
Genetic Variability of the Monkeypox Virus Clade IIb B.1

 ,
,  ,
, 
 ,
,  ,
,  ,
,  , ,
, ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
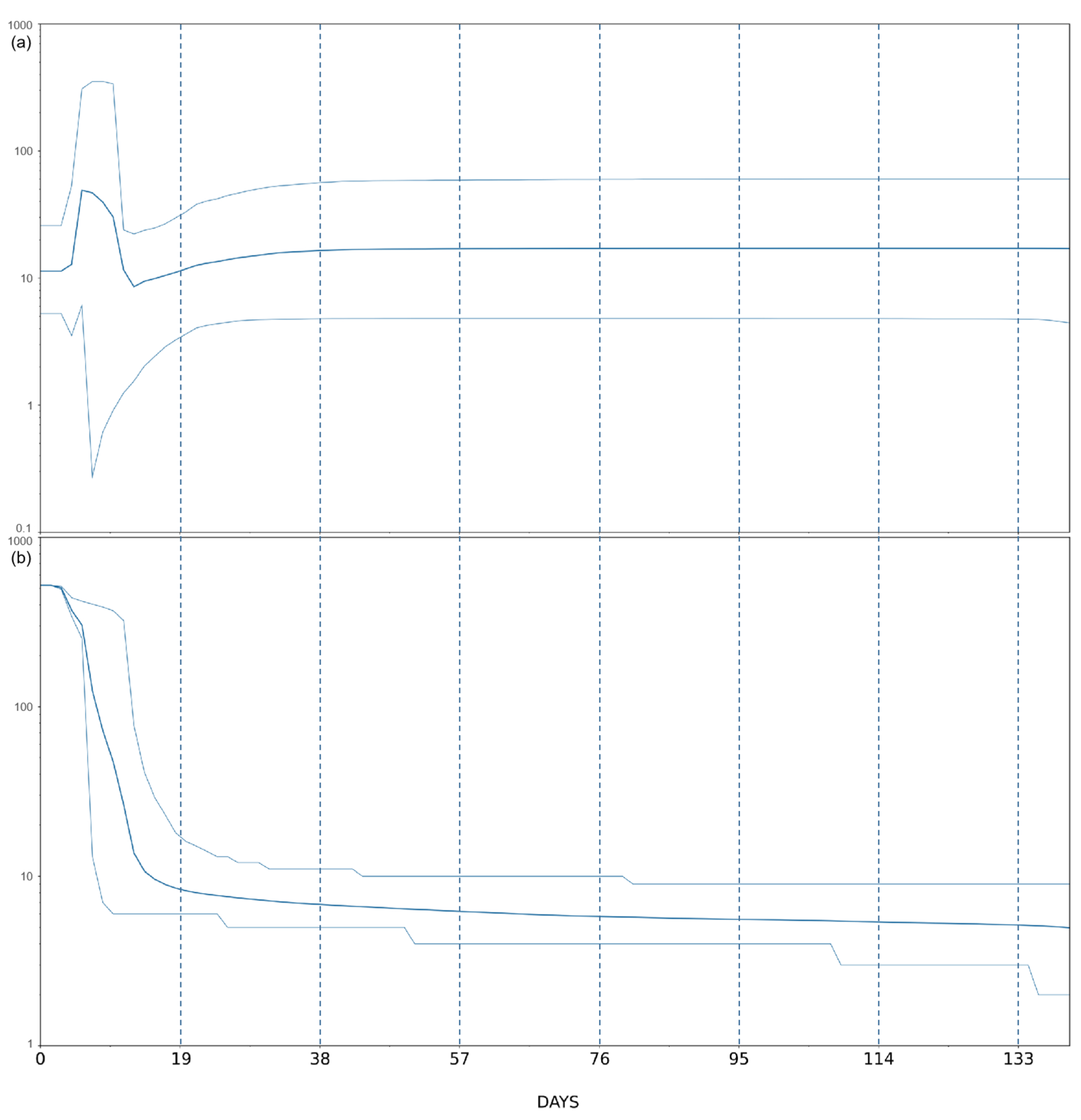
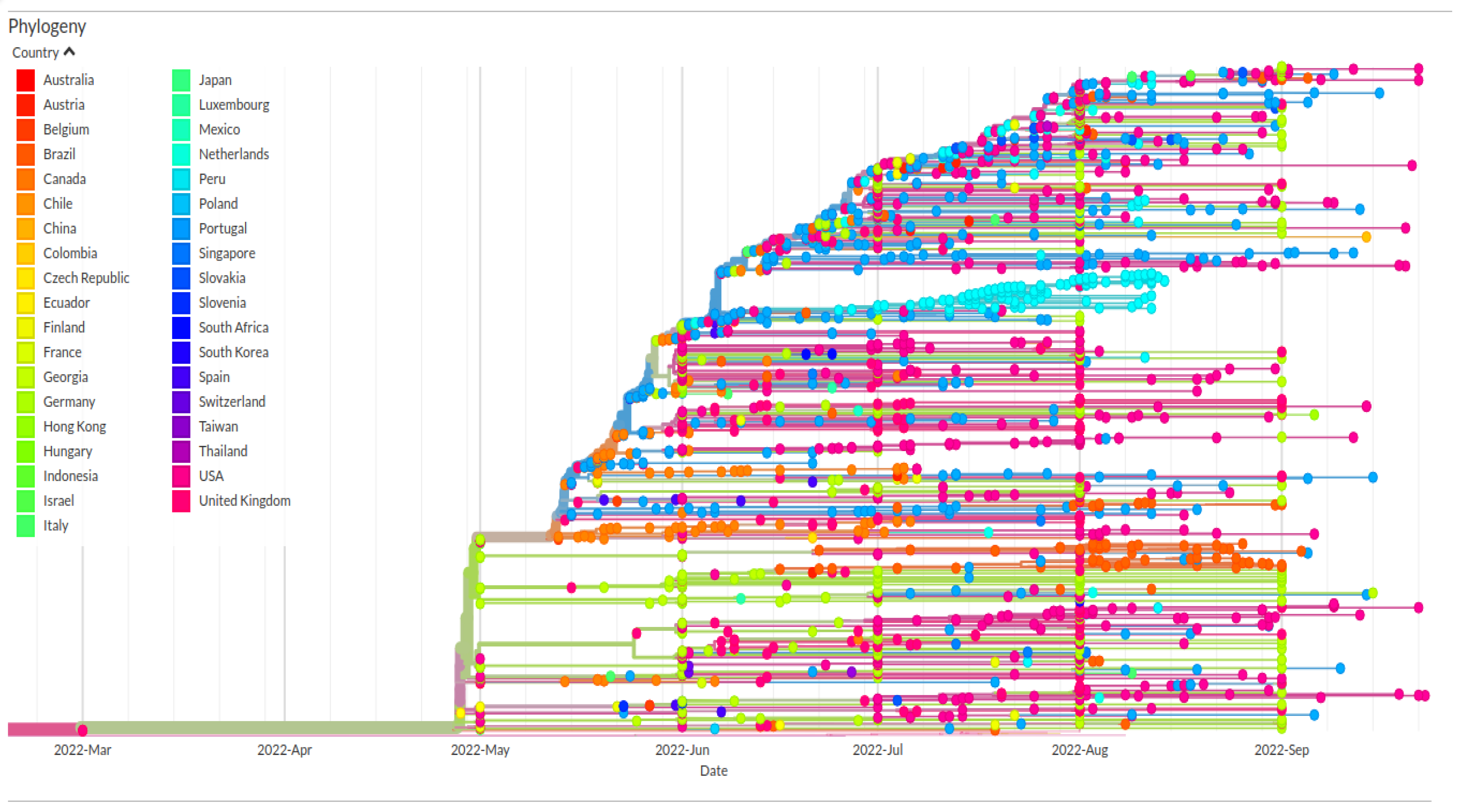
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kumar, S.; Subramaniam, G.; Karuppanan, K. Human monkeypox outbreak in 2022. J. Med. Virol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.K.; Ansari, S.; Maurya, V.K.; Kumar, S.; Jain, A.; Paweska, J.T.; Tripathi, A.K.; Abdel-Moneim, A.S. Re-emerging human monkeypox: A major public-health debacle. J. Med. Virol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Luna, N.; Ramír, A.L.; Muñoz, M.; Ballesteros, N.; Patiño, L.H.; Castañeda, S.A.; Bonilla-Aldana, D.K.; Paniz-Mondolfi, A.; Ramírez, J.D. Phylogenomic analysis of the monkeypox virus (MPXV) 2022 outbreak: Emergence of a novel viral lineage? Travel Med. Infect. Dis. 2022, 49, 102402. [Google Scholar] [CrossRef]
- Isidro, J.; Borges, V.; Pinto, M.; Sobral, D.; Santos, J.D.; Nunes, A.; Mixão, V.; Ferreira, R.; Santos, D.; Duarte, S.; et al. Phylogenomic characterization and signs of microevolution in the 2022 multi-country outbreak of monkeypox virus. Nat. Med. 2022, 28, 1569–1572. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Bruen, T.C.; Philippe, H.; Bryant, D. A simple and robust statistical test for detecting the presence of recombination. Genetics 2006, 172, 2665–2681. [Google Scholar] [CrossRef] [PubMed]
- Firth, C.; Kitchen, A.; Shapiro, B.; Suchard, M.A.; Holmes, E.C.; Rambaut, A. Using Time-Structured data to estimate evolutionary rates of Double-Stranded DNA viruses. Mol. Biol. Evol. 2010, 27, 2038–2051. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; Van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Scarpa, F.; Sanna, D.; Azzena, I.; Giovanetti, M.; Benvenuto, D.; Angeletti, S.; Ceccarelli, G.; Pascarella, S.; Casu, M.; Fiori, P.L.; et al. On the SARS-CoV-2 BA.2.75 variant: A genetic and structural point of view. J. Med. Virol. 2022. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Carvalho, L.M.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst. Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Borsetti, A.; Scarpa, F.; Maruotti, A.; Divino, F.; Ceccarelli, G.; Giovanetti, M.; Ciccozzi, M. The unresolved question on COVID-19 virus origin: The three cards game? J. Med. Virol. 2022, 94, 1257–1260. [Google Scholar] [CrossRef] [PubMed]
- Zella, D.; Giovanetti, M.; Benedetti, F.; Unali, F.; Spoto, S.; Guarino, M.; Angeletti, S.; Ciccozzi, M. The variants question: What is the problem. J. Med. Virol. 2021, 93, 6479–6485. [Google Scholar] [CrossRef] [PubMed]
- Mugosa, B.; Vujosevic, D.; Ciccozzi, M.; Valli, M.B.; Capobianchi, M.R.; Presti, A.L.; Cella, E.; Giovanetti, M.; Lai, A.; Angeletti, S.; et al. Genetic diversity of the haemagglutinin (HA) of human influenza A (H1N1) virus in Montenegro: Focus on its origin and evolution. J. Med. Virol. 2016, 88, 1905–1913. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, F.; Sanna, D.; Azzena, I.; Cossu, P.; Giovanetti, M.; Benvenuto, D.; Coradduzza, E.; Alexiev, I.; Casu, M.; Fiori, P.L.; et al. Update on the Phylodynamics of SADS-CoV. Life 2021, 11, 820. [Google Scholar] [CrossRef] [PubMed]
- Renner, D.W.; Szpara, M.L. Impacts of Genome-Wide Analyses on Our Understanding of Human Herpesvirus Diversity and Evolution. J. Virol. 2018, 92, e00908-17. [Google Scholar] [CrossRef] [PubMed]
- Delmotte, J.; Pelletier, C.; Morga, B.; Galinier, R.; Petton, B.; Lamy, J.-B.; Kaltz, O.; Avarre, J.-C.; Jacquot, M.; Montagnani, C.; et al. Genetic diversity and connectivity of the Ostreid herpesvirus 1 populations in France: A first attempt to phylogeographic inference for a marine mollusc disease. Virus Evol. 2022, 8, veac039. [Google Scholar] [CrossRef] [PubMed]
- Maruotti, A.; Böhning, D.; Rocchetti, I.; Ciccozzi, M. Estimating the undetected infections in the Monkeypox outbreak. J. Med. Virol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Jolly, B.; Scaria, V. A distinct phylogenetic cluster of Monkeypox genomes suggests an early and cryptic spread of the virus. J. Infect. 2022. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| WHO Region | Confirmed Cases | Deaths |
|---|---|---|
| African Region | 714 | 13 |
| Region of the Americas | 43,181 | 6 |
| Eastern Mediterranean Region | 64 | 1 |
| European Region | 24,737 | 4 |
| South-East Asia Region | 23 | 1 |
| Western Pacific Region | 181 | 0 |
| World | 68,900 | 25 |
| # | Countries | Number of Genomes | # | Countries | Number of Genomes |
|---|---|---|---|---|---|
| 1 | Australia | 1 | 20 | Japan | 1 |
| 2 | Austria | 1 | 21 | Luxembourg | 2 |
| 3 | Belgium | 16 | 22 | Mexico | 2 |
| 4 | Brazil | 97 | 23 | Netherlands | 31 |
| 5 | Canada | 103 | 24 | Peru | 131 |
| 6 | Chile | 1 | 25 | Poland | 1 |
| 7 | China | 1 | 26 | Portugal | 327 |
| 8 | Colombia | 14 | 27 | Singapore | 10 |
| 9 | Czech Republic | 1 | 28 | Slovakia | 12 |
| 10 | Ecuador | 1 | 29 | Slovenia | 7 |
| 11 | Finland | 2 | 30 | South Africa | 2 |
| 12 | France | 22 | 31 | South Korea | 1 |
| 13 | Georgia | 1 | 32 | Spain | 9 |
| 14 | Germany | 305 | 33 | Switzerland | 6 |
| 15 | Hong Kong | 1 | 34 | Taiwan | 2 |
| 16 | Hungary | 2 | 35 | Thailand | 4 |
| 17 | Indonesia | 1 | 36 | USA | 471 |
| 18 | Israel | 1 | 37 | United Kingdom | 171 |
| 19 | Italy | 16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scarpa, F.; Sanna, D.; Azzena, I.; Cossu, P.; Locci, C.; Angeletti, S.; Maruotti, A.; Ceccarelli, G.; Casu, M.; Fiori, P.L.; et al. Genetic Variability of the Monkeypox Virus Clade IIb B.1. J. Clin. Med. 2022, 11, 6388. https://doi.org/10.3390/jcm11216388
Scarpa F, Sanna D, Azzena I, Cossu P, Locci C, Angeletti S, Maruotti A, Ceccarelli G, Casu M, Fiori PL, et al. Genetic Variability of the Monkeypox Virus Clade IIb B.1. Journal of Clinical Medicine. 2022; 11(21):6388. https://doi.org/10.3390/jcm11216388
Chicago/Turabian StyleScarpa, Fabio, Daria Sanna, Ilenia Azzena, Piero Cossu, Chiara Locci, Silvia Angeletti, Antonello Maruotti, Giancarlo Ceccarelli, Marco Casu, Pier Luigi Fiori, and et al. 2022. "Genetic Variability of the Monkeypox Virus Clade IIb B.1" Journal of Clinical Medicine 11, no. 21: 6388. https://doi.org/10.3390/jcm11216388
APA StyleScarpa, F., Sanna, D., Azzena, I., Cossu, P., Locci, C., Angeletti, S., Maruotti, A., Ceccarelli, G., Casu, M., Fiori, P. L., Petrosillo, N., & Ciccozzi, M. (2022). Genetic Variability of the Monkeypox Virus Clade IIb B.1. Journal of Clinical Medicine, 11(21), 6388. https://doi.org/10.3390/jcm11216388