
Clinical, Histological, and Genetic Features of 25 Patients with Autosomal Dominant Progressive External Ophthalmoplegia (ad-PEO)/PEO-Plus Due to TWNK Mutations

 ,
,  , , , , and
, , , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Data
2.2. Genetic Analysis
3. Results
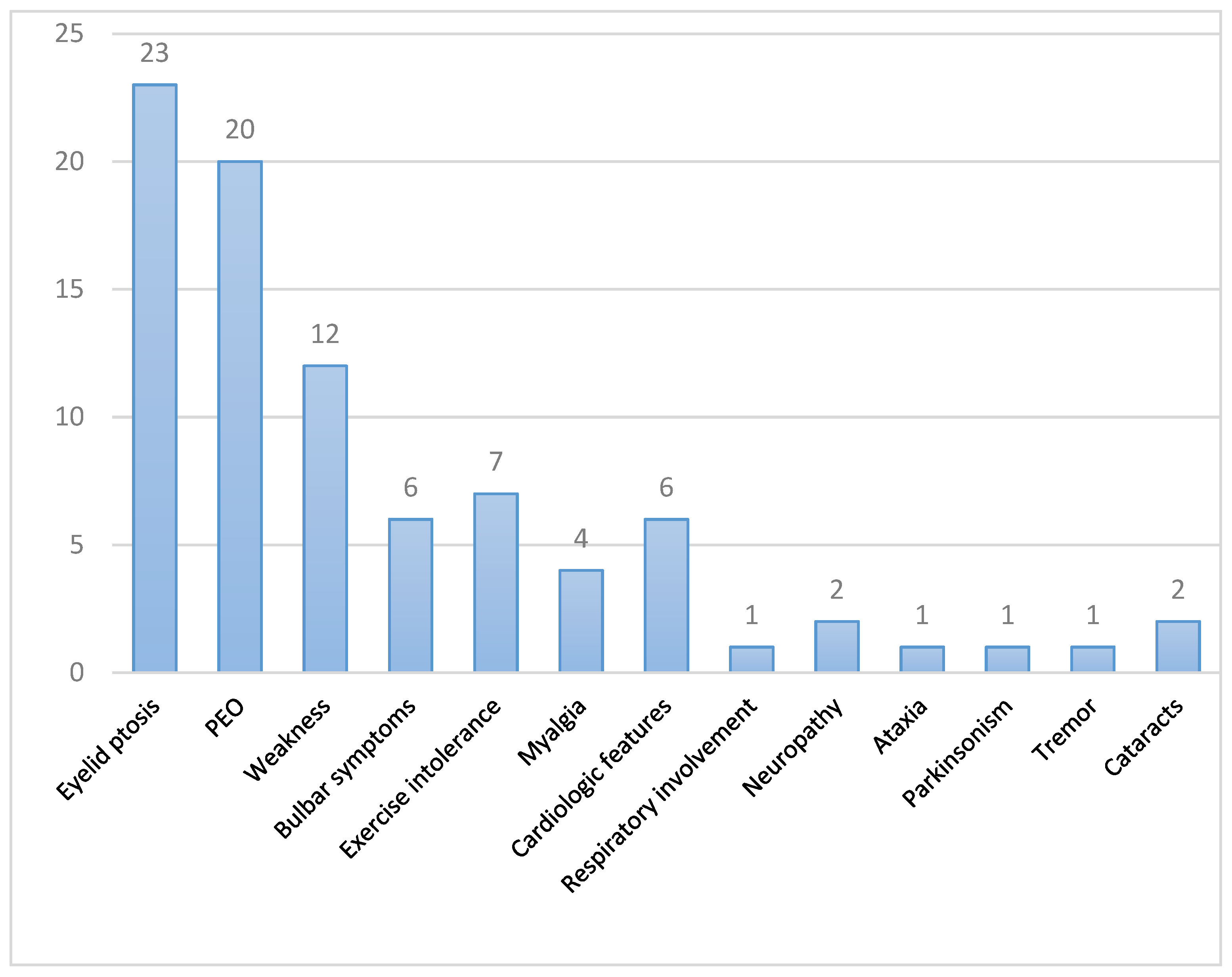
3.1. Clinical Features
3.2. Complementary Tests
3.3. Genetic Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- El-Hattab, A.W.; Craigen, W.J.; Scaglia, F. Mitochondrial DNA Maintenance Defects. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 1539–1555. [Google Scholar] [CrossRef]
- Peter, B.; Falkenberg, M. TWINKLE and Other Human Mitochondrial DNA Helicases: Structure, Function and Disease. Genes 2020, 11, 408. [Google Scholar] [CrossRef] [Green Version]
- Van Hove, J.L.K.; Cunningham, V.; Rice, C.; Ringel, S.P.; Zhang, Q.; Chou, P.-C.; Truong, C.K.; Wong, L.-J.C. Finding Twinkle in the Eyes of a 71-Year-Old Lady: A Case Report and Review of the Genotypic and Phenotypic Spectrum of TWINKLE-Related Dominant Disease. Am. J. Med. Genet. 2009, 149A, 861–867. [Google Scholar] [CrossRef]
- Fratter, C.; Gorman, G.S.; Stewart, J.D.; Buddles, M.; Smith, C.; Evans, J.; Seller, A.; Poulton, J.; Roberts, M.; Hanna, M.G.; et al. The Clinical, Histochemical, and Molecular Spectrum of PEO1 (Twinkle)-Linked AdPEO. Neurology 2010, 74, 1619–1626. [Google Scholar] [CrossRef] [Green Version]
- Martin-Negrier, M.-L.; Sole, G.; Jardel, C.; Vital, C.; Ferrer, X.; Vital, A. TWINKLE Gene Mutation: Report of a French Family with an Autosomal Dominant Progressive External Ophthalmoplegia and Literature Review: Twinkle Gene Mutation. Eur. J. Neurol. 2011, 18, 436–441. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-López, C.; García-Cárdaba, L.M.; Blázquez, A.; Serrano-Lorenzo, P.; Gutiérrez-Gutiérrez, G.; San Millán-Tejado, B.; Muelas, N.; Hernández-Laín, A.; Vílchez, J.J.; Gutiérrez-Rivas, E.; et al. Clinical, Pathological and Genetic Spectrum in 89 Cases of Mitochondrial Progressive External Ophthalmoplegia. J. Med. Genet. 2020, 57, 643–646. [Google Scholar] [CrossRef]
- Rivera, H.; Blázquez, A.; Carretero, J.; Alvarez-Cermeño, J.C.; Campos, Y.; Cabello, A.; Gonzalez-Vioque, E.; Borstein, B.; Garesse, R.; Arenas, J.; et al. Mild Ocular Myopathy Associated with a Novel Mutation in Mitochondrial Twinkle Helicase. Neuromuscul. Disord. 2007, 17, 677–680. [Google Scholar] [CrossRef]
- Clarke, L.; Fairley, S.; Zheng-Bradley, X.; Streeter, I.; Perry, E.; Lowy, E.; Tassé, A.-M.; Flicek, P. The International Genome Sample Resource (IGSR): A Worldwide Collection of Genome Variation Incorporating the 1000 Genomes Project Data. Nucleic Acids Res. 2017, 45, D854–D859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Exome Variant Server. Available online: https://evs.gs.washington.edu/EVS/ (accessed on 28 July 2021).
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Genome Aggregation Database Consortium; et al. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Peña-Chilet, M.; Roldán, G.; Perez-Florido, J.; Ortuño, F.M.; Carmona, R.; Aquino, V.; Lopez-Lopez, D.; Loucera, C.; Fernandez-Rueda, J.L.; Gallego, A.; et al. CSVS, a Crowdsourcing Database of the Spanish Population Genetic Variability. Nucleic Acids Res. 2021, 49, D1130–D1137. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving Access to Variant Interpretations and Supporting Evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT Web Server: Predicting Effects of Amino Acid Substitutions on Proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation Prediction for the Deep-Sequencing Age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Jagadeesh, K.A.; Wenger, A.M.; Berger, M.J.; Guturu, H.; Stenson, P.D.; Cooper, D.N.; Bernstein, J.A.; Bejerano, G. M-CAP Eliminates a Majority of Variants of Uncertain Significance in Clinical Exomes at High Sensitivity. Nat. Genet. 2016, 48, 1581–1586. [Google Scholar] [CrossRef]
- Choi, Y.; Chan, A.P. PROVEAN Web Server: A Tool to Predict the Functional Effect of Amino Acid Substitutions and Indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A General Framework for Estimating the Relative Pathogenicity of Human Genetic Variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, G.M.; Stone, E.A.; Asimenos, G.; NISC Comparative Sequencing Program; Green, E.D.; Batzoglou, S.; Sidow, A. Distribution and Intensity of Constraint in Mammalian Genomic Sequence. Genome Res. 2005, 15, 901–913. [Google Scholar] [CrossRef] [Green Version]
- Hubisz, M.J.; Pollard, K.S.; Siepel, A. PHAST and RPHAST: Phylogenetic Analysis with Space/Time Models. Brief. Bioinform. 2011, 12, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; ACMG Laboratory Quality Assurance Committee; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- McClelland, C.; Manousakis, G.; Lee, M.S. Progressive External Ophthalmoplegia. Curr. Neurol. Neurosci. Rep. 2016, 16, 53. [Google Scholar] [CrossRef] [PubMed]
- Orsucci, D.; Angelini, C.; Bertini, E.; Carelli, V.; Comi, G.P.; Federico, A.; Minetti, C.; Moggio, M.; Mongini, T.; Santorelli, F.M.; et al. Revisiting Mitochondrial Ocular Myopathies: A Study from the Italian Network. J. Neurol. 2017, 264, 1777–1784. [Google Scholar] [CrossRef] [PubMed]
- Virgilio, R.; Ronchi, D.; Hadjigeorgiou, G.M.; Bordoni, A.; Saladino, F.; Moggio, M.; Adobbati, L.; Kafetsouli, D.; Tsironi, E.; Previtali, S.; et al. Novel Twinkle (PEO1) Gene Mutations in Mendelian Progressive External Ophthalmoplegia. J. Neurol. 2008, 255, 1384–1391. [Google Scholar] [CrossRef] [PubMed]
- Garone, C.; Taylor, R.W.; Nascimento, A.; Poulton, J.; Fratter, C.; Domínguez-González, C.; Evans, J.C.; Loos, M.; Isohanni, P.; Suomalainen, A.; et al. Retrospective Natural History of Thymidine Kinase 2 Deficiency. J. Med. Genet. 2018, 55, 515–521. [Google Scholar] [CrossRef]
- Cohen, B.H. Mitochondrial and Metabolic Myopathies. Contin. Lifelong Learn. Neurol. 2019, 25, 1732–1766. [Google Scholar] [CrossRef]
- Domínguez-González, C.; Hernández-Laín, A.; Rivas, E.; Hernández-Voth, A.; Sayas Catalán, J.; Fernández-Torrón, R.; Fuiza-Luces, C.; García García, J.; Morís, G.; Olivé, M.; et al. Late-Onset Thymidine Kinase 2 Deficiency: A Review of 18 Cases. Orphanet J. Rare Dis. 2019, 14, 100. [Google Scholar] [CrossRef] [Green Version]
- Davis, R.L.; Liang, C.; Sue, C.M. A Comparison of Current Serum Biomarkers as Diagnostic Indicators of Mitochondrial Diseases. Neurology 2016, 86, 2010–2015. [Google Scholar] [CrossRef]
- Poulsen, N.S.; Madsen, K.L.; Hornsyld, T.M.; Eisum, A.-S.V.; Fornander, F.; Buch, A.E.; Stemmerik, M.G.; Ruiz-Ruiz, C.; Krag, T.O.; Vissing, J. Growth and Differentiation Factor 15 as a Biomarker for Mitochondrial Myopathy. Mitochondrion 2020, 50, 35–41. [Google Scholar] [CrossRef]
- Peñas, A.; Fernández-De la Torre, M.; Laine-Menéndez, S.; Lora, D.; Illescas, M.; García-Bartolomé, A.; Morales-Conejo, M.; Arenas, J.; Martín, M.A.; Morán, M.; et al. Plasma Gelsolin Reinforces the Diagnostic Value of FGF-21 and GDF-15 for Mitochondrial Disorders. Int. J. Mol. Sci. 2021, 22, 6396. [Google Scholar] [CrossRef]
- Braz, L.P.; Ng, Y.S.; Gorman, G.S.; Schaefer, A.M.; McFarland, R.; Taylor, R.W.; Turnbull, D.M.; Whittaker, R.G. Neuromuscular Junction Abnormalities in Mitochondrial Disease: An Observational Cohort Study. Neurol. Clin. Pract. 2021, 11, 97–104. [Google Scholar] [CrossRef]
- Engel, A.G.; Shen, X.-M.; Selcen, D.; Sine, S.M. Congenital Myasthenic Syndromes: Pathogenesis, Diagnosis, and Treatment. Lancet Neurol. 2015, 14, 420–434. [Google Scholar] [CrossRef] [Green Version]
- Ciafaloni, E. Myasthenia Gravis and Congenital Myasthenic Syndromes. Contin. Lifelong Learn. Neurol. 2019, 25, 1767–1784. [Google Scholar] [CrossRef] [PubMed]
- Ostos, F.; Alcantara Miranda, P.; Hernández-Laín, A.; Domínguez-González, C. Congenital Ophthalmoplegia and Late-Onset Limb Weakness Caused by MUSK Mutations. J. Clin. Neuromuscul. Dis. 2020, 21, 222–224. [Google Scholar] [CrossRef]
- Brais, B. Oculopharyngeal Muscular Dystrophy. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2011; Volume 101, pp. 181–192. ISBN 978-0-08-045031-5. [Google Scholar]
- Wong, K.T.; Dick, D.; Anderson, J.R. Mitochondrial Abnormalities in Oculopharyngeal Muscular Dystrophy. Neuromuscul. Disord. 1996, 6, 163–166. [Google Scholar] [CrossRef]
- Alonso-Jimenez, A.; Kroon, R.H.M.J.M.; Alejaldre-Monforte, A.; Nuñez-Peralta, C.; Horlings, C.G.C.; van Engelen, B.G.M.; Olivé, M.; González, L.; Verges-Gil, E.; Paradas, C.; et al. Muscle MRI in a Large Cohort of Patients with Oculopharyngeal Muscular Dystrophy. J. Neurol. Neurosurg. Psychiatry 2019, 90, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Pasnoor, M.; Dimachkie, M.M.; Farmakidis, C.; Barohn, R.J. Diagnosis of Myasthenia Gravis. Neurol. Clin. 2018, 36, 261–274. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Patient | Sex | Age (y.o) | Age at Onset | Family History | Eyelid Ptosis | CPEO | Weakness | Dysarthria/Dysphagia | Cardiac/Respiratory Involvement | Others | Previous Diagnosis and Treatment |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 65 | 30 | Yes. Nephew of pt. 23 | Yes | Yes | Inferior facial, mild cervical, proximal UL and LL | Dysarthria and dysphagia | No suggestive clinical data; normal ECG, FVC 105% | No | OPMD; no treatment |
| 2 | F | 74 | Adult | Yes | Yes | Yes | No | No | Tachycardia under study, ECG: LAFB | No | Seronegative ocular MG, no treatment |
| 3 | M | 82 | 40 | Yes Brother of pt. 7 | Yes | Yes | Mild cervical, mild proximal UL and LL | No | No suggestive clinical data | Exercise intolerance, ataxia, PNP | Seronegative MG; pyridostigmine (NR) |
| 4 | F | 79 | 60 | Yes | yes | Yes | Mild cervical, mild proximal LL | Dysphagia | Mild LVH, normal ECG; sleep apnea with CPAP indication | Myalgias, exercise intolerance | Seronegative MG; pyridostigmine and GC (NR) |
| 5 | M | 28 | 17 | No | Yes | Yes | No | No | Palpitations with normal TTE and Holter-ECG | Myalgias, exercise intolerance, subclinical PNP | Seronegative MG; no treatment |
| 6 | F | 64 | 60 | Yes | Yes | Yes | No | No | No suggestive clinical data | Episodes of acute worsening with bulbar and limb weakness | Seronegative MG; pyridostigmine (NR), acute episodes: IVIg, plasma exchange, IS (NR) |
| 7 | F | 86 | 60 | Yes Sister of pt. 3 | Yes | No | Mild proximal LL | No | Suspicion of sleep apnea; mild LVEF reduction | Essential tremor (UL, head and jaw), exercise intolerance | - |
| 8 | M | 68 | 30 | Yes | Yes | Yes | No | No | No suggestive clinical data | - | - |
| 9 | M | 36 | 25 | No | Yes | Yes | No | No | No suggestive clinical data; ECG: RBBB, normal TTE | - | Seronegative MG, OPMD; no treatment |
| 10 | F | 58 | 50 | Yes | Yes | Yes | No | No | No suggestive clinical data | - | - |
| 11 | F | 59 | 43 | Sister with epilepsy. Both parents dementia | No | No | Mild cervical, mild proximal UL and LL | No | No suggestive clinical data | Generalized myalgias at rest, cramps with statins in the past, exercise intolerance | - |
| 12 | M | 67 | Adult onset | Yes | Yes | Yes | No | No | No suggestive clinical data | - | OPMD; no treatment. |
| 13 | F | 72 | 40 | Yes | Yes | No | Mild cervical | Dysphagia | No suggestive clinical data | - | OPMD, no treatment |
| 14 | M | 54 | 40 | Yes | Yes | No | No | No | No suggestive clinical data | - | Seronegative MG. |
| 15 | F | 51 | Not reported | Yes | No | Yes | No | No | No suggestive clinical data | - | Seronegative MG |
| 16 | F | 67 | Adult onset | Yes | Yes | Yes | Proximal UL and LL | No | No suggestive clinical data | - | OPMD; no treatment. |
| 17 | F | 32 | Not reported | Yes | Yes | Yes | Cervical, proximal UL and LL | No | No suggestive clinical data | Exercise intolerance | - |
| 18 | F | 49 | 14 | Yes | Yes | Yes | Cervical, proximal and distal UL and LL | Dysphagia | No suggestive clinical data | - | - |
| 19 | F | 44 | Childhood onset | Yes | Yes | Yes | No | No | No suggestive clinical data | Occasional cramps; fatigable ocular weakness reported | Seronegative MG |
| 20 | F | 78 | 50 | Yes | Yes | Yes | Proximal at limbs | No | No suggestive clinical data | - | - |
| 21 | M | 80 | 45 | Yes Brother of pt. 22 | Yes | Yes | Orbicularis oculi, inferior facial, cervical, proximal at limbs | Dysarthria and dysphagia | No suggestive clinical data; FVC 100.9%; mild LVH | Myalgias, exercise intolerance | - |
| 22 | F | 80 | 40 | Yes Sister of pt. 21 | Yes | Yes | Orbicularis oculi, cervical, limbs | Dysphagia | FVC 57% | Parkinsonism | - |
| 23 | F | 82 | 60 | Yes Aunt of pt. 1 | Yes | No | No | No | Not reported | - | - |
| 24 | M | 55 | 48 | Yes Son of pt. 25 | Yes | Yes | No | No | Not reported | Cataracts 48 y.o | - |
| 25 | F | 76 | 64 | Yes Mother of pt. 24 | Yes | Yes | No | No | Not reported | Cataracts 50 y.o | - |
| Patient | CK (IU/L) | Lactate (mmol/L) | GDF15 (pg/mL) | EMG/ENG | Muscle MRI | Muscle Biopsy | TWNK Variant | Protein Change | Multiple mtDNA Deletions (Skeletal Muscle) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 300 | - | - | - | - | - | c.1361T>G | p.Val454Gly | - |
| 2 | 990.6 | Normal | RRF and COX negative fibers | c.1070G>C | p.Arg357Pro | Y | |||
| 3 | 68 | 3.7 | - | - | - | - | c.1070G>C | p.Arg357Pro | - |
| 4 | 21 | - | - | Normal | IL: diffuse edema of both gastrocnemius and lateral and distal portion of left soleus | Frequent RBF, 1% COX negative fibers | c.1070G>C | p.Arg357Pro | Y |
| 5 | 101 | 1.7 | 1454 | Mild SNAP amplitude reduction in both surals and superficial peroneals | IL: normal | 9% COX negative fibers | c.1121G>A | p.Arg374Gln | Y |
| 6 | 37 | - | 2727 | - | - | RRFs and 1% COX negative fibers. | c.1361T>G | p.Val454Gly | - |
| 7 | 300 | - | - | Myopathic | - | RRF, COX negative fibers, complex IV deficiency | c.1070G>C | p.Arg357Pro | Y |
| 8 | Normal | - | - | Myopathic | - | Occasional RRF | c.1361T>G | p.Val454Gly | Y |
| 9 | 305 | Normal | - | Normal | - | Frequent RBF (most of them COX negative, 3.5% of total fibers) | c.1121G>A | p.Arg374Gln | Y |
| 10 | - | - | - | - | - | - | c.1361T>G | p.Val454Gly | - |
| 11 | 224 | - | - | Moderate chronic neurogenic signs R L5 | - | Frequent subsarcolemic SDH accumulation, frequent COX negative fibers, type I fiber predominance | c.1087T>A (new description) | p.Trp363Arg | Y |
| 12 | - | - | - | Moderate myopathic signs | - | Unspecific: isolated RBF, <1% COX negative fibers | c.908G>A | p.Arg303Gln | Y |
| 13 | - | - | - | - | Diffuse fatty infiltration of paravertebral muscles, abnormal signal at both serratus anterior | RRF and frequent COX negative fibers | c.1106C>T (new description) | p.Ser369Phe | Y |
| 14 | 95 | - | - | Myopathic | - | RRF, 10% COX negative, type 1 fiber predominance | c.1070G>C | p.Arg357Pro | Y |
| 15 | Normal | - | - | - | - | Occasional RRF, 2% COX negative fibers | c.1361T>G | p.Val454Gly | Y |
| 16 | 74 | - | - | Myopathic | - | Unspecific oxidative alterations, 3 RRF | c.1361T>G | p.Val454Gly | Y |
| 17 | - | - | - | - | - | RRF, frequent COX negative fibers, some internal nuclei | c.1121 G>A | p.Arg374Gln | Y |
| 18 | - | - | - | - | - | - | c.1433T>G | p.Phe478Cys | - |
| 19 | 312 | - | - | Myopathic | - | 3% COX negative fibers | c.1084G>C | p.Arg362Pro | Y |
| 20 | Normal | Normal | - | Myopathic | - | RRF and COX negative fibers | c.1071G>C | p.Arg357Pro | Y |
| 21 | 113 | Raised | - | Myopathic | - | RRF, massive mitochondrial accumulations with SDH, loss of COX activity. Complex I and IV deficiency | c.1411T>G | p.Tyr471Asp | Y |
| 22 | 66 | - | - | - | - | - | c.1411T>G | p.Tyr471Asp | - |
| 23 | 54 | - | - | Myopathic | - | RRF, type 1 fiber predominance | c.1361T>G | p.Val454Gly | - |
| 24 | - | - | - | - | - | - | c.1070G>C | p.Arg357Pro | - |
| 25 | 61 | 3.24 | - | Normal ENG | - | RRF | c.1070G>C | p.Arg357Pro | Y |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bermejo-Guerrero, L.; de Fuenmayor-Fernández de la Hoz, C.P.; Serrano-Lorenzo, P.; Blázquez-Encinar, A.; Gutiérrez-Gutiérrez, G.; Martínez-Vicente, L.; Galán-Dávila, L.; García-García, J.; Arenas, J.; Muelas, N.; et al. Clinical, Histological, and Genetic Features of 25 Patients with Autosomal Dominant Progressive External Ophthalmoplegia (ad-PEO)/PEO-Plus Due to TWNK Mutations. J. Clin. Med. 2022, 11, 22. https://doi.org/10.3390/jcm11010022
Bermejo-Guerrero L, de Fuenmayor-Fernández de la Hoz CP, Serrano-Lorenzo P, Blázquez-Encinar A, Gutiérrez-Gutiérrez G, Martínez-Vicente L, Galán-Dávila L, García-García J, Arenas J, Muelas N, et al. Clinical, Histological, and Genetic Features of 25 Patients with Autosomal Dominant Progressive External Ophthalmoplegia (ad-PEO)/PEO-Plus Due to TWNK Mutations. Journal of Clinical Medicine. 2022; 11(1):22. https://doi.org/10.3390/jcm11010022
Chicago/Turabian StyleBermejo-Guerrero, Laura, Carlos Pablo de Fuenmayor-Fernández de la Hoz, Pablo Serrano-Lorenzo, Alberto Blázquez-Encinar, Gerardo Gutiérrez-Gutiérrez, Laura Martínez-Vicente, Lucía Galán-Dávila, Jorge García-García, Joaquín Arenas, Nuria Muelas, and et al. 2022. "Clinical, Histological, and Genetic Features of 25 Patients with Autosomal Dominant Progressive External Ophthalmoplegia (ad-PEO)/PEO-Plus Due to TWNK Mutations" Journal of Clinical Medicine 11, no. 1: 22. https://doi.org/10.3390/jcm11010022
APA StyleBermejo-Guerrero, L., de Fuenmayor-Fernández de la Hoz, C. P., Serrano-Lorenzo, P., Blázquez-Encinar, A., Gutiérrez-Gutiérrez, G., Martínez-Vicente, L., Galán-Dávila, L., García-García, J., Arenas, J., Muelas, N., Hernández-Laín, A., Domínguez-González, C., & Martín, M. A. (2022). Clinical, Histological, and Genetic Features of 25 Patients with Autosomal Dominant Progressive External Ophthalmoplegia (ad-PEO)/PEO-Plus Due to TWNK Mutations. Journal of Clinical Medicine, 11(1), 22. https://doi.org/10.3390/jcm11010022