
Founder Effects in Hereditary Hemorrhagic Telangiectasia



Abstract
1. Introduction
1.1. Definition
1.2. Founder Effects in Population Isolates
1.3. Pioneer Reports for Founder Effects in Hereditary Hemorrhagic Telangiectasia
2. Study Design
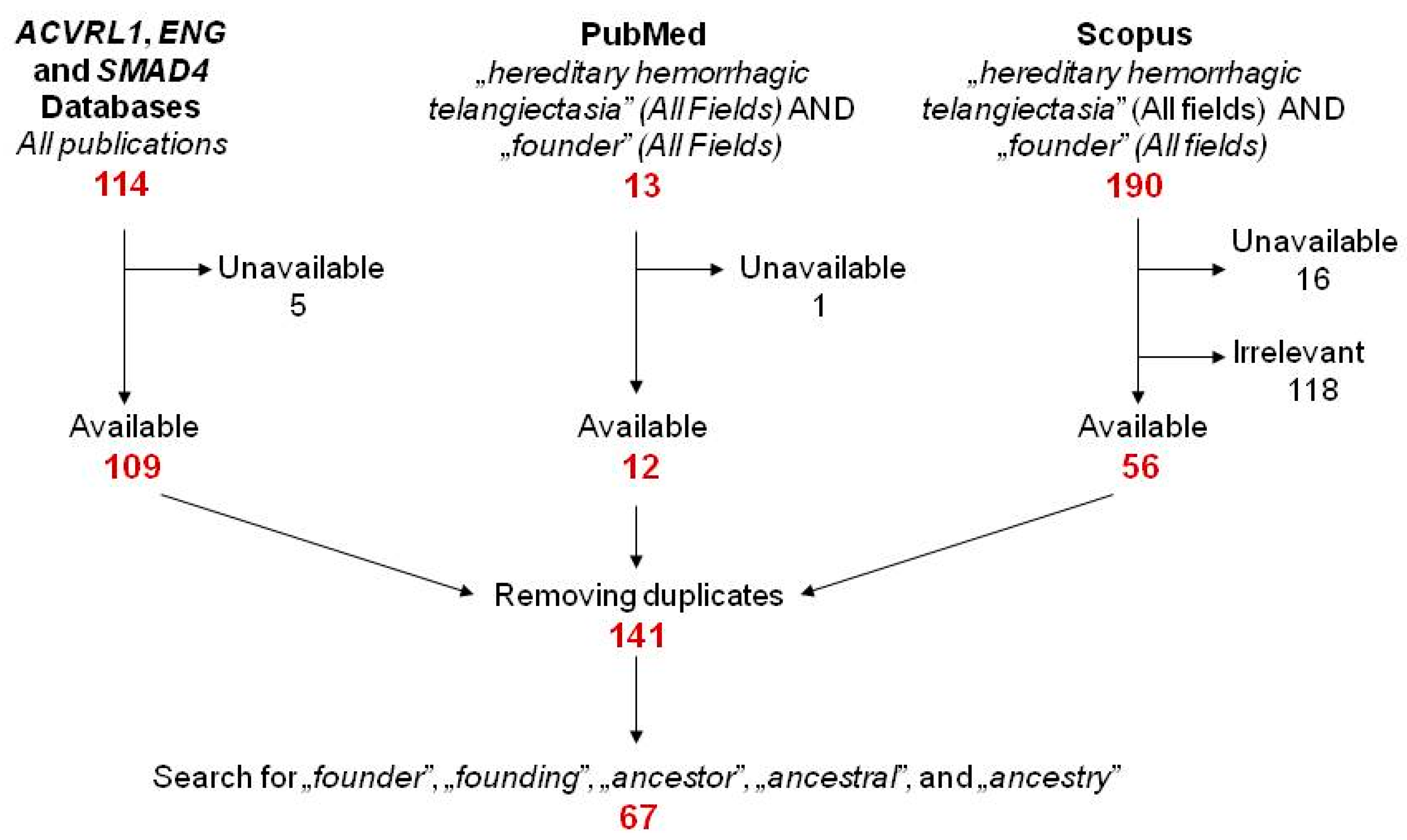
2.1. Literature Search
2.2. An Arbitrary Grading System to Assess Evidences for Founder Effect in HHT
3. The Overview of Potential Founders
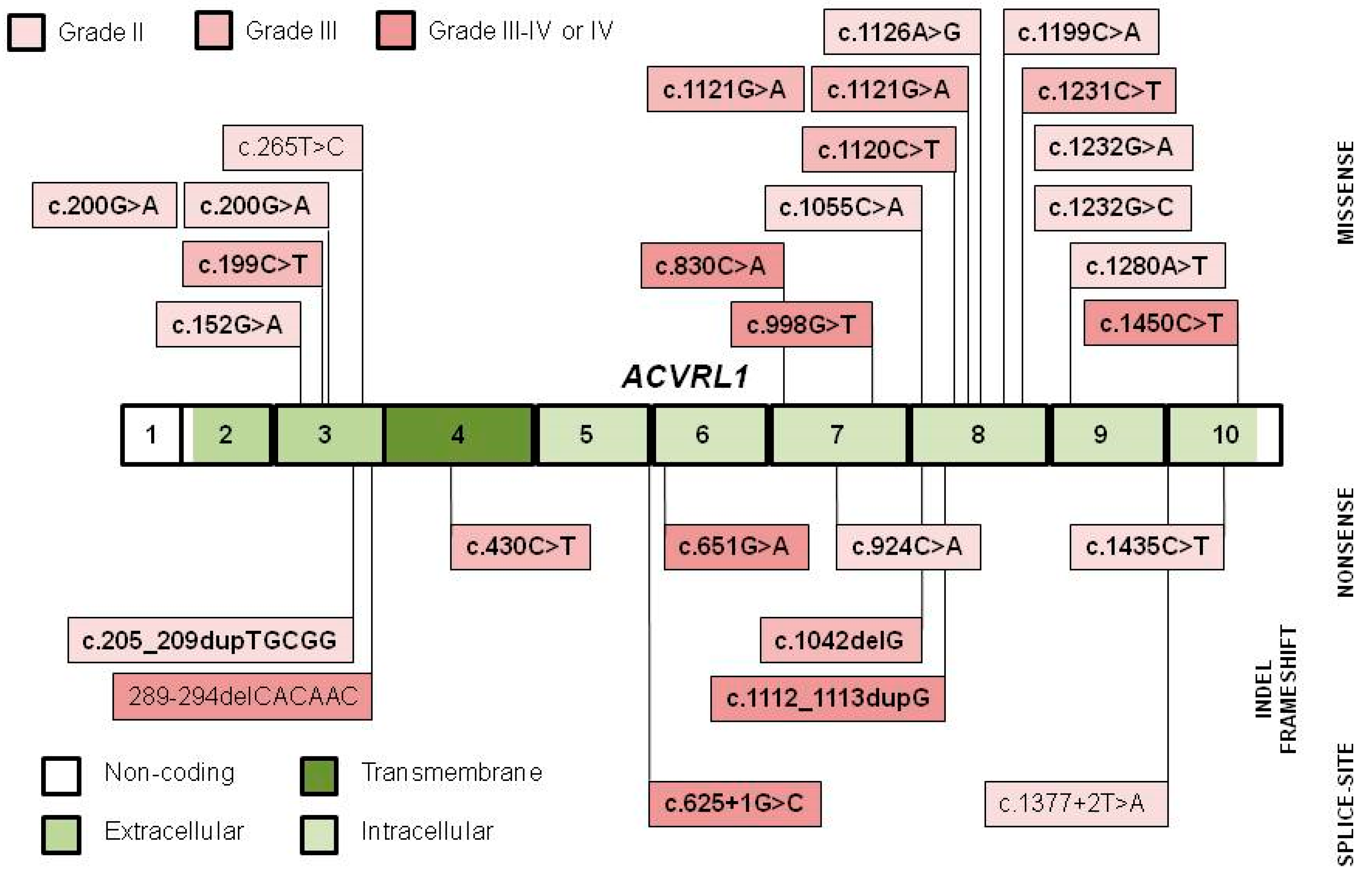
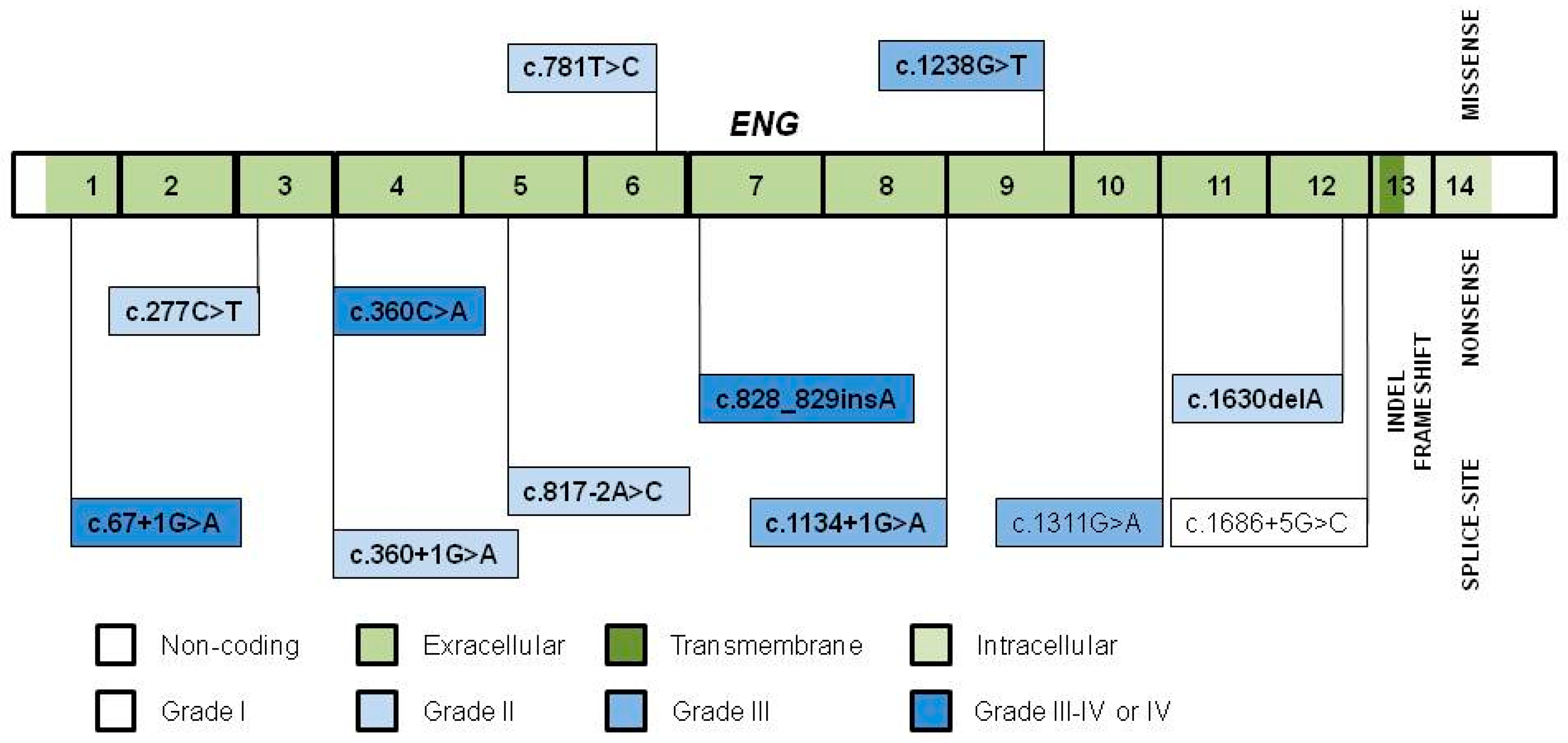
3.1. Variant Distribution, Type and Pathogenicity
3.2. Grade IV Founder Variants
3.3. Potentially Grade IV Founder Variants
3.4. Tracing the Founder Event
3.5. Mutation Age
3.6. The Contribution of Genealogy
3.7. How Can an Autosomal Dominant Disease Like HHT Result in a Founder Effect?
4. The Significance of Founder Effects
5. Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Slatkin, M. A Population-Genetic Test of Founder Effects and Implications for Ashkenazi Jewish Diseases. Am. J. Hum. Genet. 2004, 75, 282–293. [Google Scholar] [CrossRef]
- Arcos-Burgos, M.; Muenke, M. Genetics of population isolates. Clin. Genet. 2002, 61, 233–247. [Google Scholar] [CrossRef]
- Payne, M.; Rupar, C.A.; Siu, G.M.; Siu, V.M. Amish, Mennonite, and Hutterite Genetic Disorder Database. Paediatr. Child Health 2011, 16, e23–e24. [Google Scholar] [CrossRef]
- Ostrer, H.; Skorecki, K. The population genetics of the Jewish people. Hum. Genet. 2012, 132, 119–127. [Google Scholar] [CrossRef]
- Francomano, C.A.; McKusick, V.A.; Biesecker, L.G. Medical genetic studies in the Amish: Historical perspective. Am. J. Med. Genet. 2003, 121C, 1–4. [Google Scholar] [CrossRef]
- Charrow, J. Ashkenazi Jewish genetic disorders. Fam. Cancer 2004, 3, 201–206. [Google Scholar] [CrossRef]
- Orton, N.C.; Innes, A.M.; Chudley, A.E.; Bech-Hansen, N.T. Unique disease heritage of the Dutch-German Mennonite population. Am. J. Med. Genet. 2008, 146A, 1072–1087. [Google Scholar] [CrossRef]
- Pisano, M.; Cossu, A.; Persico, I.; Palmieri, G.; Angius, A.; Casu, G.; Palomba, G.; Sarobba, M.G.; Ossu Rocca, P.C.; Dedola, M.F.; et al. Identification of a founder BRCA2 mutation in Sardinia. Br. J. Cancer 2000, 82, 553–559. [Google Scholar] [CrossRef]
- Dagan, E.; Gershoni-Baruch, R.; Kurolap, A.; Fried, G. Early onset breast cancer in Ashkenazi women carriers of founder BRCA1/2 mutations: Beyond 10 years of follow-up. Eur. J. Cancer Care 2016, 26, e12594. [Google Scholar] [CrossRef]
- Wallace, S.E.; Bean, L.J.H. Resources for Genetics Professionals—Genetic Disorders Associated with Founder Variants Common in the Druze Population; Adam, M.P., Ardinger, H.H., Pagon, R.A., Eds.; GeneReviews®: Seattle, WA, USA, 2019. Available online: https://www.ncbi.nlm.nih.gov/books/NBK549466/ (accessed on 25 February 2021).
- Borst, A.J.; Nakano, T.A.; Blei, F.; Adams, D.M.; Duis, J. A Primer on a Comprehensive Genetic Approach to Vascular Anomalies. Front. Pediatr. 2020, 8. [Google Scholar] [CrossRef]
- About Rare Diseases. Available online: https://www.12/consor/cgi-bin/Education_AboutRareDiseases.php?lng=EN (accessed on 24 February 2021).
- McDonald, J.; Wooderchak-Donahue, W.; Van Sant Webb, C.; Whitehead, K.; Stevenson, D.A.; Bayrak-Toydemir, P. Hereditary hemorrhagic telangiectasia: Genetics and molecular diagnostics in a new era. Front. Genet. 2015, 6, 1–8. [Google Scholar] [CrossRef]
- Sharathkumar, A.A.; Shapiro, A. Hereditary haemorrhagic telangiectasia. Haemophilia 2008, 14, 1269–1280. [Google Scholar] [CrossRef] [PubMed]
- McAllister, K.A.; Grogg, K.M.; Johnson, D.W.; Gallione, C.J.; Baldwin, M.A.; Jackson, C.E.; Helmbold, E.A.; Markel, D.S.; McKinnon, W.C.; Murrell, J. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat. Genet. 1994, 8, 345–351. [Google Scholar] [CrossRef]
- Johnson, D.W.; Berg, L.N.; Baldwin, M.A.; Gallione, C.J.; Marondel, I.; Yoon, S.J.; Stenzel, T.T.; Speer, M.; Pericak-Vance, M.A.; Diamond, A.; et al. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat. Genet. 1996, 13, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Plauchu, H.; De Chadarévian, J.-P.; Bideau, A.; Robert, J.-M. Age-related clinical profile of hereditary hemorrhagic telangiectasia in an epidemiologically recruited population. Am. J. Med. Genet. 1989, 32, 291–297. [Google Scholar] [CrossRef]
- Jessurun, G.A.; Nossent, J.C. Cerebrovascular accidents at a young age in Rendu-Osler-Weber disease; a survey in the Netherlands Antilles. Ned. Tijdschr. Geneeskd. 1992, 136, 428–431. [Google Scholar] [PubMed]
- Porteous, M.E.; Burn, J.; Proctor, S.J. Hereditary haemorrhagic telangiectasia: A clinical analysis. J. Med. Genet. 1992, 29, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Guttmacher, A.E.; McKinnon, W.C.; Upton, M.D. Hereditary hemorrhagic telangiectasia: A disorder in search of the genetics community. Am. J. Med. Genet. 1994, 52, 252–253. [Google Scholar] [CrossRef] [PubMed]
- Kjeldsen, A.D.; Vase, P.; Green, A. Hereditary haemorrhagic telangiectasia: A population-based study of prevalence and mortality in Danish patients. J. Intern. Med. 1999, 245, 31–39. [Google Scholar] [CrossRef]
- Bideau, A.; Brunet, G.; Heyer, E.; Plauchu, H.; Robert, J.-M. An abnormal concentration of cases of Rendu-Osler disease in the Valserine valley of the French Jura: A genealogical and demographic study. Ann. Hum. Biol. 1992, 19, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Gallione, C.J.; Scheessele, E.A.; Reinhardt, D.; Duits, A.J.; Berg, J.N.; Westermann, C.J.J.; Marchuk, D.A. Two common endoglin mutations in families with hereditary hemorrhagic telangiectasia in the Netherlands Antilles: Evidence for a founder effect. Hum. Genet. 2000, 107, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Lesca, G.; Plauchu, H.; Coulet, F.; Lefebvre, S.; Plessis, G.; Odent, S.; Rivière, S.; Leheup, B.; Goizet, C.; Carette, M.-F.; et al. Molecular screening ofALK1/ACVRL1andENGgenes in hereditary hemorrhagic telangiectasia in France. Hum. Mutat. 2004, 23, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Lesca, G.; Genin, E.; Blachier, C.; Olivieri, C.; Coulet, F.; Brunet, G.; Dupuis-Girod, S.; Buscarini, E.; Soubrier, F.; Calender, A.; et al. Hereditary hemorrhagic telangiectasia: Evidence for regional founder effects of ACVRL1 mutations in French and Italian patients. Eur. J. Hum. Genet. 2008, 16, 742–749. [Google Scholar] [CrossRef]
- ENG Database. Available online: https://arup.utah.edu/database/ENG/ENG_display.php (accessed on 7 February 2021).
- ACVRL1 Database. Available online: https://arup.utah.edu/database/ACVRL1/ACVRL1_display.php (accessed on 7 February 2021).
- SMAD4 Dabase. Available online: https://arup.utah.edu/database/SMAD4/SMAD4_display.php (accessed on 7 February 2021).
- Olivieri, C.; Pagella, F.; Semino, L.; Lanzarini, L.; Valacca, C.; Pilotto, A.; Corno, S.; Scappaticci, S.; Manfredi, G.; Buscarini, E.; et al. Analysis of ENG and ACVRL1 genes in 137 HHT Italian families identifies 76 different mutations (24 novel). Comparison with other European studies. J. Hum. Genet. 2007, 52, 820–829. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Giordano, P.; Nigro, A.; Lenato, G.M.; Guanti, G.; Suppressa, P.; Lastella, P.; De Mattia, D.; Sabba, C. Screening for children from families with Rendu-Osler-Weber disease: From geneticist to clinician. J. Thromb. Haemost. 2006, 4, 1237–1245. [Google Scholar] [CrossRef]
- Ricard, N.; Bidart, M.; Mallet, C.; Lesca, G.; Giraud, S.; Prudent, R.; Feige, J.-J.; Bailly, S. Functional analysis of the BMP9 response of ALK1 mutants from HHT2 patients: A diagnostic tool for novel ACVRL1 mutations. Blood 2010, 116, 1604–1612. [Google Scholar] [CrossRef]
- Olivieri, C.; Mira, E.; Delù, G.; Pagella, F.; Zambelli, A.; Malvezzi, L.; Buscarini, E.; Danesino, C. Identification of 13 new mutations in the ACVRL1 gene in a group of 52 unselected Italian patients affected by hereditary haemorrhagic telangiectasia. J. Med. Genet. 2002, 39, e39. [Google Scholar] [CrossRef]
- Abdalla, S.A.; Cymerman, U.; Rushlow, D.; Chen, N.; Stoeber, G.P.; Lemire, E.G.; Letarte, M. Novel mutations and polymorphisms in genes causing hereditary hemorrhagic telangiectasia. Hum. Mutat. 2005, 25, 320–321. [Google Scholar] [CrossRef]
- Kuehl, H.K.; Caselitz, M.; Hasenkamp, S.; Wagner, S.; El-Harith, H.A.; Manns, M.P.; Stuhrmann, M. Hepatic manifestation is associated with ALK1 in hereditary hemorrhagic telangiectasia: Identification of five novel ALK1 and one novel ENG mutations. Hum. Mutat. 2005, 25, 320. [Google Scholar] [CrossRef]
- Ha, M.; Kim, Y.J.; Kwon, K.A.; Hahm, K.B.; Kim, M.J.; Kim, D.K.; Lee, Y.J.; Oh, S.P. Gastric angiodysplasia in a hereditary hemorrhagic telangiectasia type 2 patient. World J. Gastroenterol. 2012, 18, 1840–1844. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Yang, Q.-H.; Liu, D.; Liu, Q.-Q.; Eyries, M.; Wen, L.; Jiang, X.; Yuan, P.; Zhang, R.; Soubrier, F.; et al. Clinical and genetic characteristics of Chinese patients with hereditary haemorrhagic telangiectasia-associated pulmonary hypertension. Eur. J. Clin. Investig. 2013, 43, 1016–1024. [Google Scholar] [CrossRef]
- Berg, J.N.; Gallione, C.J.; Stenzel, T.T.; Johnson, D.W.; Allen, W.P.; Schwartz, C.E.; Jackson, C.E.; Porteous, M.E.; Marchuk, D.A. The activin receptor-like kinase 1 gene: Genomic structure and mutations in hereditary hemorrhagic telangiectasia type 2. Am. J. Hum. Genet. 1997, 61, 60–67. [Google Scholar] [CrossRef]
- Schulte, C.; Geisthoff, U.; Lux, A.; Kupka, S.; Zenner, H.P.; Blin, N.; Pfister, M. High frequency of ENG and ALK1/ACVRL1 mutations in German HHT patients. Hum. Mutat. 2005, 25, 595. [Google Scholar] [CrossRef] [PubMed]
- Lenato, G.M.; Lastella, P.; Di Giacomo, M.C.; Resta, N.; Suppressa, P.; Pasculli, G.; Sabbà, C.; Guanti, G. DHPLC-based mutation analysis of ENG and ALK-1 genes in HHT Italian population. Hum. Mutat. 2006, 27, 213–214. [Google Scholar] [CrossRef][Green Version]
- Zhao, Y.; Zhang, Y.; Wang, X.; Zhang, L. Variant analysis in Chinese families with hereditary hemorrhagic telangiectasia. Mol. Genet. Genom. Med. 2019, 7, e893. [Google Scholar] [CrossRef]
- Major, T.; Gindele, R.; Szabó, Z.; Jóni, N.; Kis, Z.; Bora, L.; Bárdossy, P.; Rácz, T.; Karosi, T.; Bereczky, Z. A herediter haemorrhagiás teleangiectasia (Osler–Weber–Rendu-kór) genetikai diagnosztikája. Orv. Hetil. 2019, 160, 710–719. [Google Scholar] [CrossRef]
- Abdalla, S.A.; Cymerman, U.; Johnson, R.M.; Deber, C.M.; Letarte, M. Disease-associated mutations in conserved residues of ALK-1 kinase domain. Eur. J. Hum. Genet. 2003, 11, 279–287. [Google Scholar] [CrossRef]
- Brusgaard, K.; Kjeldsen, A.; Poulsen, L.; Moss, H.; Vase, P.; Rasmussen, K.; Kruse, T.A.; Hørder, M. Mutations in endoglin and in activin receptor-like kinase 1 among Danish patients with hereditary haemorrhagic telangiectasia. Clin. Genet. 2004, 66, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Komiyama, M.; Ishiguro, T.; Yamada, O.; Morisaki, H.; Morisaki, T. Hereditary hemorrhagic telangiectasia in Japanese patients. J. Hum. Genet. 2014, 59, 37–41. [Google Scholar] [CrossRef]
- Major, T.; Gindele, R.; Szabó, Z.; Alef, T.; Thiele, B.; Bora, L.; Kis, Z.; Bárdossy, P.; Rácz, T.; Havacs, I.; et al. Evidence for the founder effect of a novel ACVRL1 splice-site mutation in Hungarian hereditary hemorrhagic telangiectasia families. Clin. Genet. 2016, 90, 466–467. [Google Scholar] [CrossRef]
- Major, T.; Gindele, R.; Szabó, Z.; Kis, Z.; Bora, L.; Jóni, N.; Bárdossy, P.; Rácz, T.; Bereczky, Z. The Stratified Population Screening of Hereditary Hemorrhagic Telangiectasia. Pathol. Oncol. Res. 2020, 26, 2783–2788. [Google Scholar] [CrossRef]
- Heimdal, K.; Dalhus, B.; Rødningen, O.K.; Kroken, M.; Eiklid, K.; Dheyauldeen, S.; Røysland, T.; Andersen, R.; Kulseth, M.A. Mutation analysis in Norwegian families with hereditary hemorrhagic telangiectasia: Founder mutations inACVRL1. Clin. Genet. 2015, 89, 182–186. [Google Scholar] [CrossRef]
- Bossler, A.D.; Richards, J.; George, C.; Godmilow, L.; Ganguly, A. Novel mutations in ENG and ACVRL1 identified in a series of 200 individuals undergoing clinical genetic testing for hereditary hemorrhagic telangiectasia (HHT): Correlation of genotype with phenotype. Hum. Mutat. 2006, 27, 667–675. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.E.; Miller, F.J.; Hallam, S.E.; Nelson, L.; Marchuk, D.A.; Ward, K.J. Clinical manifestations in a large hereditary hemorrhagic telangiectasia (HHT) type 2 kindred. Am. J. Med. Genet. 2000, 93, 320–327. [Google Scholar] [CrossRef]
- Abdalla, S.A.; Geisthoff, U.W.; Bonneau, D.; Plauchu, H.; McDonald, J.; Kennedy, S.; Faughnan, M.E.; Letarte, M. Visceral manifestations in hereditary haemorrhagictelangiectasia type 2. J. Med. Genet. 2003, 40, 494–502. [Google Scholar] [CrossRef]
- Letteboer, T.G.W.; Zewald, R.A.; Kamping, E.J.; de Haas, G.; Mager, J.J.; Snijder, R.J.; Lindhout, D.; Hennekam, F.A.M.; Westermann, C.J.J.; Ploos van Amstel, J.K. Hereditary hemorrhagic telangiectasia: ENG and ALK-1 mutations in Dutch patients. Hum. Genet. 2004, 116, 8–16. [Google Scholar] [CrossRef]
- Smoot, L.B.; Obler, D.; McElhinney, D.B.; Boardman, K.; Wu, B.-L.; Lip, V.; Mullen, M.P. Clinical features of pulmonary arterial hypertension in young people with an ALK1 mutation and hereditary haemorrhagic telangiectasia. Arch. Dis. Child. 2009, 94, 506–511. [Google Scholar] [CrossRef]
- Bayrak-Toydemir, P.; McDonald, J.; Markewitz, B.; Lewin, S.; Miller, F.; Chou, L.; Gedge, F.; Tang, W.; Coon, H.; Mao, R. Genotype-phenotype correlation in hereditary hemorrhagic telangiectasia: Mutations and manifestations. Am. J. Med. Genet. 2006, 140, 463–470. [Google Scholar] [CrossRef]
- Kjeldsen, A.D.; Brusgaard, K.; Poulsen, L.; Kruse, T.; Rasmussen, K.; Green, A.; Vase, P. Mutations in theALK-1 gene and the phenotype of hereditary hemorrhagic telangiectasia in two large Danish families. Am. J. Med. Genet. 2001, 98, 298–302. [Google Scholar] [CrossRef]
- Harrison, R.E.; Flanagan, J.A.; Sankelo, M.; Abdalla, S.A.; Rowell, J.; Machado, R.D.; Elliott, C.G.; Robbins, I.M.; Olschewski, H.; McLaughlin, V.; et al. Molecular and functional analysis identifies ALK-1 as the predominant cause of pulmonary hypertension related to hereditary haemorrhagic telangiectasia. J. Med. Genet. 2003, 40, 865–871. [Google Scholar] [CrossRef]
- Sanz-Rodriguez, F.; Fernandez-L, A.; Zarrabeitia, R.; Perez-Molino, A.; Ramírez, J.R.; Coto, E.; Bernabeu, C.; Botella, L.M. Mutation analysis in Spanish patients with hereditary hemorrhagic telangiectasia: Deficient endoglin up-regulation in activated monocytes. Clin. Chem. 2004, 50, 2003–2011. [Google Scholar] [CrossRef]
- Wehner, L.-E.; Folz, B.; Argyriou, L.; Twelkemeyer, S.; Teske, U.; Geisthoff, U.; Wernerb, J.A.; Engel, W.; Nayernia, K. Mutation analysis in hereditary haemorrhagic telangiectasia in Germany reveals 11 novel ENG and 12 novel ACVRL1/ALK1 mutations. Clin. Genet. 2006, 69, 239–245. [Google Scholar] [CrossRef]
- Gedge, F.; McDonald, J.; Phansalkar, A.; Chou, L.S.; Calderon, F.; Mao, R.; Lyon, E.; Bayrak-Toydemir, P. Clinical and analytical sensitivities in hereditary hemorrhagic telangiectasia testing and a report of de novo mutations. J. Mol. Diagn. 2007, 9, 258–265. [Google Scholar] [CrossRef]
- Fontalba, A.; Fernandez-L, A.; García-Alegria, E.; Albiñana, V.; Garrido-Martin, E.M.; Blanco, F.J.; Zarrabeitia, R.; Perez-Molino, A.; Bernabeu-Herrero, M.E.; Ojeda, M.-L.; et al. Mutation study of Spanish patients with Hereditary Hemorrhagic Telangiectasia. BMC Med. Genet. 2008, 9. [Google Scholar] [CrossRef]
- Trembath, R.C.; Thomson, J.R.; Machado, R.D.; Morgan, N.V.; Atkinson, C.; Winship, I.; Simonneau, G.; Galie, N.; Loyd, J.E.; Humbert, M.; et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N. Engl. J. Med. 2001, 345, 325–334. [Google Scholar] [CrossRef]
- Lin, W.D.; Wu, J.Y.; Hsu, H.B.; Tsai, F.J.; Lee, C.C.; Tsai, C.H. Mutation analysis of a family with hereditary hemorrhagic telangiectasia associated with hepatic arteriovenous malformation. J. Formos. Med. Assoc. 2001, 100, 817–819. [Google Scholar]
- Abdalla, S.A.; Gallione, C.J.; Barst, R.J.; Horn, E.M.; Knowles, J.A.; Marchuk, D.A.; Letarte, M.; Morse, J.H. Primary pulmonary hypertension in families with hereditary haemorrhagic telangiectasia. Eur. Respir. J. 2004, 23, 373–377. [Google Scholar] [CrossRef]
- Yan, Z.M.; Fan, Z.P.; Du, J.; Hua, H.; Xu, Y.Y.; Wang, S.L. A novel mutation in ALK-1 causes hereditary hemorrhagic telangiectasia type 2. J. Dent. Res. 2006, 85, 705–710. [Google Scholar] [CrossRef]
- Lesca, G.; Burnichon, N.; Raux, G.; Tosi, M.; Pinson, S.; Marion, M.J.; Babin, E.; Gilbert-Dussardier, B.; Rivière, S.; Goizet, C.; et al. French Rendu-Osler Network. Distribution of ENG and ACVRL1 (ALK1) mutations in French HHT patients. Hum. Mutat. 2006, 27, 598. [Google Scholar] [CrossRef]
- Kjeldsen, A.D.; Moller, T.R.; Brusgaard, K.; Vase, P.; Andersen, P.E. Clinical symptoms according to genotype amongst patients with hereditary haemorrhagic telangiectasia. J. Intern. Med. 2005, 258, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Tørring, P.M.; Brusgaard, K.; Ousager, L.B.; Andersen, P.E.; Kjeldsen, A.D. National mutation study among Danish patients with hereditary haemorrhagic telangiectasia. Clin. Genet. 2013, 86, 123–133. [Google Scholar] [CrossRef]
- Pece, N.; Vera, S.; Cymerman, U.; White, R.I., Jr.; Wrana, J.L.; Letarte, M. Mutant endoglin in hereditary hemorrhagic telangiectasia type 1 is transiently expressed intracellularly and is not a dominant negative. J. Clin. Investig. 1997, 100, 2568–2579. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Kim, S.T.; Lee, H.D.; Lee, K.Y.; Seo, J.; Lee, J.B.; Lee, Y.J.; Oh, S.P. Clinical and genetic analyses of three Korean families with hereditary hemorrhagic telangiectasia. BMC Med. Genet. 2011, 12, 130. [Google Scholar] [CrossRef] [PubMed]
- Dakeishi, M.; Shioya, T.; Wada, Y.; Shindo, T.; Otaka, K.; Manabe, M.; Nozaki, J.; Inoue, S.; Koizumi, A. Genetic epidemiology of hereditary hemorrhagic telangiectasia in a local community in the northern part of Japan. Hum. Mutat. 2002, 19, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Shovlin, C.L.; Hughes, J.M.B.; Scott, J.; Seidman, C.E.; Seidman, J.G. Characterization of Endoglin and Identification of Novel Mutations in Hereditary Hemorrhagic Telangiectasia. Am. J. Hum. Genet. 1997, 61, 68–79. [Google Scholar] [CrossRef]
- Wooderchak-Donahue, W.L.; McDonald, J.; O’Fallon, B.; Upton, P.D.; Li, W.; Roman, B.L.; Young, S.; Plant, P.; Fulop, G.T.; Langa, C.; et al. BMP9 Mutations Cause a Vascular-Anomaly Syndrome with Phenotypic Overlap with Hereditary Hemorrhagic Telangiectasia. Am. J. Hum. Genet. 2013, 93, 530–537. [Google Scholar] [CrossRef]
- Hernandez, F.; Huether, R.; Carter, L.; Johnston, T.; Thompson, J.; Gossage, J.R.; Chao, E.; Elliott, A.M. Mutations in RASA1 and GDF2 identified in patients with clinical features of hereditary hemorrhagic telangiectasia. Hum. Genome Var. 2015, 2, 15040. [Google Scholar] [CrossRef]
- Brunet, G.; Lesca, G.; Génin, E.; Dupuis-Girod, S.; Bideau, A.; Plauchu, H. Thirty Years of Research into Rendu-Osler-Weber Disease in France: Historical Demography, Population Genetics and Molecular Biology. Population 2009, 273–291. [Google Scholar] [CrossRef]
- Westermann, C.J.J.; Rosina, A.F.; de Vries, v.; de Coteau, P.A. The prevalence and manifestations of hereditary hemorrhagic telangiectasia in the Afro-Caribbean population of the Netherlands Antilles: A family screening. Am. J. Med. Genet. 2003, 116, 324–328. [Google Scholar] [CrossRef]
- Canzonieri, C.; Ornati, F.; Matti, E.; Chu, F.; Manfredi, G.; Olivieri, C.; Buscarini, E.; Pagella, F. Hereditary haemorrhagic telangiectasia in North African and sub-Saharan patients. S. Afr. Med. J. 2014, 104, 256–257. [Google Scholar] [CrossRef] [PubMed]
- Hirano, K.; Yamashita, S.; Nakajima, N.; Arai, T.; Maruyama, T.; Yoshida, Y.; Ishigami, M.; Sakai, N.; Kameda-Takemura, K.; Matsuzawa, Y. Genetic cholesteryl ester transfer protein deficiency is extremely frequent in the Omagari area of Japan. Marked hyperalphalipoproteinemia caused by CETP gene mutation is not associated with longevity. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1053–1059. [Google Scholar] [CrossRef]
- Heutink, P.; Haitjema, T.; Breedveld, G.J.; Janssen, B.; Sandkuijl, L.A.; Bontekoe, C.J.; Westerman, C.J.; Oostra, B.A. Linkage of hereditary haemorrhagic telangiectasia to chromosome 9q34 and evidence for locus heterogeneity. J. Med. Genet. 1994, 31, 933–936. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Laplante, B. From France to the Church: The Generalization of Parish Registers in the Catholic Countries. J. Fam. Hist. 2018, 44, 24–51. [Google Scholar] [CrossRef]
- Donaldson, J.W.; McKeever, T.M.; Hall, I.P.; Hubbard, R.B.; Fogarty, A.W. Complications and mortality in hereditary hemorrhagic telangiectasia: A population-based study. Neurology 2015, 84, 1886–1893. [Google Scholar] [CrossRef]
- de Gussem, E.M.; Edwards, C.P.; Hosman, A.E.; Westermann, C.J.; Snijder, R.J.; Faughnan, M.E.; Mager, J.J. Life expextancy of parents with Hereditary Haemorrhagic Telangiectasia. Orphanet J. Rare Dis. 2016, 22, 11–46. [Google Scholar] [CrossRef]
- Brakensiek, K.; Frye-Boukhriss, H.; Mälzer, M.; Abramowicz, M.; Bahr, M.J.; von Beckerath, N.; Bergmann, C.; Caselitz, M.; Holinski-Feder, E.; Muschke, P.; et al. Detection of a significant association between mutations in the ACVRL1 gene and hepatic involvement in German patients with hereditary haemorrhagic telangiectasia. Clin. Genet. 2008, 74, 171–777. [Google Scholar] [CrossRef]
- Faughnan, M.E.; Palda, V.A.; Garcia-Tsao, G.; Geisthoff, U.W.; McDonald, J.; Proctor, D.D.; Spears, J.; Brown, D.H.; Buscarini, E.; Chesnutt, M.S.; et al. HHT Foundation International—Guidelines Working Group. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J. Med. Genet. 2011, 48, 73–87. [Google Scholar] [CrossRef]
- Faughnan, M.E.; Mager, J.J.; Hetts, S.W.; Palda, V.A.; Lang-Robertson, K.; Buscarini, E.; Deslandres, E.; Kasthuri, R.S.; Lausman, A.; Poetker, D.; et al. Second International Guidelines for the Diagnosis and Management of Hereditary Hemorrhagic Telangiectasia. Ann. Intern. Med. 2020, 15, 989–1001. [Google Scholar] [CrossRef]
- Bernabeu, C.; Bayrak-Toydemir, P.; McDonald, J.; Letarte, M. Potential Second-Hits in Hereditary Hemorrhagic Telangiectasia. J. Clin. Med. 2020, 9, 3571. [Google Scholar] [CrossRef]
- Nishida, T.; Faughnan, M.E.; Krings, T.; Chakinala, M.; Gossage, J.R.; Young, W.L.; Kim, H.; Pourmohamad, T.; Henderson, K.J.; Schrum, S.D.; et al. Brain arteriovenous malformations associated with hereditary hemorrhagic telangiectasia: Gene-phenotype correlations. Am. J. Med. Genet. 2012, 158A, 2829–2834. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Grade | Description |
|---|---|
| I | Identical mutation in apparently unrelated families in a HHT center |
| II | Shared area of ancestry/residence (2–4) OR Shared core haplotype (2–4) OR Genealogical evidence of common ancestry (2–4) |
| III | Shared area of ancestry/residence (≥5) OR Shared core haplotype (≥5) OR Genealogical evidence of common ancestry (≥5) OR shared core haplotype (2–4) AND shared area of ancestry/residence (2–4) |
| IV | Grade 3 AND a locally still-prevalent mutation |
| Location | ACVRL1 Variant | Type | Classi- Fication | Population | No. of Families | Founder Grade | Comment | Reference | Independent Reference |
|---|---|---|---|---|---|---|---|---|---|
| Ex 3 | c.152G>A, p.Cys51Tyr | M | P | Italian (Pavia–-Crema Center) | 2 | II | Shared area of ancestry | [29] | [31,32] |
| c.199C>T, p.Arg67Trp | M | P | Italian (Pavia–Crema Center) | 2 | III | Shared haplotype AND area of ancestry | [33] | [34,35,36,37] | |
| 2 | II | Shared area of ancestry | [29] | ||||||
| German | 2 | I | Recurrent | [35] | |||||
| c.200G>A, p.Arg67Gln | M | P | French and Italian | 3 | II | Shared haplotype 1 | [25] | [38,39] | |
| French | 2 | II | Shared haplotype 2 | [25] | |||||
| Italian (Pavia–Crema Center) | 3 | II | Shared area of ancestry | [29] | |||||
| Italian (Bari Center) | 4 | I | Recurrent | [31] | |||||
| 3 | I | Recurrent | [40] | ||||||
| Han Chinese | 2 | I | Recurrent | [41] | |||||
| c.205_209dupTGCGG p.Asn71Alafs*53 | FS | P | Italian | 2 | II | Shared area of ancestry | [29] | ||
| c.265 T>C, p.Cys89Arg | M | LP | Hungarian (Nógrád County) | 3 | II 2 | Genealogy | [42] | ||
| 289-294delCACAAC p.His97_Asn98del | D | LP 1 | Italian (Pavia–Crema Center) | 2 | III | Shared haplotype AND area of ancestry | [33] | ||
| Italian (Bergamo County) | 10 | III-IV 2 | Shared area of residence. Prevalent? 4 | [29] | |||||
| Ex 4 | c.430C>T, p.Arg144* | N | P | French | 2 | II | Shared haplotype | [24] | [31,41,43,44,45] |
| French and Italian | 7 | III 2 | Shared haplotype. Age estimate: 22 gen | [25] | |||||
| Italian | 4 | I | Recurrent | [29] | |||||
| Int 5 | c.625+1G>C | SS | P | Hungarian (Heves County) | 7 | IV 2 | Shared area of residence, shared haplotype, genealogy, prevalent 4 | [46,47] | |
| Ex 6 | c.651G>A, p.Trp217* | N | P | Norwegian (Østfold County and West Sweden) | 7 | IV 2 | Shared area of ancestry, shared haplotype, prevalent 4 | [48] | [49] |
| Ex 7 | c.830C>A, p.Thr277Lys | M | P | Norwegian (Rana, Nordland County) | 13 | IV 2 | Shared area of ancestry, shared haplotype, prevalent 4 | [48] | |
| c.924C>A, p.Cys308* | N | P | Italian (Pavia–Crema Center) | 2 | II | Shared area of ancestry | [29] | [38,40] | |
| c.998G>T, p.Ser333Ile | M | P | American (Utah, US) | 5 | III-IV | Genealogy. Prevalent? 4 | [50] | [40] | |
| American (Toronto Center) | +1 | Area of ancestry in Utah, US4 | [43,51] | ||||||
| c.1042delG, p.Asp348Thrfs*6 | FS | P | Dutch | 7 | III 2 | Genealogy in 5 families 4 | [52] | ||
| Ex 8 | c.1055C>A, p.Ala352Asp | M | P | American (Massachusetts, US) | 2 | II 2 | Shared haplotype | [53] | |
| Location | ACVRL1 Variant | Type | Classi- Fication | Population | No. of Families | Founder Grade | Comment | Reference | Independent Reference |
| Ex 8 | c.1112_1113dupG p.Thr372Hisfs*20 | FS | P | French (Valserine Valley, Jura County) | 17 | IV 2 | Shared area of ancestry/residence, shared haplotype, prevalent 4 | [24] | |
| French | 35 | IV 2 | Shared area of ancestry/residence, shared haplotype, prevalent. Age estimate: 13 gen 4 | [25] | |||||
| European and North American | +1? | Area of ancestry in the Rhône-Alpes region, France 4 | [51] | ||||||
| American (Utah, US) | +1 | Area of ancestry in Ain, France 4 | [54] | ||||||
| c.1120C>T, p.Arg374Trp | M | P | French and Italian | 6 | III 2 | Shared haplotype. Age estimate: 11 gen 4 | [25] | [35,40,55,56,57,58] | |
| American (Ontario, Canada) | 2 | I | Recurrent | [34,43] | |||||
| Dutch | 3 | I | Recurrent | [52] | |||||
| American | 2 | I | Recurrent | [54] | |||||
| Italian (Bari Center) | 2 | I | Recurrent | [38] | |||||
| Italian (Pavia–Crema Center) | 3 | II | Shared area of ancestry | [29] | |||||
| Han Chinese | 2 | I 3 | Recurrent | [39] | |||||
| c.1121G>A, p.Arg374Gln | M | P | French (Deux-Sevres County) | 3 | III 2 | Shared haplotype 1 AND area of ancestry 4 | [25] | [29,37,56,59,60] | |
| European and North American | +3? | II | Shared area of ancestry in Parthenay, Deux-Sevres County, France. Age estimate: 4 gen 4 | [51] | |||||
| French (Northeast France) | 3 | III 2 | Shared haplotype 2 AND area of ancestry 4 | [25] | |||||
| c.1126A>G, p.Met376Val | M | P | French | 3 | II | Shared haplotype | [25] | [59] | |
| c.1199C>A, p.Ala400Asp | M | P | Italian (Pavia–Crema Center) | 2 | II | Shared area of ancestry | [29] | ||
| c.1231C>T, p.Arg411Trp | M | P | French | 7 | III 2 | Shared haplotype | [24] | [35,61] | |
| 9 | III 2 | Shared haplotype. Age estimate: 15 gen | [25] | ||||||
| American (Ontario, Canada) | 2 | I | Recurrent | [34] | |||||
| Dutch | 2 | I | Recurrent | [52] | |||||
| German | 3 | I | Recurrent | [41] | |||||
| c.1232G>A, p.Arg411Gln | M | P | North American | 2 | II | Shared area of ancestry | [51] | [38,40,56,60,62] | |
| French | 2 | II | Shared haplotype | [25] | |||||
| American (Utah, US) | 2 | I | Recurrent | [54] | |||||
| Italian (Pavia–Crema Center) | 2 | II | Shared area of ancestry | [29] | |||||
| Han Chinese | 3 | I 3 | Recurrent | [39] | |||||
| c.1232G>C, p.Arg411Pro | M | P | French | 2 | II | Shared haplotype | [24] | [59] | |
| Location | ACVRL1 Variant | Type | Classi- Fication | Population | No. of Families | Founder Grade | Comment | Reference | Independent Reference |
| Ex 9 | c.1280A>T, p.Asp427Val | M | P 1 | French | 2 | II | Shared haplotype | [25] | [38] |
| Int 9 | c.1377+2T>A | SS | LP | Hungarian (Heves and Borsod Counties) | 2 | II | Genealogy | [42] | |
| Ex 10 | c.1435C>T, p.Arg479* | N | P | French | 2 | II | Shared haplotype | [24] | [29,37,38,52,59,60,63,64] |
| 2 | II | Shared haplotype | [25] | ||||||
| Japanese (West Japan) | 2 | I | Recurrent | [45] | |||||
| c.1450C>T, p.Arg484Trp | M | P | Norwegian (Nordland County) | 5 | III-IV 2 | Shared haplotype. Prevalent? 4 | [48] | [25,29,52,61] | |
| Italian (Bari Center) | 2 | I | Recurrent | [38] | |||||
| Location | ENG Variant | Type | Classi- Fication | Population | No. of Families | Founder Grade | Comment | Reference | Independent Reference |
| Int 1 | c.67+1G>A | SS | P | Netherlands Antillean | 7 | IV 2 | Shared haplotype, prevalent 4 | [23] | |
| Dutch | +1 | [52] | |||||||
| Ex 3 | c.277C>T, p.Arg93* | N | P | Italian | 3 | II | Shared area of ancestry | [29] | [44,52,59,65] |
| Norwegian (Southeast) | 5 | I | Recurrent. Haplotype analysis showed different haplotypes 4 | [48] | |||||
| c.360C>A, p.Tyr120* | N | P | Danish (Funen County) | 7 | IV 2 | shared haplotype, shared area of residence, prevalent. Age estimate: 13–14 gen 4 | [44] | ||
| Danish (Funen county) | 7 | IV 2 | Shared area of residence, prevalent 4 | [66] | |||||
| Danish (Nationwide) | 13 | IV 2 | Shared area of residence, prevalent 4 | [67] | |||||
| Int 3 | c.360+1G>A | SS | P | Italian (Pavia–Crema Center) | 2 | II | Shared area of ancestry | [29] | [41,52,54,68,69] |
| Ex 6 | c.781T>C, p.Trp261Arg | M | P 1 | Dutch | 8 | II | Genealogy in 3 families 4 | [52] | |
| Int 6 | c.817-2 A>C | SS | P | Hungarian (Heves and Borsod Counties) | 2 | II 2 | Shared area of ancestry, genealogy 4 | [42] | |
| Ex 7 | c.828_829insA, p.Tyr277Ilefs*57 | FS | P | Japanese (Akita, County A, Japan) | 2 | III-IV 2 | Shared haplotype AND area of residence. Prevalent? 4 | [70] | |
| Int 7 | c.1134+1G>A | SS | P | English (South England) | 2 | III | Shared haplotype AND area of residence | [71] | |
| Ex 9 | c.1238G>T, p.Gly413Val | M | P 1 | Netherlands Antillean and Dutch | 3 | III 2 | Shared haplotype AND area of ancestry 4 | [23] | |
| Dutch | +1? | [52] | |||||||
| Int 10 | c.1311G>A, p.Arg437Arg | SS | VUS 1 | Dutch | 5 | III 2 | Genealogy 4 | [52] | |
| Ex 12 | c.1630delA, p.Thr544Profs*8 | FS | P | American (Ontario, Canada) | 2 | II | Shared area of ancestry | [34] | |
| Int 12 | c.1686+5G>C | SS | LP 1 | Spanish | 3 | I 3 | Recurrent | [60] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Major, T.; Gindele, R.; Balogh, G.; Bárdossy, P.; Bereczky, Z. Founder Effects in Hereditary Hemorrhagic Telangiectasia. J. Clin. Med. 2021, 10, 1682. https://doi.org/10.3390/jcm10081682
Major T, Gindele R, Balogh G, Bárdossy P, Bereczky Z. Founder Effects in Hereditary Hemorrhagic Telangiectasia. Journal of Clinical Medicine. 2021; 10(8):1682. https://doi.org/10.3390/jcm10081682
Chicago/Turabian StyleMajor, Tamás, Réka Gindele, Gábor Balogh, Péter Bárdossy, and Zsuzsanna Bereczky. 2021. "Founder Effects in Hereditary Hemorrhagic Telangiectasia" Journal of Clinical Medicine 10, no. 8: 1682. https://doi.org/10.3390/jcm10081682
APA StyleMajor, T., Gindele, R., Balogh, G., Bárdossy, P., & Bereczky, Z. (2021). Founder Effects in Hereditary Hemorrhagic Telangiectasia. Journal of Clinical Medicine, 10(8), 1682. https://doi.org/10.3390/jcm10081682