.M.; Miguel, J.S.; Gutierrez, N.C. Effects of IL-8 Up-Regulation on Cell Survival and Osteoclastogenesis in Multiple Myeloma. Am. J. Clin. Pathol. 2016, 186, 2171–2182. [Google Scholar] [CrossRef]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Müller-Newen, G.; Schaper, F. Principles of Interleukin (IL)-6-type Cytokine Signalling and Its Regulation. Biochem. J. 2003, 374 Pt 1, 1–20. [Google Scholar] [CrossRef]
- Hongting, L.J.; Ng, B.; Lim, W. Interleukin-11: A Potential Biomarker and Molecular Therapeutic Target in Non-Small Cell Lung Cancer. Cells 2022, 11, 2257. [Google Scholar]
- Cook, S.A.; Schafer, S. Hiding in Plain Sight: Interleukin-11 Emerges as a Master Regulator of Fibrosis, Tissue Integrity, and Stromal Inflammation. Annu. Rev. Med. 2020, 71, 263–276. [Google Scholar] [CrossRef]
- Zhao, M.; Liu, Y.; Liu, R.; Qi, J.; Hou, Y.; Chang, J.; Ren, L. Upregulation of IL-11, an IL-6 Family Cytokine, Promotes Tumor Progression and Correlates with Poor Prognosis in Non-Small Cell Lung Cancer. Cell. Physiol. Biochem. 2018, 45, 2213–2224. [Google Scholar] [CrossRef]
- Giuliani, N.; Ferretti, M.; Bolzoni, M.; Storti, P.; Lazzaretti, M.; Dalla Palma, B.; Bonomini, S.; Martella, E.; Agnelli, L.; Neri, A.; et al. Increased Osteocyte Death in Multiple Myeloma Patients: Role in Myeloma-Induced Osteoclast Formation. Leukemia 2012, 26, 1391–1401. [Google Scholar] [CrossRef]
- De Raeve, H.; Vanderkerken, K. Immunomodulatory Drugs as a Therapy for Multiple Myeloma. Curr. Pharm. Biotechnol. 2006, 7, 415–421. [Google Scholar] [CrossRef]
- Dias, S.; Boyd, R.; Balkwill, F. IL-12 regulates VEGF and MMPs in a murine breast cancer model. Int. J. Cancer 1998, 78, 361–365. [Google Scholar] [CrossRef]
- Mirlekar, B.; Pylayeva-Gupta, Y. IL-12 Family Cytokines in Cancer and Immunotherapy. Cancers 2021, 13, 167. [Google Scholar] [CrossRef]
- Trinchieri, G.; Pflanz, S.; Kastelein, R.A. The IL-12 family of heterodimeric cytokines: New players in the regulation of T cell responses. Immunity 2003, 19, 641–644. [Google Scholar] [CrossRef]
- Giuliani, N.; Airoldi, I. Novel Insights into the Role of Interleukin-27 and Interleukin-23 in Human Malignant and Normal Plasma Cells. Clin. Cancer Res. 2011, 17, 6963–6970. [Google Scholar] [CrossRef] [PubMed]
- Carbotti, G.; Barisione, G.; Airoldi, I.; Mezzanzanica, D.; Bagnoli, M.; Ferrero, S.; Petretto, A.; Fabbi, M.; Ferrini, S. IL-27 induces the expression of IDO and PD-L1 in human cancer cells. Oncotarget 2015, 6, 43267–43280. [Google Scholar] [CrossRef] [PubMed]
- Dondero, A.; Casu, B.; Bellora, F.; Vacca, A.; De Luisi, A.; Frassanito, M.A.; Cantoni, C.; Gaggero, S.; Olive, D.; Moretta, A.; et al. NK Cells and Multiple Myeloma-Associated Endothelial Cells: Molecular Interactions and Influence of IL-27. Oncotarget 2017, 8, 35088–35102. [Google Scholar] [CrossRef] [PubMed]
- Tsirakis, G.; Pappa, C.A.; Kolovou, A.; Kokonozaki, M.; Neonakis, I.; Alexandrakis, M.G. Clinical significance of interleukin-22 in multiple myeloma. Hematology 2015, 20, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Berardi, S.; Caivano, A.; Ria, R.; Nico, B.; Savino, R.; Terracciano, R.; De Tullio, G.; Ferrucci, A.; De Luisi, A.; Moschetta, M.; et al. Four proteins governing overangiogenic endothelial cell phenotype in patients with multiple myeloma are plausible therapeutic targets. Oncogene 2012, 31, 2258–2269. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, M.; Turkbey, B.; Tan, E.; Kemp, T.J.; Pinto, L.A.; Berg, A.R.; Korde, N.; Minter, A.R.; Weiss, B.M.; Mena, E.; et al. Bone marrow angiogenesis in myeloma and its precursor disease: A prospective clinical trial. Leukemia 2014, 28, 413–416. [Google Scholar] [CrossRef]
- Solimando, A.G.; Da Vià, M.C.; Leone, P.; Borrelli, P.; Croci, G.A.; Tabares, P.; Brandl, A.; Di Lernia, G.; Bianchi, F.P.; Tafuri, S.; et al. Halting the vicious cycle within the multiple myeloma ecosystem: Blocking JAM-A on bone marrow endothelial cells restores angiogenic homeostasis and suppresses tumor progression. Haematologica 2021, 106, 1943–1956. [Google Scholar] [CrossRef]
- Ryan, V.L.; Flinchum, R.; Brown, N.; Ramsey, J.; Riccitelli, S.; Heuck, C.; Barlogie, B.; Shaughnessy, J.D., Jr. Translating a gene expression signature for multiple myeloma prognosis into a robust high-throughput assay for clinical use. BMC Med. Genom. 2014, 7, 25. [Google Scholar]
- Zhang, J.; Sattler, M.; Tonon, G.; Grabher, C.; Lababidi, S.; Zimmerhackl, A.; Raab, M.S.; Vallet, S.; Zhou, Y.; Cartron, M.-A.; et al. Targeting Angiogenesis via a c-Myc/Hypoxia-Inducible Factor-1α–Dependent Pathway in Multiple Myeloma. Cancer 2009, 69, 5082–5090. [Google Scholar] [CrossRef]
- Katoh, O.; Tauchi, H.; Kawaishi, K.; Kimura, A.; Satow, Y. Expression of the vascular endothelial growth factor (VEGF) receptor gene, KDR, in hematopoietic cells and inhibitory effect of VEGF on apoptotic cell death caused by ionizing radiation. Cancer Res. 1995, 55, 5687–5692. [Google Scholar]
- Braun, T.; Carvalho, G.; Fabre, C.; Grosjean, J.; Fenaux, P.; Kroemer, G. Targeting NF-kappaB in hematologic malignancies. Cell Death Differ. 2006, 13, 748–758. [Google Scholar] [CrossRef]
- Podar, K.; Tai, Y.-T.; Davies, F.; Lentzsch, S.; Sattler, M.; Hideshima, T.; Lin, B.K.; Gupta, D.; Shima, Y.; Chauhan, D.; et al. Vascular endothelial growth factor triggers signaling cascades mediating multiple myeloma cell growth and migration. Blood 2001, 98, 428–435. [Google Scholar] [CrossRef]
- Saltarella, I.; Morabito, F.; Giuliani, N.; Terragna, C.; Omedè, P.; Palumbo, A.; Bringhen, S.; De Paoli, L.; Martino, E.; LaRocca, A.; et al. Prognostic or predictive value of circulating cytokines and angiogenic factors for initial treatment of multiple myeloma in the GIMEMA MM0305 randomized controlled trial. J. Hematol. Oncol. 2019, 12, 4. [Google Scholar] [CrossRef]
- Mehta, A.K.; Gracias, D.T.; Croft, M. TNF activity and T cells. Cytokine 2018, 101, 14–18. [Google Scholar] [CrossRef]
- Bladè, J.; Filella, X.; Montoto, S.; Bosch, F.; Rosiñol, L.; Coca, F.; Giné, E.; Nadal, E.; Aymerich, M.; Rozman, M.; et al. Clinical relevance of interleukin 6 and tumor necrosis factor-alpha serum levels in monoclonal gammopathy of undetermined significance. Blood 1997, 90, 351. [Google Scholar]
- Roy, P.; Sarkar, U.A.; Basak, S. TheNF-κB Activating Pathways in Multiple Myeloma. Biomedicines 2018, 6, 59. [Google Scholar] [CrossRef]
- Dai, Y.; Pei, X.Y.; Rahmani, M.; Conrad, D.H.; Dent, P.; Grant, S. Interruption of the NF-kappaB pathway by Bay 11-7082 promotes UCN-01-mediated mitochondrial dysfunction and apoptosis in human multiple myeloma cells. Blood 2004, 103, 2761–2770. [Google Scholar] [CrossRef]
- Manier, S.; Sacco, A.; Leleu, X.; Ghobrial, I.M.; Roccaro, A.M. Bone Marrow Microenvironment in MultipleMyeloma Progression. BioMed Res. Int. 2012, 2012, 157496. [Google Scholar] [CrossRef]
- Greco, C. Reduction of serum IGF-1 levels in patients affected with monoclonal gammopathies of undetermined significance or multiple myeloma. Comparison with bFGF, VEGF and κ-ras gene mutation. J. Exp. Clin. Cancer Res. 2009, 28, 35. [Google Scholar] [CrossRef][Green Version]
- Vacca, A.; Ribatti, D. Bone marrow angiogenesis in multiple myeloma. Leukemia 2006, 20, 193–199. [Google Scholar] [CrossRef]
- Vacca, A. Angiogenesis in multiple myeloma. Chem. Immunol. Allergy 2014, 99, 180–196. [Google Scholar]
- Ria, R.; Piccoli, C.; Cirulli, T.; Falzetti, F.; Mangialardi, G.; Guidolin, D.; Tabilio, A.; Di Renzo, N.; Guarini, A.; Ribatti, D.; et al. Endothelial differentiation of hematopoietic stem and progenitor cells from patients with multiple myeloma. Clin. Cancer Res. 2008, 14, 1678–1685. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Nico, B.; Vacca, A. Importance of the bone marrow microenvironment in inducing the angiogenic response in multiple myeloma. Oncogene 2006, 25, 4257–4266. [Google Scholar] [CrossRef] [PubMed]
- Vacca, A. Bone marrow angiogenesis in patients with active multiple myeloma. Semin. Oncol. 2001, 28, 543–550. [Google Scholar] [CrossRef]
- Ting, K.R.; Henry, M.; Meiller, J.; Larkin, A.; Clynes, M.; Meleady, P.; Bazou, D.; Dowling, P.; O’Gorman, P. Novel panel of protein biomarkers to predict response to bortezomib-containing induction regimens in multiple myeloma patients. BBA Clin. 2017, 8, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Belloni, D.; Marcatti, M.; Ponzoni, M.; Ciceri, F.; Veschini, L.; Corti, A.; Cappio, F.C.; Ferrarini, M.; Ferrero, E. Angiopoietin-2 in bone marrow milieu promotes multiple myeloma-associated angiogenesis. Exp. Cell Res. 2015, 330, 1–12. [Google Scholar] [CrossRef]
- Giuliani, N.; Colla, S.; Lazzaretti, M.; Sala, R.; Roti, G.; Mancini, C.; Bonomini, S.; Lunghi, P.; Hojden, M.; Genestreti, G.; et al. Proangiogenic properties of human myeloma cells: Production of angiopoietin-1 and its potential relationship to myeloma-induced angiogenesis. Blood 2003, 102, 638–645. [Google Scholar] [CrossRef]
- Pappa, C.A.; Tsirakis, G.; Samiotakis, P.; Tsigaridaki, M.; Alegakis, A.; Goulidaki, N.; Alexandrakis, M.G. Serum levels of angiopoietin-2 are associated with the growth of multiple myeloma. Cancer Investig. 2013, 31, 385–389. [Google Scholar] [CrossRef]
- Podar, K.; Anderson, K.C. The pathophysiologic role of VEGF in hematologic malignancies: Therapeutic implications. Blood 2005, 105, 13831395. [Google Scholar] [CrossRef]
- Bilalis, A.; Pouliou, E.; Roussou, M.; Papanikolaou, A.; Tassidou, A.; Economopoulos, T.; Terpos, E. Increased expression of platelet derived growth factor receptor β on trephine biopsies correlates with advanced myeloma. J. Balk. Union Oncol. 2017, 22, 1032–1037. [Google Scholar]
- Terpos, E.; Anargyrou, E.; Katodritou, E.; Kastritis, E.; Papatheodorou, A.; Christoulas, D.; Pouli, A.; Michalis, E.; Delimpasi, S.; Gkotzamanidou, M.; et al. Greek myeloma study group, Greece. Circulating angiopoietin-1 to angiopoietin-2 ratio is an independent prognostic factor for survival in newly diagnosed patients with multiple myeloma who received therapy with novel antimyeloma agents. Int. J. Cancer 2012, 130, 735–742. [Google Scholar] [CrossRef]
- Coluccia, A.M.L.; Cirulli, T.; Neri, P.; Mangieri, D.; Colanardi, M.C.; Gnoni, A.; Di Renzo, N.; Dammacco, F.; Tassone, P.; Ribatti, D.; et al. Validation of PDGFRβ and c-Src tyrosine kinases as tumor/vessel targets in patients with multiple myeloma: Preclinical efficacy of the novel, orally available inhibitor dasatinib. Blood 2008, 12, 1346–1356. [Google Scholar] [CrossRef]
- Tsirakis, G.; Pappa, C.A.; Kanellou, P.; Stratinaki, M.A.; Xekalou, A.; Psarakis, F.E.; Sakellaris, G.; Alegakis, A.; Stathopoulos, E.N.; Alexandrakis, M.G. Role of platelet-derived growth factor-AB in tumour growth and angiogenesis in relation with other angiogenic cytokines in multiple myeloma. Hematol. Oncol. 2012, 30, 131–136. [Google Scholar] [CrossRef]
- Ferrucci, A.; Moschetta, M.; Frassanito, M.A.; Berardi, S.; Catacchio, I.; Ria, R.; Racanelli, V.; Caivano, A.; Solimando, A.G.; Vergara, D.; et al. A HGF/cMET Autocrine Loop Is Operative in Multiple Myeloma Bone Marrow Endothelial Cellsand May Represent a Novel Therapeutic Target. Clin. Cancer Res. 2014, 20, 5796–5807. [Google Scholar] [CrossRef]
- Moschetta, M.; Kawano, Y.; Podar, K. Targeting the Bone Marrow Microenvironment. Cancer Treat. Res. 2016, 169, 63–102. [Google Scholar]
- Palumbo, A.; Anderson, K. Multiple myeloma. N. Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef]
- Balakumaran, A.; Robey, P.G.; Fedarko, N.; Landgren, O. Bone marrow microenvironment in myelomagenesis: Its potential role in early diagnosis. Expert Rev. Mol. Diagn. 2010, 10, 465–480. [Google Scholar] [CrossRef]
- Gnoni, A.; Brunetti, O. Immune system and bone microenvironment: Rationale for targeted cancer therapies. Oncotarget 2020, 11, 480–487. [Google Scholar]
- Terpos, E.; Ntanasis-Stathopoulos, I.; Gavriatopoulou, M.; Dimopoulos, M.A. Pathogenesis of bone disease in multiple myeloma: From bench to bedside. Blood Cancer J. 2018, 8, 7. [Google Scholar] [CrossRef]
- Jurczyszyn, A.; Zebzda, A.; Czepiel, J.; Gdula-Argasińska, J.; Perucki, W.; Skotnicki, A.B.; Majka, M. The analysis of the relationship between multiple myeloma cells and their microenvironment. J. Cancer 2015, 6, 160–168. [Google Scholar] [CrossRef][Green Version]
- Giuliani, N.; Colla, S.; Morandi, F.; Lazzaretti, M.; Sala, R.; Bonomini, S.; Grano, M.; Colucci, S.; Svaldi, M.; Rizzoli, V. Myeloma cells block RUNX2/CBFA1 activity in human bone marrow osteoblast progenitors and inhibit osteoblast formation and differentiation. Blood 2005, 106, 2472–2483. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, G.B.; Gupta, V.A.; Vertino, P.M.; Boise, L.H. Cell of Origin and Genetic Alterations in the Pathogenesis of Multiple Myeloma. Front. Immunol. 2019, 10, 1121. [Google Scholar]
- Barille-Nion, S.; Bataille, R. New insights in myeloma-induced osteolysis. Leuk. Lymphoma 2003, 44, 1463–1467. [Google Scholar] [CrossRef] [PubMed]
- Lentzsch, S.; Gries, M.; Janz, M.; Bargou, R.; Dörken, B.; Mapara, M.Y. Macrophage inflammatory protein 1-α (MIP-1 α) triggers migration and signaling cascades mediating survival and proliferation in multiple myeloma (MM) cells. Blood 2003, 101, 3568–3573. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.E.; Brown, J.E.; Lawson, M.A.; Chantry, A.D. Myeloma Bone Disease: The Osteoblast in the Spotlight. J. Clin. Med. 2021, 10, 3973. [Google Scholar] [CrossRef]
- Calvani, N.; Cafforio, P.; Silvestris, F.; Dammacco, F. Functional osteoclast-like transformation of cultured human myeloma cell lines. Br. J. Haematol. 2005, 130, 926–938. [Google Scholar] [CrossRef]
- Sun, L.; Iqbal, J.; Dolgilevich, S.; Yuen, T.; Wu, X.; Moonga, B.S.; Adebanjo, O.A.; Bevis, P.J.R.; Lund, F.; Huang, C.L.; et al. Disordered osteoclast formation and function in a CD38 (ADP-ribosyl cyclase)-deficient mouse establishes an essential role for CD38 in bone resorption. FASEB J. 2003, 17, 369–375. [Google Scholar] [CrossRef]
- An, G.; Acharya, C.; Feng, X.; Wen, K.; Zhong, M.; Zhang, L.; Munshi, N.C.; Qiu, L.; Tai, Y.-T.; Anderson, K.C. Osteoclasts promote immune suppressive microenvironment in multiple myeloma: Therapeutic implication. Blood 2016, 128, 1590–1603. [Google Scholar] [CrossRef]
- Horenstein, A.L.; Quarona, V.; Toscani, D.; Costa, F.; Chillemi, A.; Pistoia, V.; Giuliani, N.; Malavasi, F. Adenosine generated in the bone marrow niche through a CD38-mediated pathway correlates with progression of human myeloma. Mol. Med. 2016, 22, 694–704. [Google Scholar] [CrossRef]
- Saltarella, I.; Desantis, V.; Melaccio, A.; Solimando, A.G.; Lamanuzzi, A.; Ria, R.; Storlazzi, C.T.; Mariggiò, M.A.; Vacca, A.; Frassanito, M.A. Mechanisms of Resistance to Anti-CD38 Daratumumab in Multiple Myeloma. Cells 2020, 9, 167. [Google Scholar] [CrossRef]
- Hagen, P.; Zhang, J.; Barton, K. High-risk disease in newly diagnosed multiple myeloma: Beyond the R-ISS and IMWG definitions. Blood Cancer J. 2022, 12, 83. [Google Scholar] [CrossRef]
- De la Puente, P.; Muz, B.; Azab, F.; Luderer, M.; Azab, A.K. Molecularly Targeted Therapies in Multiple Myeloma. Leuk. Res. Treat. 2014, 976567, 8. [Google Scholar] [CrossRef]
- Zheng, Z.; Liu, T.; Zheng, J.; Hu, J. Clarifying the molecular mechanism associated with carfilzomib resistance in human multiple myeloma using microarray gene expression profile and genetic interaction network. Oncol. Targets Ther. 2017, 10, 1327–1334. [Google Scholar] [CrossRef]
- Furukawa, Y.; Kikuchi, J. Epigenetic mechanisms of cell adhesion-mediated drug resistance in multiple myeloma. Int. J. Hematol. 2016, 104, 281–292. [Google Scholar] [CrossRef]
- Kortüm, K.M.; Mai, E.K.; Hanafiah, N.H.; Shi, C.-X.; Zhu, Y.-X.; Bruins, L.; Barrio, S.; Jedlowski, P.; Merz, M.; Xu, J.; et al. Targeted sequencing of refractory myeloma reveals a high incidence of mutations in CRBN and Ras pathway genes. Blood 2016, 128, 1226–1233. [Google Scholar] [CrossRef]
- Bellone, S.; Roque, D.; Cocco, E.; Gasparrini, S.; Bortolomai, I.; Buza, N.; Abu-Khalaf, M.; Silasi, D.-A.; Ratner, E.; Azodi, M.; et al. Downregulation of membrane complement inhibitors CD55 and CD59 by siRNA sensitizes uterine serous carcinoma overexpressing Her2/neu to complement and antibody-dependent cell cytotoxicity in vitro: Implications for trastuzumab-based immunotherapy. Br. J. Cancer 2012, 106, 1543–1550. [Google Scholar] [CrossRef]
- Kankeu, L.; Sirpilla, O.; Sakemura, R.; Siegler, R.L.; Kenderian, S.S. CAR T cell therapy and the tumor microenvironment: Current challenges and opportunities. Mol. Ther. Oncolytics 2022, 25, 69–77. [Google Scholar] [CrossRef]
- Thakurta, A.; Pierceall, W.E.; Amatangelo, M.D.; Flynt, E.; Agarwal, A. Developing next generation immunomodulatory drugs and their combinations in multiple myeloma. Oncotarget 2021, 12, 1555–1563. [Google Scholar] [CrossRef]
- Robak, P.; Drozdz, I.; Szemraj, J. Robak Drug resistance in multiple myeloma. Cancer Treat. Rev. 2018, 70, 199–208. [Google Scholar] [CrossRef]
- Ria, R.; Vacca, A. Bone Marrow Stromal T. Cells-Induced Drug Resistance in Multiple Myeloma. Int. J. Mol. Sci. 2020, 21, 613. [Google Scholar] [CrossRef]
- Suzuki, K. Treatment Strategies Considering Micro-Environment and Clonal Evolution in Multiple Myeloma. Cancers 2021, 13, 215. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M. Mechanisms of cancer drug resistance. Annu. Rev. Med. 2002, 53, 615–627. [Google Scholar] [CrossRef] [PubMed]
- De Santis, V.; Saltarella, I.; Lamanuzzi, A.; Melaccio, A.; Solimando, A.G.; Mariggiò, M.A.; Racanelli, V.; Paradiso, A.; Vacca, A.; Frassanito, M.A. MicroRNAs-Based Nano-Strategies as New Therapeutic Approach in Multiple Myeloma to Overcome Disease Progression and Drug Resistance. Int. J. Mol. Sci. 2020, 21, 3084. [Google Scholar] [CrossRef] [PubMed]
- Damiano, J.S.; Cress, A.E.; Hazlehurst, L.A.; Shtil, A.; Dalton, W.S. Cell adhesion mediated drug resistance (CAM-DR): Role of integrins and resistance to apoptosis in human myeloma cell lines. Blood 1999, 93, 1658–1667. [Google Scholar] [CrossRef] [PubMed]
- Fu, J. Cx43 expressed on bone marrow stromal cells plays an essential role in multiple myeloma cell survival and drug resistance. Arch. Med. Sci. 2017, 13, 236–245. [Google Scholar] [CrossRef]
- Sanz-Rodriguez, F.; Teixido, J. VLA-4-dependent myeloma cell adhesion. Leuk. Lymphoma 2001, 41, 239–245. [Google Scholar] [CrossRef]
- Cheung, W.C.; Van, N.B. Distinct IL-6 signal transduction leads to growth arrest and death in B cells or growth promotion and cell survival in myeloma cells. Leukemia 2002, 16, 1182–1188. [Google Scholar] [CrossRef]
- Abdi, J.; Chen, G.; Chang, H. Drug resistance in multiple myeloma: Latest findings and new concepts on molecular mechanisms. Oncotarget 2013, 4, 2186–2207. [Google Scholar] [CrossRef]
- Ria, R.; Catacchio, I.; Berardi, S.; De Luisi, A.; Caivano, A.; Piccoli, C.; Ruggieri, V.; Frassanito, M.A.; Ribatti, D.; Nico, B.; et al. HIF-1α of bone marrow endothelial cells implies relapse and drug resistance in patients with multiple myeloma and may act as a therapeutic target. Clin. Cancer Res. 2014, 20, 847–858. [Google Scholar] [CrossRef]
- Katz, B.Z. Adhesion molecules-The lifelines of multiple myeloma cells. Semin. Cancer Biol. 2010, 20, 186–195. [Google Scholar] [CrossRef]
- Vacca, A.; Di Loreto, M.; Ribatti, D.; Di Stefano, R.; Gadaleta-Caldarola, G.; Iodice, G.; Caloro, D.; Dammacco, F. Bone marrow of patients with active multiple myeloma: Angiogenesis and plasma cell adhesion molecules LFA-1, VLA-4, LAM-1, and CD44. Am. J. Hematol. 1995, 50, 9–14. [Google Scholar] [CrossRef]
- Barker, H.F.; Ball, J.; Drew, M.; Hamilton, M.S.; Franklin, I.M. The role of adhesion molecules in multiple myeloma. Leuk. Lymphoma 1992, 8, 189–196. [Google Scholar] [CrossRef]
- Hideshima, T.; Bergsagel, P.L.; Kuehl, W.M.; Anderson, K.C. Advances in biology of multiple myeloma: Clinical applications. Blood 2004, 104, 607–618. [Google Scholar] [CrossRef]
- Noborio-Hatano, K.; Kikuchi, J.; Takatoku, M.; Shimizu, R.; Wada, T.; Ueda, M.; Nobuyoshi, M.; Oh, I.; Sato, K.; Suzuki, T.; et al. Bortezomib overcomes cell-adhesion-mediated drug resistance through downregulation of VLA-4 expression in multiple myeloma. Oncogene 2009, 28, 231–242. [Google Scholar] [CrossRef]
- Pulido, R.; Elices, M.J.; Campanero, M.R.; Osborn, L.; Schiffer, S.; García-Pardo, A.; Lobb, R.; E Hemler, M.; Sánchez-Madrid, F. Functional evidence for three distinct and independently inhibitable adhesion activities mediated by the human integrin VLA-4. Correlation with distinct alpha 4 epitopes. J. Biol. Chem. 1991, 266, 10241–10245. [Google Scholar] [CrossRef]
- Roccaro, A.; Hideshima, T.; Richardson, P.; Russo, D.; Ribatti, D.; Vacca, A.; Dammacco, F.; Anderson, K. Bortezomib as an antitumor agent. Curr. Pharm. Biothechnol. 2006, 7, 441–448. [Google Scholar] [CrossRef]
- Hideshima, T.; Chauhan, D.; Richardson, P.; Mitsiades, C.; Mitsiades, N.; Hayashi, T.; Munshi, N.; Dang, L.; Castro, A.; Palombella, V. NF-kappa B as a therapeutic target in multiple myeloma. J. Biol. Chem. 2002, 277, 16639–16647. [Google Scholar] [CrossRef]
- Ribatti, D.; Vacca, A. Novel Therapeutic Approaches Targeting Vascular Endothelial Growth Factor and its receptors in hematological malignancies. Curr. Cancer Drug Targets 2005, 5, 573–578. [Google Scholar] [CrossRef]
- Segeren, M.C.; Sonneveld, P.; van der Holt, B.; Vellenga, E.; Croockewit, A.J.; Verhoef, G.E.G.; Cornelissen, J.J.; Schaafsma, M.R.; Marivan Oers, M.H.J.; Wijermans, P.W. Overall and event-free survival are not improved by the use of myeloablative therapy following intensified chemotherapy in previously untreated patients with multiple myeloma: A prospective randomized phase 3 study. Blood 2003, 101, 2144–2151. [Google Scholar] [CrossRef] [PubMed]
- D’Amato, R.J. Thalidomide is an inhibitor of angiogemesis. Proc. Natl. Acad. Sci. USA 1994, 91, 4082–4085. [Google Scholar] [CrossRef]
- Bhutani, M.; Foureau, D.M.; Atrash, S.; Voorhees, P.M.; Usmani, S.Z. Extramedullary multiple myeloma. Leukemia 2020, 34, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Da Vià, M.C.; Solimando, A.G.; Garitano-Trojaola, A.; Barrio, S.; Munawar, U.; Strifler, S.; Haertle, L.; Rhodes, N.; Teufel, E.; Vogt, C.; et al. CIC Mutation as a Molecular Mechanism of Acquired Resistance to Combined BRAF-MEK Inhibition in Extramedullary Multiple Myeloma with Central Nervous System Involvement. Oncologist 2019, 25, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Neri, P.; Bahlis, N.J.; Lonial, S. New Strategies in Multiple Myeloma: Immunotherapy as a Novel Approach to Treat Patients With Multiple Myeloma. Clin. Cancer Res. 2016, 22, 5959–5965. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Frampton, J.E. Isatuximab: A Review of Its Use in Multiple Myeloma. Target Oncol. 2021, 16, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Bardenstein, D.S.; Cheyer, C.; Okada, N.; Morgan, B.P.; Medof, M.E. Cell surface regulators of complement, 5I2 antigen, and CD59, in the rat eye and adnexal tissues. Investig. Ophthalmol. Vis. Sci. 1999, 40, 519–524. [Google Scholar]
- Gavriatopoulu, M.; Ntanasis-Stathopoulos, J.; Dimopoulos, M.A.; Terpos, E. Anti-BCMA Antibodies in the Future Management of Multiple Myeloma. Expert Rev. Anticancer Ther. 2019, 19, 319–326. [Google Scholar] [CrossRef]
- Larysa, S.; Dardac, A.; Madduri, D.; Richard, S.; Richter, J. B-cell maturation antigen (BCMA) in multiple myeloma: The new frontier of targeted therapies. Ther. Adv. Hematol. 2021, 12, 1–16. [Google Scholar]
- Gomes-Silva, D.; Ramos, C.A. Cancer Immunotherapy Using CAR-T Cells: From the Research Bench to the Assembly Line. Biotechnol. J. 2018, 13, 1700097. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, Z.; Dang, Q.; Xu, H.; Lv, J.; Li, H.; Han, X. Immunosuppression in tumor immune microenvironment and its optimization from CAR-T cell therapy. Theranostics 2022, 12, 6273–6290. [Google Scholar] [CrossRef]
- Borja, P.; María-Victoria, M.; González-Calle, V. Anti-BCMA CAR T-cell Therapy: Changing the Natural History of Multiple Myeloma. Hemasphere 2022, 6, e691. [Google Scholar]
- Kleber, M. BCMA in Multiple Myeloma—A Promising Key to Therapy. J. Clin. Med. 2021, 10, 4088. [Google Scholar] [CrossRef]
- Neumeister, P.; Schulz, E.; Pansy, K.; Szmyra, M.; Deutsch, A.J. Targeting the Microenvironment for Treating Multiple Myeloma. Int. J. Mol. Sci. 2022, 23, 7627. [Google Scholar] [CrossRef]
- Chen, L.; Willis, S.N.; Wei, A.; Smith, B.J.; Fletcher, J.I.; Hinds, M.G.; Colman, P.M.; Day, C.L.; Adams, J.M.; Huang, D.C. Differential Targeting of Prosurvival Bcl-2 Proteins by Their BH3-Only Ligands Allows Complementary Apoptotic Function. Mol. Cell 2005, 17, 393–403. [Google Scholar] [CrossRef]
- Shehata, M.; Schnabl, S.; Demirtas, D.; Hilgarth, M.; Hubmann, R.; Ponath, E.; Badrnya, S.; Lehner, C.; Hoelbl, A.; Duechler, M.; et al. Reconstitution of PTEN Activity by CK2 Inhibitors and Interference with the PI3-K/Akt Cascade Counteract the Antiapoptotic Effect of Human Stromal Cells in Chronic Lymphocytic Leukemia. Blood 2010, 116, 2513–2521. [Google Scholar] [CrossRef]
- Di Marzo, L.; Desantis, V.; Solimando, A.G.; Ruggieri, S.; Annese, T.; Nico, B.; Fumarulo, R.; Vacca, A.; Frassanito, M.A. Microenvironment Drug Resistance in Multiple Myeloma: Emerging New Players. Oncotarget 2016, 7, 60698–60711. [Google Scholar] [CrossRef]
- Sutton, V.R.; Davis, J.E.; Cancilla, M.; Johnstone, R.; Ruefli, A.A.; Sedelies, K.; Browne, K.A.; Trapani, J. Initiation of Apoptosis by Granzyme B Requires Direct Cleavage of Bid, but Not Direct Granzyme B–Mediated Caspase Activation. J. Exp. Med. 2000, 192, 1403–1414. [Google Scholar] [CrossRef]
- Catalán, E.; Jaime-Sánchez, P.; Aguilo, N.; Simon, M.M.; Froelich, C.J.; Pardo, J. Mouse Cytotoxic T Cell-Derived Granzyme B Activates the Mitochondrial Cell Death Pathway in a Bim-Dependent Fashion. J. Biol. Chem. 2015, 290, 6868–6877. [Google Scholar] [CrossRef]
- Cullen, S.P.; Brunet, M.; Martin, S.J. Granzymes in Cancer and Immunity. Cell Death Differ. 2010, 17, 616–623. [Google Scholar] [CrossRef]
- Ewen, C.L.; Kane, K.P.; Bleackley, R.C. A Quarter Century of Granzymes. Cell Death Differ. 2011, 19, 28–35. [Google Scholar] [CrossRef]
- Holthof, L.C.; Mutis, T. Challenges for Immunotherapy in Multiple Myeloma: Bone Marrow Microenvironment-Mediated Immune Suppression and Immune Resistance. Cancers 2020, 12, 988. [Google Scholar] [CrossRef]
- Giallongo, C.; Tibullo, D.; Camiolo, G.; Parrinello, N.L.; Romano, A.; Puglisi, F.; Barbato, A.; Conticello, C.; Lupo, G.; Anfuso, C.D. TLR4 Signaling Drives Mesenchymal Stromal Cells Commitment to Promote Tumor Microenvironment Transformation in Multiple Myeloma. Cell Death Dis. 2019, 10, 704. [Google Scholar] [CrossRef] [PubMed]
- André, T.; Najar, M.; Stamatopoulos, B.; Pieters, K.; Pradier, O.; Bron, D.; Meuleman, N.; Lagneaux, L. Immune Impairments in Multiple Myeloma Bone Marrow Mesenchymal Stromal Cells. Cancer Immunol. Immunother. 2015, 64, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Franssen, L.E.; Mutis, T.; Lokhorst, H.M.; van de Donk, N.W.J.C. Immunotherapy in Myeloma: How Far Have We Come? Ther. Adv. Hematol. 2019, 10, 2040620718822660. [Google Scholar] [CrossRef] [PubMed]
- Görgün, G.T.; Whitehill, G.; Anderson, J.L.; Hideshima, T.; Maguire, C.; Laubach, J.; Raje, N.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. Tumor-Promoting Immune-Suppressive Myeloid-Derived Suppressor Cells in the Multiple Myeloma Microenvironment in Humans. Blood 2013, 121, 2975–2987. [Google Scholar] [CrossRef] [PubMed]
- Di Nicola, M.; Carlo-Stella, C.; Magni, M.; Milanesi, M.; Longoni, P.D.; Matteucci, P.; Grisanti, P.; Gianni, A.M. Human Bone Marrow Stromal Cells Suppress T-Lymphocyte Proliferation Induced by Cellular or Nonspecific Mitogenic Stimuli. Blood 2002, 99, 3838–3843. [Google Scholar] [CrossRef]
- Hwang, W.; Jung, K.; Jeon, Y.; Yun, S.; Kim, T.W.; Choi, I. Knockdown of the Interleukin-6 Receptor Alpha Chain of Dendritic Cell Vaccines Enhances the Therapeutic Potential against IL-6 Producing Tumors. Vaccine 2010, 29, 34–44. [Google Scholar] [CrossRef]
- Ohno, Y.; Kitamura, H.; Takahashi, N.; Ohtake, J.; Kaneumi, S.; Sumida, K.; Homma, S.; Kawamura, H.; Minagawa, N.; Shibasaki, S.; et al. IL-6 down-Regulates HLA Class II Expression and IL-12 Production of Human Dendritic Cells to Impair Activation of Antigen-Specific CD4+ T Cells. Cancer Immunol. Immunother. 2016, 65, 193–204. [Google Scholar] [CrossRef]
- Sumida, K.; Wakita, D.; Narita, Y.; Masuko, K.; Terada, S.; Watanabe, K.; Satoh, T.; Kitamura, H.; Nishimura, T. Anti-IL-6 Receptor MAb Eliminates Myeloid-Derived Suppressor Cells and Inhibits Tumor Growth by Enhancing T-Cell Responses. Eur. J. Immunol. 2012, 42, 2060–2072. [Google Scholar] [CrossRef]
- Holthof, L.C.; Van der Horst, H.J.; Poels, R.; van der Schans, J.T.; Gelderloos, A.T.; Li, F.; Lokhorst, H.; Zweegman, S.; Themeli, M.; Van de Donk, N.W.J.C.; et al. The Impact and Modulation of Microenvironment-Induced Immune Resistance Against CAR T Cell and Antibody Treatments in Multiple Myeloma. Blood 2019, 134, 137. [Google Scholar] [CrossRef]
- Frerichs, K.A.; Nagy, N.A.; Lindenbergh, P.L.; Bosman, P.; Soto, J.M.; Broekmans, M.; Groen, R.W.J.; Themeli, M.; Nieuwenhuis, L.; Stege, C.; et al. CD38-Targeting Antibodies in Multiple Myeloma: Mechanisms of Action and Clinical Experience. Expert Rev. Clin. Immunol. 2018, 14, 197–206. [Google Scholar] [CrossRef]
- Lee, A.H.S.; E Gillett, C.; Ryder, K.; Fentiman, I.S.; Miles, D.W.; Millis, R.R. Different patterns of inflammation and prognosis in invasive carcinoma of the breast. Histopathology 2006, 48, 692–701. [Google Scholar] [CrossRef]
- Boissinot, M.; Vilaine, M.; Hermouet, S. The hepatocyte growth factor (HGF)/ met axis: A neglected target in the treatment of chronic myeloproliferative neoplasms? Cancers 2014, 6, 1631–1669. [Google Scholar] [CrossRef]
- Lust, J.A.; Lacy, M.Q.; Zeldenrust, S.R.; Witzig, T.E.; Moon-Tasson, L.L.; Dinarello, C.A.; Donovan, K.A. Reduction in C-reactive protein indicates successful targeting of the IL-1/IL-6 axis resulting in improved survival in early stage multiple myeloma. Am. J. Hematol. 2016, 91, 571–574. [Google Scholar] [CrossRef]
- Scavelli, C.; Di Pietro, G.; Cirulli, T.; Coluccia, M.; Boccarelli, A.; Giannini, T.; Mangialardi, G.; Bertieri, R.; Coluccia, A.M.L.; Ribatti, D.; et al. Zoledronic acid affects ocer-angiogenic phenotype of endothelial cells in patients with multiple myeloma. Mol. Cancer Ther. 2007, 6 Pt 1, 3256–3262. [Google Scholar] [CrossRef]
- Ria, R.; Reale, A.; Moschetta, M.; Mangialardi, G.; Dammacco, F.; Vacca, A. A retrospective study of skeletal and disease-free survival benefits of zoledronic acid therapy in patients with multiple myeloma treated with novel agents. Int. J. Clin. Exp. Med. 2013, 6, 30–38. [Google Scholar]
- Ribatti, D.; Nico, B.; Mangieri, D.; Maruotti, N.; Longo, V.; Vacca, A.; Cantatore, F.P. Neridronate inhibits angiogenesis in vitro and in vivo. Clin. Rheumatol. 2007, 26, 1094–1098. [Google Scholar] [CrossRef]
- Allegra, A. Nanoparticles in oncology: The new theragnostic molecules. Anti-Cancer Agents Med. Chem. 2011, 11, 669–686. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).