
Comprehensive RNA Expression Analysis Revealed Biological Functions of Key Gene Sets and Identified Disease-Associated Cell Types Involved in Rat Traumatic Brain Injury

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Acquisition and Preprocessing
2.2. WGCNA
2.3. GO and KEGG Enrichment Analysis
2.4. Computational Analysis of snRNA Seq Datasets
2.5. Gene Set Enrichment Analysis
2.6. Gene Set Variation Analysis
2.7. Statistical Analysis
3. Results
3.1. WGCNA Identified Key Modules Related to Sample Traits in TBI
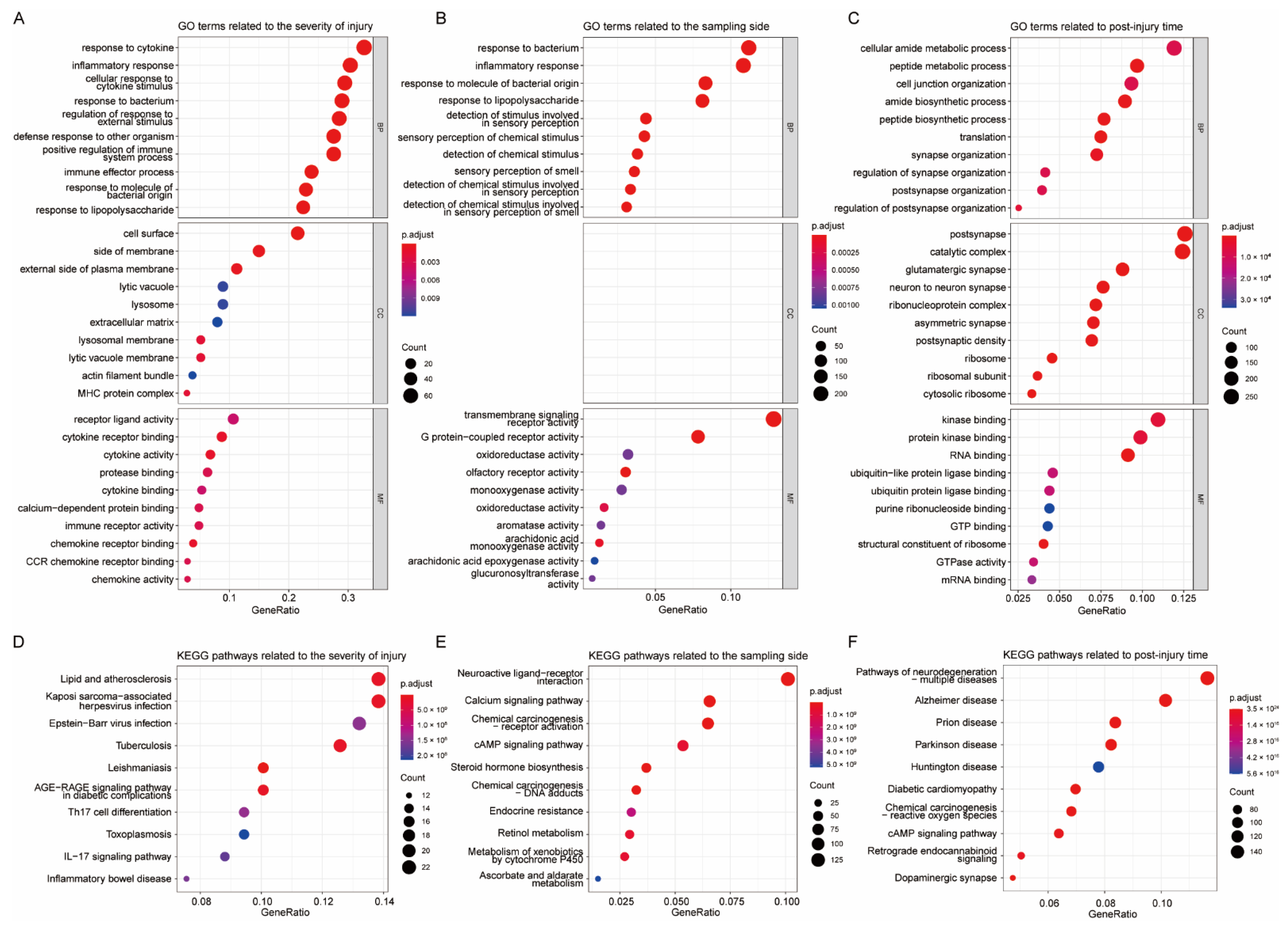
3.2. Function Enrichment Analysis of Key Modules
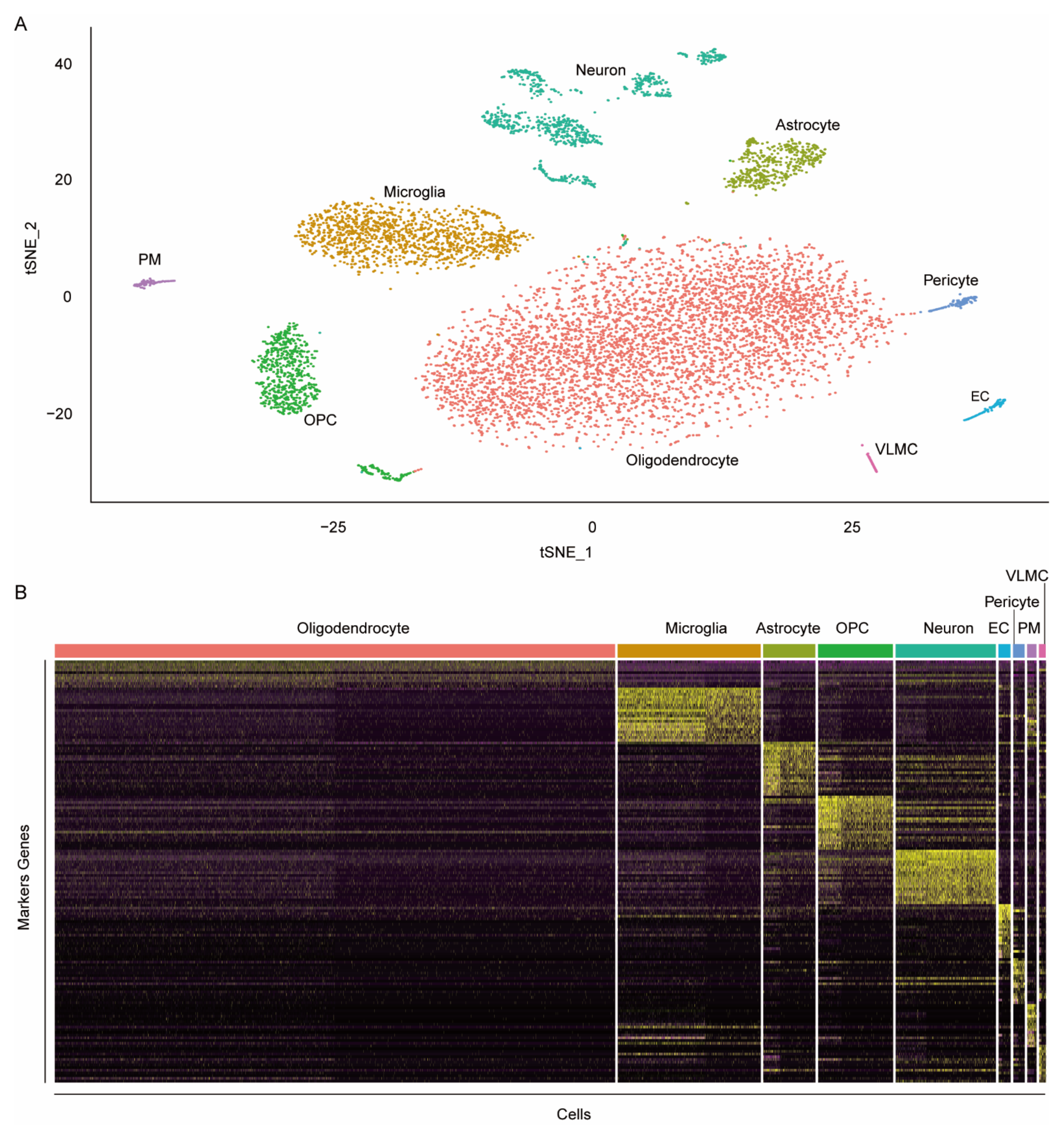
3.3. Identification of the Markers of Rat Brain Cell Types
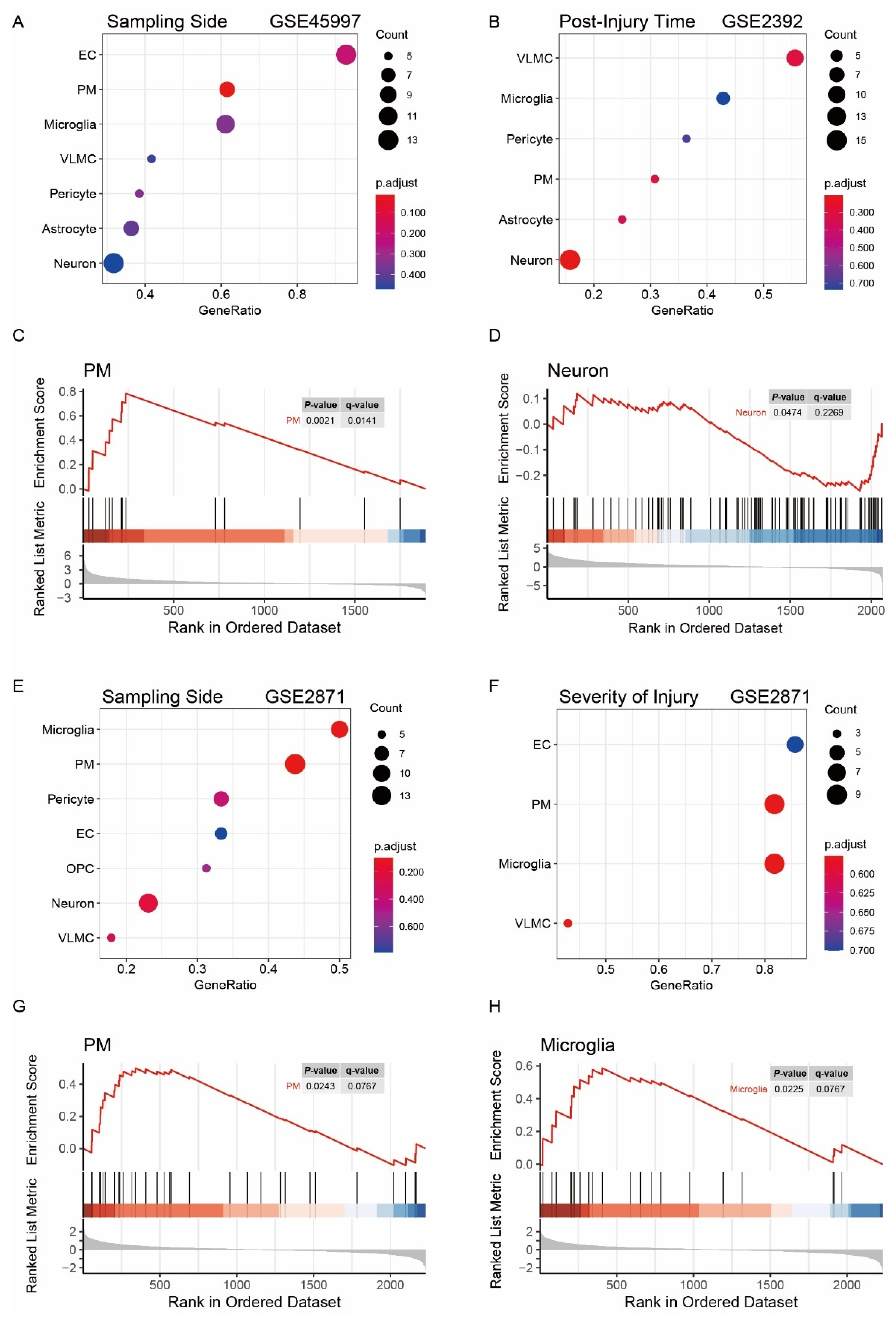
3.4. Characterization of Key Cell Types Associated with Traits
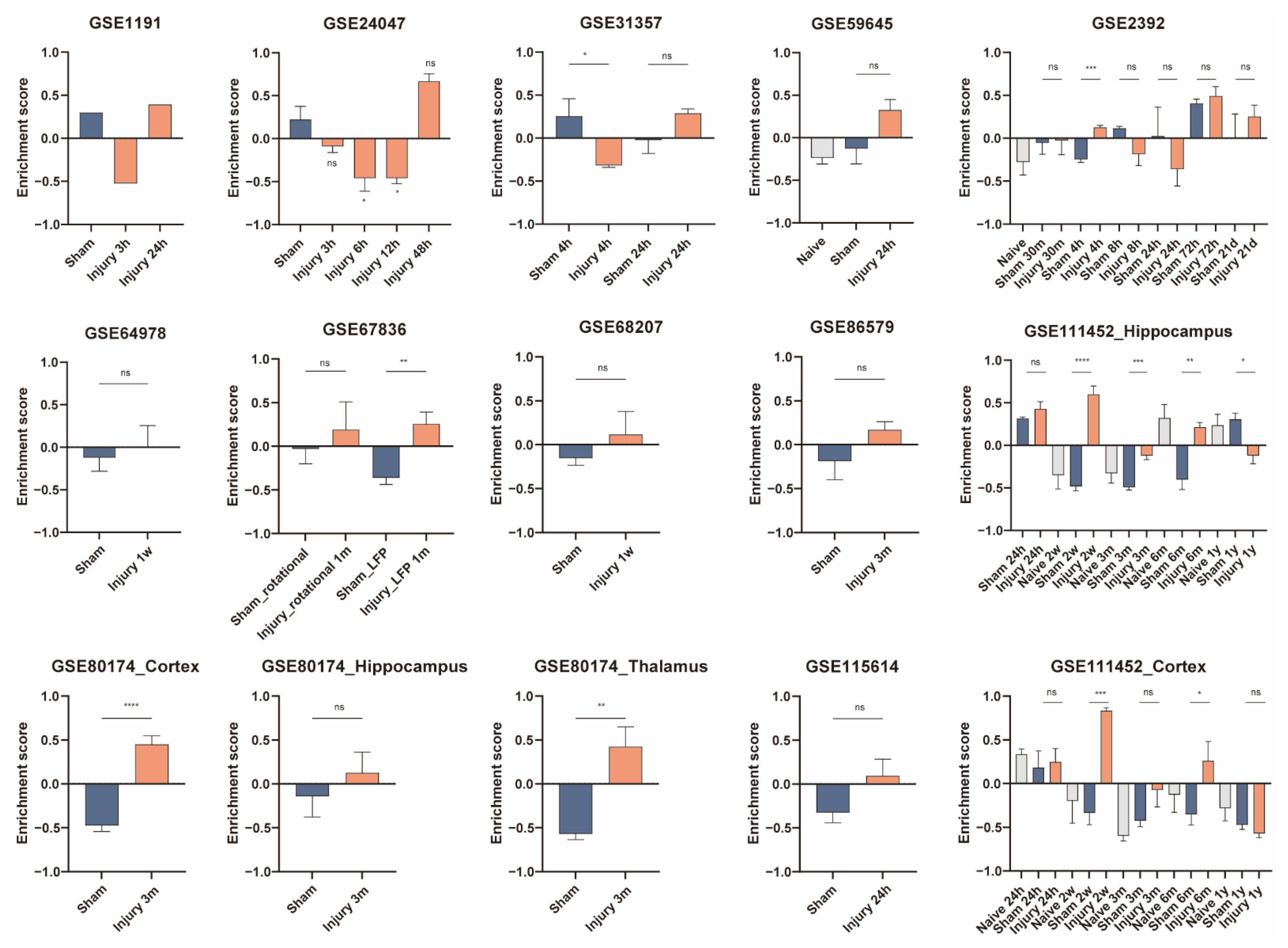
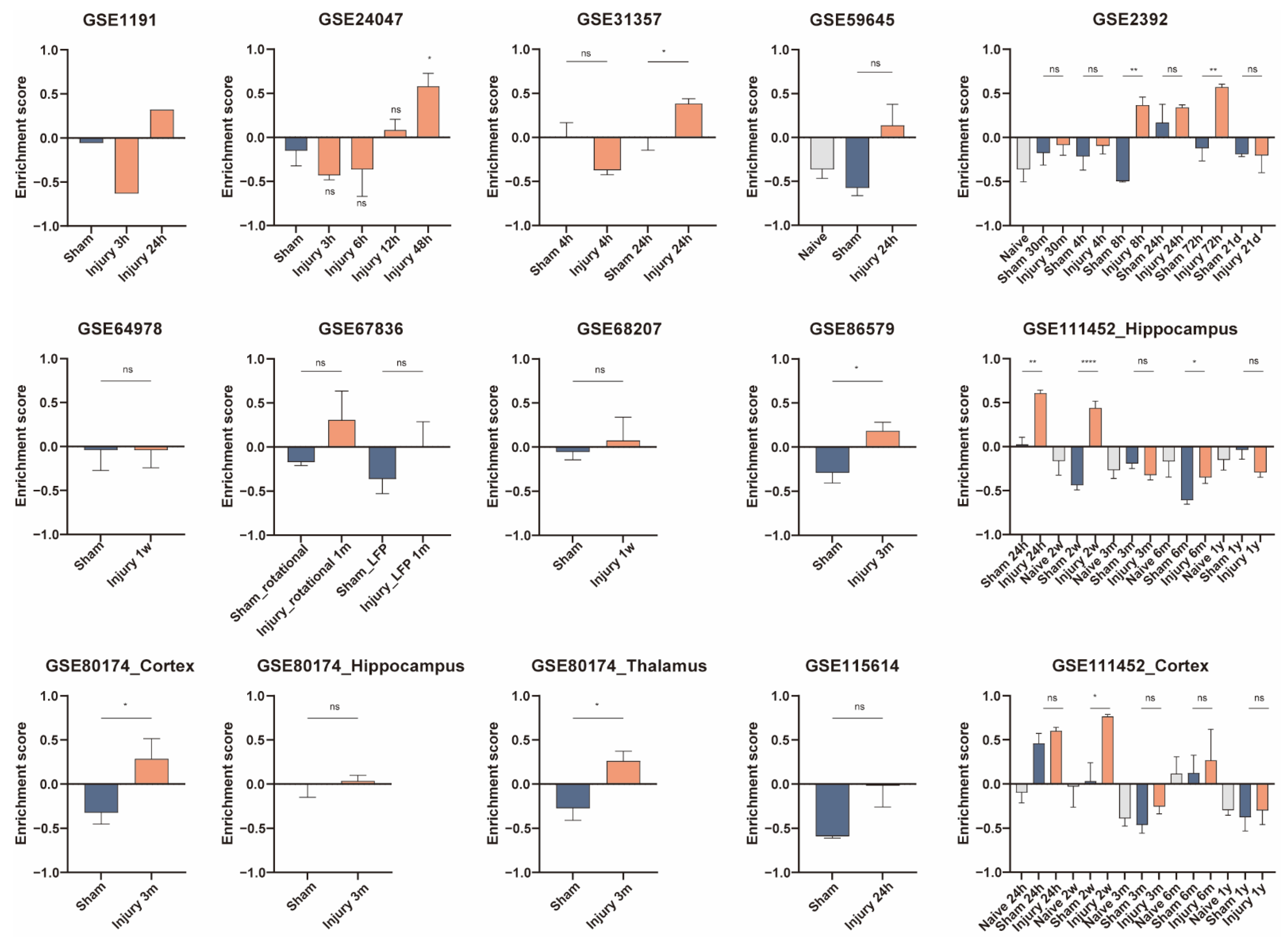
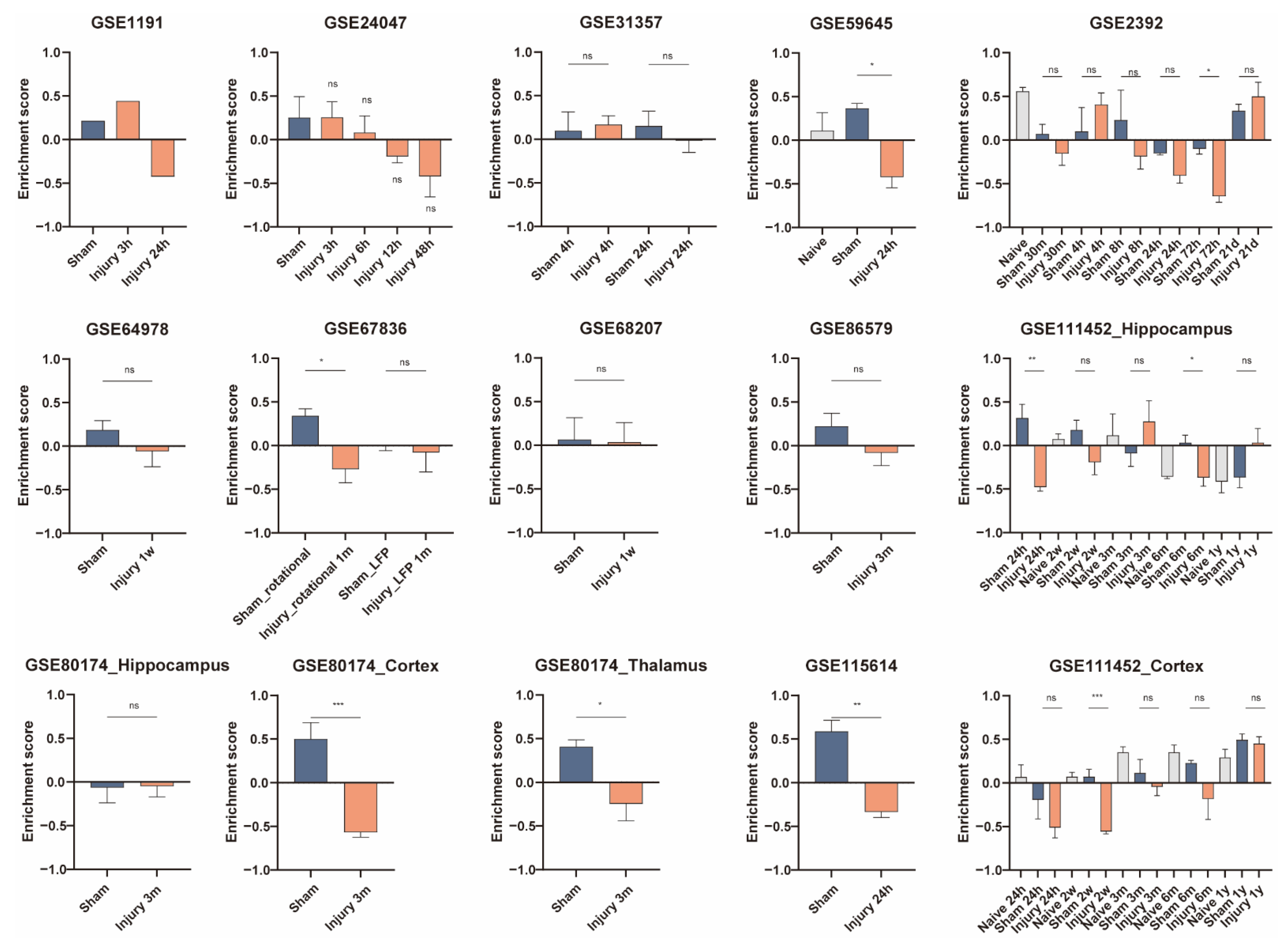
3.5. Validation of Cell Activity after Traumatic Brain Injury
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Andriessen, T.M.; Jacobs, B.; Vos, P.E. Clinical characteristics and pathophysiological mechanisms of focal and diffuse traumatic brain injury. J. Cell Mol. Med. 2010, 14, 2381–2392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bains, M.; Hall, E.D. Antioxidant therapies in traumatic brain and spinal cord injury. Biochim. Biophys. Acta 2012, 1822, 675–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fann, J.R.; Ribe, A.R.; Pedersen, H.S.; Fenger-Gron, M.; Christensen, J.; Benros, M.E.; Vestergaard, M. Long-term risk of dementia among people with traumatic brain injury in denmark: A population-based observational cohort study. Lancet Psychiatry 2018, 5, 424–431. [Google Scholar] [CrossRef]
- Jassam, Y.N.; Izzy, S.; Whalen, M.; McGavern, D.B.; El Khoury, J. Neuroimmunology of traumatic brain injury: Time for a paradigm shift. Neuron 2017, 95, 1246–1265. [Google Scholar] [CrossRef] [Green Version]
- Maas, A.I.R.; Menon, D.K.; Adelson, P.D.; Andelic, N.; Bell, M.J.; Belli, A.; Bragge, P.; Brazinova, A.; Buki, A.; Chesnut, R.M.; et al. Traumatic brain injury: Integrated approaches to improve prevention, clinical care, and research. Lancet Neurol. 2017, 16, 987–1048. [Google Scholar] [CrossRef] [Green Version]
- Simon, D.W.; McGeachy, M.J.; Bayir, H.; Clark, R.S.; Loane, D.J.; Kochanek, P.M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 171–191. [Google Scholar] [CrossRef] [Green Version]
- The Lancet Neurology. The future research path of traumatic brain injury. Lancet Neurol. 2022, 21, 295. [Google Scholar] [CrossRef]
- Xiong, Y.; Mahmood, A.; Chopp, M. Animal models of traumatic brain injury. Nat. Rev. Neurosci. 2013, 14, 128–142. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Horvath, S. A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, 17. [Google Scholar] [CrossRef]
- Ma, S.; Sun, S.; Geng, L.; Song, M.; Wang, W.; Ye, Y.; Ji, Q.; Zou, Z.; Wang, S.; He, X.; et al. Caloric restriction reprograms the single-cell transcriptional landscape of rattus norvegicus aging. Cell 2020, 180, 984–1001.e22. [Google Scholar] [CrossRef]
- Babikian, T.; Prins, M.L.; Cai, Y.; Barkhoudarian, G.; Hartonian, I.; Hovda, D.A.; Giza, C.C. Molecular and physiological responses to juvenile traumatic brain injury: Focus on growth and metabolism. Dev. Neurosci. 2010, 32, 431–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natale, J.E.; Ahmed, F.; Cernak, I.; Stoica, B.; Faden, A.I. Gene expression profile changes are commonly modulated across models and species after traumatic brain injury. J. Neurotrauma 2003, 20, 907–927. [Google Scholar] [CrossRef] [PubMed]
- White, T.E.; Ford, G.D.; Surles-Zeigler, M.C.; Gates, A.S.; Laplaca, M.C.; Ford, B.D. Gene expression patterns following unilateral traumatic brain injury reveals a local pro-inflammatory and remote anti-inflammatory response. BMC Genom. 2013, 14, 282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matzilevich, D.A.; Rall, J.M.; Moore, A.N.; Grill, R.J.; Dash, P.K. High-density microarray analysis of hippocampal gene expression following experimental brain injury. J. Neurosci. Res. 2002, 67, 646–663. [Google Scholar] [CrossRef] [PubMed]
- Shojo, H.; Kaneko, Y.; Mabuchi, T.; Kibayashi, K.; Adachi, N.; Borlongan, C.V. Genetic and histologic evidence implicates role of inflammation in traumatic brain injury-induced apoptosis in the rat cerebral cortex following moderate fluid percussion injury. Neuroscience 2010, 171, 1273–1282. [Google Scholar] [CrossRef]
- Shojo, H.; Borlongan, C.V.; Mabuchi, T. Genetic and histological alterations reveal key role of prostaglandin synthase and cyclooxygenase 1 and 2 in traumatic brain injury-induced neuroinflammation in the cerebral cortex of rats exposed to moderate fluid percussion injury. Cell Transpl. 2017, 26, 1301–1313. [Google Scholar] [CrossRef]
- Hellmich, H.L.; Rojo, D.R.; Micci, M.A.; Sell, S.L.; Boone, D.R.; Crookshanks, J.M.; DeWitt, D.S.; Masel, B.E.; Prough, D.S. Pathway analysis reveals common pro-survival mechanisms of metyrapone and carbenoxolone after traumatic brain injury. PLoS ONE 2013, 8, e53230. [Google Scholar] [CrossRef]
- Sell, S.L.; Boone, D.R.; Weisz, H.A.; Cardenas, C.; Willey, H.E.; Bolding, I.J.; Micci, M.A.; Falduto, M.T.; Torres, K.E.O.; DeWitt, D.S.; et al. Microrna profiling identifies a novel compound with antidepressant properties. PLoS ONE 2019, 14, e0221163. [Google Scholar] [CrossRef]
- Meng, Q.; Zhuang, Y.; Ying, Z.; Agrawal, R.; Yang, X.; Gomez-Pinilla, F. Traumatic brain injury induces genome-wide transcriptomic, methylomic, and network perturbations in brain and blood predicting neurological disorders. eBioMedicine 2017, 16, 184–194. [Google Scholar] [CrossRef] [Green Version]
- Paban, V.; Ogier, M.; Chambon, C.; Fernandez, N.; Davidsson, J.; Risling, M.; Alescio-Lautier, B. Molecular gene expression following blunt and rotational models of traumatic brain injury parallel injuries associated with stroke and depression. J. Transl. Sci. 2016, 2, 330–339. [Google Scholar] [CrossRef]
- Lipponen, A.; Paananen, J.; Puhakka, N.; Pitkanen, A. Analysis of post-traumatic brain injury gene expression signature reveals tubulins, nfe2l2, nfkb, cd44, and s100a4 as treatment targets. Sci. Rep. 2016, 6, 31570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puhakka, N.; Bot, A.M.; Vuokila, N.; Debski, K.J.; Lukasiuk, K.; Pitkanen, A. Chronically dysregulated notch1 interactome in the dentate gyrus after traumatic brain injury. PLoS ONE 2017, 12, e0172521. [Google Scholar] [CrossRef] [PubMed]
- Boone, D.R.; Weisz, H.A.; Willey, H.E.; Torres, K.E.O.; Falduto, M.T.; Sinha, M.; Spratt, H.; Bolding, I.J.; Johnson, K.M.; Parsley, M.A.; et al. Traumatic brain injury induces long-lasting changes in immune and regenerative signaling. PLoS ONE 2019, 14, e0214741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for rna-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. Wgcna: An r package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Hanzelmann, S.; Castelo, R.; Guinney, J. Gsva: Gene set variation analysis for microarray and rna-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Mejia, J.J.; Meng, E.C.; Pico, A.R.; MacParland, S.A.; Ketela, T.; Pugh, T.J.; Bader, G.D.; Morris, J.H. Evaluation of methods to assign cell type labels to cell clusters from single-cell rna-sequencing data. F1000Research 2019, 8, 296. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. Clusterprofiler: An r package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., 3rd; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., 3rd; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive integration of single-cell data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. Kegg: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [Green Version]
- Kan, K.J.; Guo, F.; Zhu, L.; Pallavi, P.; Sigl, M.; Keese, M. Weighted gene co-expression network analysis reveals key genes and potential drugs in abdominal aortic aneurysm. Biomedicines 2021, 9, 546. [Google Scholar] [CrossRef]
- Russo, M.V.; McGavern, D.B. Inflammatory neuroprotection following traumatic brain injury. Science 2016, 353, 783–785. [Google Scholar] [CrossRef] [Green Version]
- Mallah, K.; Couch, C.; Alshareef, M.; Borucki, D.; Yang, X.; Alawieh, A.; Tomlinson, S. Complement mediates neuroinflammation and cognitive decline at extended chronic time points after traumatic brain injury. Acta Neuropathol. Commun. 2021, 9, 72. [Google Scholar] [CrossRef]
- Shi, K.; Zhang, J.; Dong, J.F.; Shi, F.D. Dissemination of brain inflammation in traumatic brain injury. Cell Mol. Immunol. 2019, 16, 523–530. [Google Scholar] [CrossRef]
- Brett, B.L.; Gardner, R.C.; Godbout, J.; Dams-O’Connor, K.; Keene, C.D. Traumatic brain injury and risk of neurodegenerative disorder. Biol. Psychiatry 2022, 91, 498–507. [Google Scholar] [CrossRef]
- Uryu, K.; Chen, X.H.; Martinez, D.; Browne, K.D.; Johnson, V.E.; Graham, D.I.; Lee, V.M.; Trojanowski, J.Q.; Smith, D.H. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp. Neurol. 2007, 208, 185–192. [Google Scholar] [CrossRef] [Green Version]
- Schaffert, J.; LoBue, C.; White, C.L.; Chiang, H.S.; Didehbani, N.; Lacritz, L.; Rossetti, H.; Dieppa, M.; Hart, J.; Cullum, C.M. Traumatic brain injury history is associated with an earlier age of dementia onset in autopsy-confirmed alzheimer’s disease. Neuropsychology 2018, 32, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Perry, V.H.; Teeling, J. Microglia and macrophages of the central nervous system: The contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin. Immunopathol. 2013, 35, 601–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergink, V.; Gibney, S.M.; Drexhage, H.A. Autoimmunity, inflammation, and psychosis: A search for peripheral markers. Biol. Psychiatry 2014, 75, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Borst, K.; Dumas, A.A.; Prinz, M. Microglia: Immune and non-immune functions. Immunity 2021, 54, 2194–2208. [Google Scholar] [CrossRef] [PubMed]
- Corps, K.N.; Roth, T.L.; McGavern, D.B. Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol. 2015, 72, 355–362. [Google Scholar] [CrossRef] [Green Version]
- Roth, T.L.; Nayak, D.; Atanasijevic, T.; Koretsky, A.P.; Latour, L.L.; McGavern, D.B. Transcranial amelioration of inflammation and cell death after brain injury. Nature 2014, 505, 223–228. [Google Scholar] [CrossRef] [Green Version]
- Willis, E.F.; MacDonald, K.P.A.; Nguyen, Q.H.; Garrido, A.L.; Gillespie, E.R.; Harley, S.B.R.; Bartlett, P.F.; Schroder, W.A.; Yates, A.G.; Anthony, D.C.; et al. Repopulating microglia promote brain repair in an il-6-dependent manner. Cell 2020, 180, 833–846.e816. [Google Scholar] [CrossRef]
- Faraco, G.; Park, L.; Anrather, J.; Iadecola, C. Brain perivascular macrophages: Characterization and functional roles in health and disease. J. Mol. Med. 2017, 95, 1143–1152. [Google Scholar] [CrossRef] [Green Version]
- Hawkes, C.A.; McLaurin, J. Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc. Natl. Acad. Sci. USA 2009, 106, 1261–1266. [Google Scholar] [CrossRef] [Green Version]
- Thanopoulou, K.; Fragkouli, A.; Stylianopoulou, F.; Georgopoulos, S. Scavenger receptor class b type i (sr-bi) regulates perivascular macrophages and modifies amyloid pathology in an alzheimer mouse model. Proc. Natl. Acad. Sci. USA 2010, 107, 20816–20821. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Guo, R.; Zhang, F. Brain perivascular macrophages: Recent advances and implications in health and diseases. CNS Neurosci. Ther. 2019, 25, 1318–1328. [Google Scholar] [CrossRef] [PubMed]
- Szmydynger-Chodobska, J.; Strazielle, N.; Gandy, J.R.; Keefe, T.H.; Zink, B.J.; Ghersi-Egea, J.F.; Chodobski, A. Posttraumatic invasion of monocytes across the blood-cerebrospinal fluid barrier. J. Cereb. Blood Flow Metab. 2012, 32, 93–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semple, B.D.; Bye, N.; Rancan, M.; Ziebell, J.M.; Morganti-Kossmann, M.C. Role of ccl2 (mcp-1) in traumatic brain injury (tbi): Evidence from severe tbi patients and ccl2-/- mice. J. Cereb. Blood Flow Metab. 2010, 30, 769–782. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.L.; Niemi, E.C.; Wang, S.H.; Lee, C.C.; Bingham, D.; Zhang, J.; Cozen, M.L.; Charo, I.; Huang, E.J.; Liu, J.; et al. Ccr2 deficiency impairs macrophage infiltration and improves cognitive function after traumatic brain injury. J. Neurotrauma 2014, 31, 1677–1688. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, R.; Lu, H.; Butovsky, O.; Ohno, N.; Rietsch, A.M.; Cialic, R.; Wu, P.M.; Doykan, C.E.; Lin, J.; Cotleur, A.C.; et al. Differential roles of microglia and monocytes in the inflamed central nervous system. J. Exp. Med. 2014, 211, 1533–1549. [Google Scholar] [CrossRef] [Green Version]
- Hoffe, B.; Holahan, M.R. Hyperacute excitotoxic mechanisms and synaptic dysfunction involved in traumatic brain injury. Front. Mol. Neurosci. 2022, 15, 831825. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset ID | TBI | Time | Tissue | Sample Number Included |
|---|---|---|---|---|
| GSE1911 [14] | CCI | 3 h, 24 h | hippocampus | 3 |
| GSE2392 [12] | Moderate FPI | 30 min, 4 h, 8 h, 24 h, 3 d, 3 w | perilesional cortex | 39 |
| GSE2871 [11] | Mild and severe FPI | 4 h, 24 h | parietal cortex and hippocampus, ipsilateral and contralateral | 47 |
| GSE24047 [15,16] | FPI | 3 h, 6 h, 12 h, 48 h | lateral cortex | 16 |
| GSE31357 [17] | TBI | 4 h, 24 h | hippocampus | 16 |
| GSE45997 [13] | CCI | 24 h | ipsilateral and contralateral brain | 9 |
| GSE59645 [18] | TBI | 24 h | hippocampus | 8 |
| GSE64978 [19] | FPI | 1 w | hippocampus | 10 |
| GSE67836 [20] | Rot-TBI and FPI | 1 m | frontal cortex | 13 |
| GSE68207 [19] | FPI | 1 w | Hippocampus | 8 |
| GSE80174 [21] | TBI | 3 m | perilesional cortex, dorsal hippocampus, ipsilateral thalamus | 30 |
| GSE86579 [22] | FPI | 3 m | hippocampus | 11 |
| GSE111452 [23] | FPI | 24 h, 2 w, 3 m, 6 m, 1 y | hippocampus, cortex | 113 |
| GSE115614 [18] | TBI | 24 h | hippocampus | 5 |
| GSE137869 [10] | - | - | brain | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, Q.; Song, M.; Zhao, R.; Han, X.; Deng, L.; Xue, H.; Li, W.; Li, G. Comprehensive RNA Expression Analysis Revealed Biological Functions of Key Gene Sets and Identified Disease-Associated Cell Types Involved in Rat Traumatic Brain Injury. J. Clin. Med. 2022, 11, 3437. https://doi.org/10.3390/jcm11123437
Tang Q, Song M, Zhao R, Han X, Deng L, Xue H, Li W, Li G. Comprehensive RNA Expression Analysis Revealed Biological Functions of Key Gene Sets and Identified Disease-Associated Cell Types Involved in Rat Traumatic Brain Injury. Journal of Clinical Medicine. 2022; 11(12):3437. https://doi.org/10.3390/jcm11123437
Chicago/Turabian StyleTang, Qilin, Mengmeng Song, Rongrong Zhao, Xiao Han, Lin Deng, Hao Xue, Weiguo Li, and Gang Li. 2022. "Comprehensive RNA Expression Analysis Revealed Biological Functions of Key Gene Sets and Identified Disease-Associated Cell Types Involved in Rat Traumatic Brain Injury" Journal of Clinical Medicine 11, no. 12: 3437. https://doi.org/10.3390/jcm11123437
APA StyleTang, Q., Song, M., Zhao, R., Han, X., Deng, L., Xue, H., Li, W., & Li, G. (2022). Comprehensive RNA Expression Analysis Revealed Biological Functions of Key Gene Sets and Identified Disease-Associated Cell Types Involved in Rat Traumatic Brain Injury. Journal of Clinical Medicine, 11(12), 3437. https://doi.org/10.3390/jcm11123437