
Brain Glucose Metabolism in Health, Obesity, and Cognitive Decline—Does Insulin Have Anything to Do with It? A Narrative Review



Abstract
1. Introduction
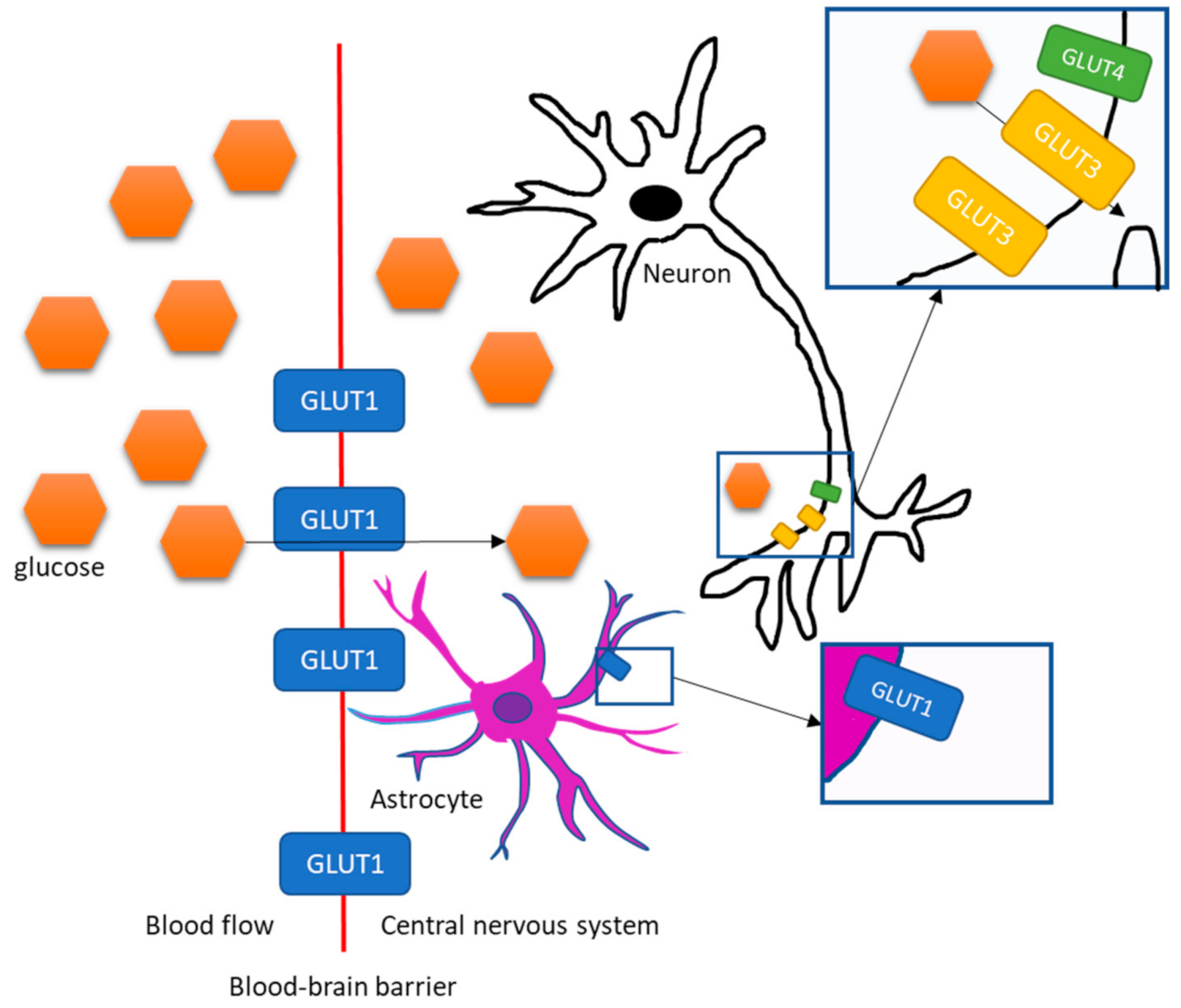
2. Transportation of Glucose and Insulin into the Brain
3. Central Insulin Actions
4. Brain Glucose Metabolism in Alzheimer’s Disease, Mild Cognitive Impairment (MCI), Insulin Resistance and Obesity in the Fasting State Measured with [18F]-FDG-PET
4.1. Alzheimer’s Disease
4.2. Mild Cognitive Impairment
4.3. Systemic Insulin Resistance
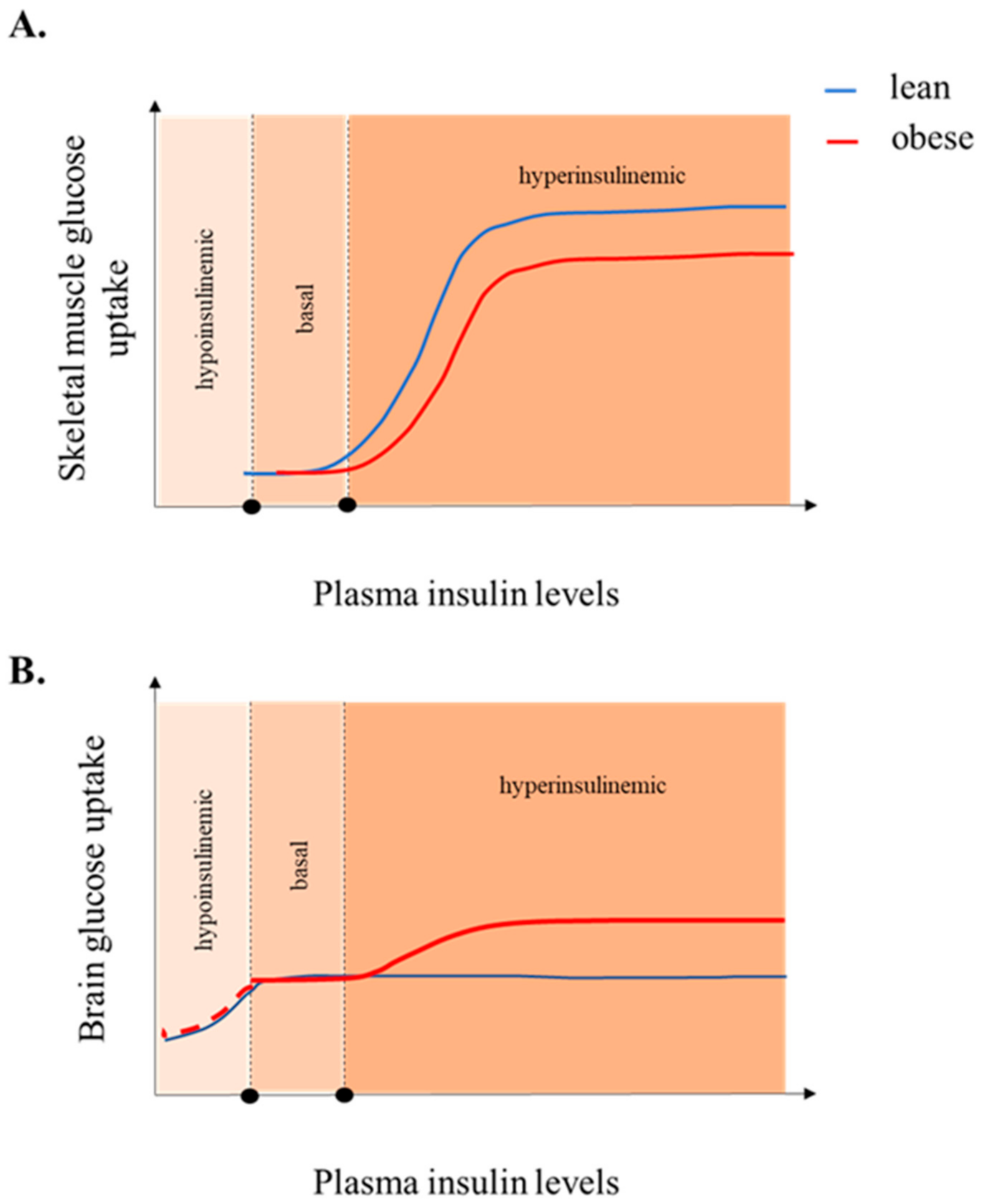
4.4. Obesity

{kind=link}
{kind=link}
| N | Age (Years) | Subjects’ Status | BGU | Method Used | Reference | |
|---|---|---|---|---|---|---|
| fasting | ||||||
| AD § | 548 | 50–85 | 199 AD, 114 MCI, 98 FTD, 27 DLB, 110 controls | decreased | 3-D Z scores | Mosconi et al. [71] |
| MCI | 96 | 69 ± 4.6 | 8 MCI, 66 AD, 22 controls | decreased | count ratio | Minoshima et al. [80] |
| MCI | 47 | 73 ± 8.1 | 17 MCI, 17 AD, 13 controls | decreased | SUV ratio | Bailly et al. [83] |
| MCI | 27 | 66 ± 10 | 10 MCI, 9 AD, 8 controls | increased/decreased | quantification * | Ashraf et al. [77] |
| MCI | 63 | 76.9 ± 5.8 | 20 MCI, 19 AD, 24 controls | increased/decreased | quantification * | Croteau et al. [78] |
| Aging | 205 | 20–82 | cognitively normal adults | decreased | count ratio | Goyal et al. [96] |
| Obesity | 29 168 222 | 45 ± 9 74 ± 6 74 ± 6 | cognitively unimpaired cognitively unimpaired cognitively unimpaired | unchanged increased increased | quantification * SUV ratio SPM T-ratio | Tuulari et al. [22] Pegueroles et al. [91] Sala et al. [92] |
| IR | 29 150 | 74 ± 7 61 ± 6 | cognitively normal cognitively normal with family history of AD | decreased decreased | counts count ratio | Baker et al. [18] Willette et al. [89] |
| euglycemic insulin clamp | ||||||
| AD | - | - | - | - | - | - |
| MCI | - | - | - | - | - | - |
| Aging | 194 | 20–80 | cognitively unimpaired | decreased | quantification * | Rebelos et al. [21] |
| Obesity | 34 | 45 ± 9 | obese and lean | increased | quantification * | Tuulari et al. [22] |
| IR | 194 | 20–80 | cognitively unimpaired | increased | quantification * | Rebelos et al. [21] |
5. Insulin-Stimulated BGU
6. Attempts to Define Central Insulin Resistance
7. Increased Brain Glucose Uptake and Brain Insulin Resistance, Two Sides of the Same Coin?
8. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Kelly, T.; Yang, W.; Chen, C.-S.; Reynolds, K.; He, J. Global burden of obesity in 2005 and projections to 2030. Int. J. Obes. 2008, 32, 1431–1437. [Google Scholar] [CrossRef] [PubMed]
- Whiting, D.R.; Guariguata, L.; Weil, C.; Shaw, J. IDF diabetes atlas: Global estimates of the prevalence of diabetes for 2011 and 2030. Diabetes Res. Clin. Pract. 2011, 94, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Ott, A.; Stolk, R.P.; van Harskamp, F.; Pols, H.A.; Hofman, A.; Breteler, M.M. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 1999, 53, 1937–1942. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.J.; Raji, C.A.; Becker, J.T.; Lopez, O.L.; Huller, L.H.; Hua, X.; Lee, S.; Hibar, D.; Dinov, I.D.; Stein, J.L.; et al. Obesity is linked with lower brain volume in 700 AD and MCI patients. Neurobiol. Aging 2010, 31, 1326–1339. [Google Scholar] [CrossRef] [PubMed]
- Reaven, G.M. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 1988, 37, 1595–1607. [Google Scholar] [CrossRef] [PubMed]
- Talbot, K.; Wang, H.-Y.; Kazi, H.; Han, L.-Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef]
- Ekblad, L.L.; Rinne, J.O.; Puukka, P.; Laine, H.; Ahtiluoto, S.; Sulkava, R.; Viitanen, M.; Jula, A. Insulin resistance predicts cognitive decline: An 11-year follow-up of a nationally representative adult population sample. Diabetes Care 2017, 40, 751–758. [Google Scholar] [CrossRef]
- Kuusisto, J.; Koivisto, K.; Mykkänen, L.; Helkala, E.L.; Vanhanen, M.; Hanninen, T.; Pyorala, K.; Riekkinen, P.; Laakso, M. Essential hypertension and cognitive function. The role of hyperinsulinemia. Hypertension 1993, 22, 771–779. [Google Scholar] [CrossRef]
- Rönnemaa, E.; Zethelius, B.; Sundelöf, J.; Sundstrom, J.; Degerman-Gunnanrosn, M.; Berne, C.; Lannfelt, L.; KIlander, L. Impaired insulin secretion increases the risk of Alzheimer disease. Neurology 2008, 71, 1065–1071. [Google Scholar] [CrossRef]
- Peila, R.; Rodriguez, B.L.; White, L.R.; Launer, L.J. Fasting insulin and incident dementia in an elderly population of Japanese-American men. Neurology 2004, 63, 228–233. [Google Scholar] [CrossRef]
- Mehlig, K.; Lapidus, L.; Thelle, D.S.; Waern, M.; Zetterberg, H.; Bjorkelund, C.; Skoog, I.; Lissner, L. Low fasting serum insulin and dementia in nondiabetic women followed for 34 years. Neurology 2018, 91, e427–e435. [Google Scholar] [CrossRef]
- Latva-Rasku, A.; Honka, M.-J.; Stančáková, A.; Koistinen, H.A.; Kuusisto, J.; Guan, L.; Manning, A.K.; Stringham, H.; Gloyn, A.L.; Lindgren, C.M.; et al. A partial loss-of-function variant in akt2 is associated with reduced insulin-mediated glucose uptake in multiple insulin-sensitive tissues: A genotype-based callback positron emission tomography study. Diabetes 2018, 67, 334–342. [Google Scholar] [CrossRef]
- Rebelos, E.; Nummenmaa, L.; Dadson, P.; Latva-Rasku, A.; Nuutila, P. Brain insulin sensitivity is linked to body fat distribution-the positron emission tomography perspective. Eur. J. Nucl. Med. Mol. Imaging 2020. [Google Scholar] [CrossRef]
- Rebelos, E.; Immonen, H.; Bucci, M.; Hannukainen, J.C.; Nummenmaa, L.; Honka, M.-J.; Soinio, M.; Salminen, P.; Ferrannini, E.; Iozzo, P.; et al. Brain glucose uptake is associated with endogenous glucose production in obese patients before and after bariatric surgery and predicts metabolic outcome at follow-up. Diabetes Obes. Metab. 2019, 21, 218–226. [Google Scholar] [CrossRef]
- Kullmann, S.; Valenta, V.; Wagner, R.; Tschritter, O.; Machann, J.; Haring, H.-U.; Preissl, H.; Fritsche, A.; Heni, M. Brain insulin sensitivity is linked to adiposity and body fat distribution. Nat. Commun. 2020, 11, 1841. [Google Scholar] [CrossRef]
- Willette, A.A.; Modanlo, N.; Kapogiannis, D. Insulin resistance predicts medial temporal hypermetabolism in mild cognitive impairment conversion to Alzheimer disease. Diabetes 2015, 64, 1933–1940. [Google Scholar] [CrossRef]
- Willette, A.A.; Johnson, S.C.; Birdsill, A.C.; Sager, M.A.; Christian, B.; Baker, L.D.; Craft, S.; Oh, J.; Statz, E.; Hermann, B.P.; et al. Insulin resistance predicts brain amyloid deposition in late middle-aged adults. Alzheimers Dement. 2015, 11, 504–510. [Google Scholar] [CrossRef]
- Baker, L.D.; Cross, D.J.; Minoshima, S.; Belongia, D.; Watson, G.S.; Craft, S. Insulin resistance and Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Arch. Neurol. 2011, 68, 51–57. [Google Scholar] [CrossRef]
- Ekblad, L.L.; Johansson, J.; Helin, S.; Viitanen, M.; Laine, H.; Puukka, P.; Jula, A.; Rinne, J.O. Midlife insulin resistance, APOE genotype, and late-life brain amyloid accumulation. Neurology 2018, 90, e1150–e1157. [Google Scholar] [CrossRef]
- Craft, S.; Raman, R.; Chow, T.W.; Rafii, M.S.; Sun, C.-K.; Rissman, R.A.; Donohue, M.C.; Brewer, J.B.; Jenkins, C.; Harless, K.; et al. Safety, efficacy, and feasibility of intranasal insulin for the treatment of mild cognitive impairment and Alzheimer Disease Dementia: A randomized clinical trial. JAMA Neurol. 2020, 77, 1099–1109. [Google Scholar] [CrossRef]
- Rebelos, E.; Bucci, M.; Karjalainen, T.; Oikonen, V.; Bertoldo, A.; Hannukainen, J.C.; Virtanen, K.A.; Latva-Rasku, A.; Hirvonen, J.; Heinonen, I.; et al. Insulin resistance is associated with enhanced brain glucose uptake during euglycemic hyperinsulinemia: A large-scale PET cohort. Diabetes Care 2021, 44, 1–7. [Google Scholar] [CrossRef]
- Tuulari, J.J.; Karlsson, H.K.; Hirvonen, J.; Hannukainen, J.C.; Bucci, M.; Helmio, M.; Ovaska, J.; Soinio, M.; Salminen, P.; Savisto, N.; et al. Weight loss after bariatric surgery reverses insulin-induced increases in brain glucose metabolism of the morbidly obese. Diabetes 2013, 62, 2747–2751. [Google Scholar] [CrossRef]
- Benarroch, E.E. Brain glucose transporters: Implications for neurologic disease. Neurology 2014, 82, 1374–1379. [Google Scholar] [CrossRef]
- Deng, D.; Yan, N. GLUT, SGLT, and SWEET: Structural and mechanistic investigations of the glucose transporters. Protein Sci. 2016, 25, 546–558. [Google Scholar] [CrossRef]
- Mink, J.W.; Blumenschine, R.J.; Adams, D.B. Ratio of central nervous system to body metabolism in vertebrates: Its constancy and functional basis. Am. J. Physiol. 1981, 241, R203–R212. [Google Scholar] [CrossRef]
- Clarke, D.W.; Boyd, F.T.J.; Kappy, M.S.; Raizada, M.K. Insulin binds to specific receptors and stimulates 2-deoxy-D-glucose uptake in cultured glial cells from rat brain. J. Biol. Chem. 1984, 259, 11672–11675. [Google Scholar] [CrossRef]
- Huang, S.; Czech, M.P. The GLUT4 glucose transporter. Cell Metab. 2007, 5, 237–252. [Google Scholar] [CrossRef] [PubMed]
- Vannucci, S.J.; Koehler-Stec, E.M.; Li, K.; Reynolds, T.H.; Clark, R.; Simpson, I.A. GLUT4 glucose transporter expression in rodent brain: Effect of diabetes. Brain Res. 1998, 797, 1–11. [Google Scholar] [CrossRef]
- Lin, H.V.; Ren, H.; Samuel, V.T.; Lee, H.-Y.; Lu, T.Y.; Shulman, G.I.; Accili, D. Diabetes in mice with selective impairment of insulin action in Glut4-expressing tissues. Diabetes 2011, 60, 700–709. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Yan, S.; Zhang, B.; Lu, T.Y.; Arancio, O.; Accili, D. Glut4 expression defines an insulin-sensitive hypothalamic neuronal population. Mol. Metab. 2014, 3, 452–459. [Google Scholar] [CrossRef]
- Kotani, K.; Peroni, O.D.; Minokoshi, Y.; Boss, O.; Kahn, B.B. GLUT4 glucose transporter deficiency increases hepatic lipid production and peripheral lipid utilization. J. Clin. Investig. 2004, 114, 1666–1675. [Google Scholar] [CrossRef]
- Lauro, D.; Kido, Y.; Castle, A.L.; Zarnowski, M.J.; Hayashi, H.; Ebina, Y.; Accili, D. Impaired glucose tolerance in mice with a targeted impairment of insulin action in muscle and adipose tissue. Nat. Genet. 1998, 20, 294–298. [Google Scholar] [CrossRef]
- Reno, C.M.; Puente, E.C.; Sheng, Z.; Daphna-Iken, D.; Bree, A.J.; Routh, V.H.; Kahn, B.B.; Fisher, S.J. Brain GLUT4 knockout mice have impaired glucose tolerance, decreased insulin sensitivity, and impaired hypoglycemic counterregulation. Diabetes 2017, 66, 587–597. [Google Scholar] [CrossRef]
- Banks, W.A.; Owen, J.B.; Erickson, M.A. Insulin in the brain: There and back again. Pharmacol. Ther. 2012, 136, 82–93. [Google Scholar] [CrossRef]
- Banks, W.A. The source of cerebral insulin. Eur. J. Pharmacol. 2004, 490, 5–12. [Google Scholar] [CrossRef]
- Csajbók, É.A.; Tamás, G. Cerebral cortex: A target and source of insulin? Diabetologia 2016, 59, 1609–1615. [Google Scholar] [CrossRef]
- Mazucanti, C.H.; Liu, Q.-R.; Lang, D.; Huang, N.; O’Connell, J.F.; Camandola, S.; Egan, J.M. Release of insulin produced by the choroid plexis is regulated by serotonergic signaling. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Nemoto, T.; Toyoshima-Aoyama, F.; Yanagita, T.; Maruta, T.; Fujita, H.; Koshida, T.; Yonaha, T.; Wada, A.; Sawaguchi, A.; Murakami, M. New insights concerning insulin synthesis and its secretion in rat hippocampus and cerebral cortex: Amyloid-β1–42-induced reduction of proinsulin level via glycogen synthase kinase-3β. Cell. Signal. 2014, 26, 253–259. [Google Scholar] [CrossRef]
- Mehran, A.E.; Templeman, N.M.; Brigidi, G.S.; Lim, G.E.; Chu, K.-Y.; Hu, X.; Botezelli, J.D.; Asadi, A.; Hoffman, B.G.; Kieffer, T.J.; et al. Hyperinsulinemia drives diet-induced obesity independently of brain insulin production. Cell Metab. 2012, 16, 723–737. [Google Scholar] [CrossRef]
- Israel, P.A.; Park, C.R.; Schwartz, M.W.; Green, P.K.; Sipols, A.J.; Woods, S.C.; Porte, D., Jr.; Figlewicz, D.P. Effect of diet-induced obesity and experimental hyperinsulinemia on insulin uptake into CSF of the rat. Brain Res. Bull. 1993, 30, 571–575. [Google Scholar] [CrossRef]
- Kaiyala, K.J.; Prigeon, R.L.; Kahn, S.E.; Woods, S.C.; Schwartz, M.W. Obesity induced by a high-fat diet is associated with reduced brain insulin transport in dogs. Diabetes 2000, 49, 1525–1533. [Google Scholar] [CrossRef]
- Banks, W.A.; Jaspan, J.B.; Kastin, A.J. Effect of diabetes mellitus on the permeability of the blood-brain barrier to insulin. Peptides 1997, 18, 1577–1584. [Google Scholar] [CrossRef]
- Urayama, A.; Banks, W.A. Starvation and triglycerides reverse the obesity-induced impairment of insulin transport at the blood-brain barrier. Endocrinology 2008, 149, 3592–3597. [Google Scholar] [CrossRef]
- Schwartz, M.W.; Figlewicz, D.F.; Kahn, S.E.; Baskin, D.G.; Greenwood, M.R.; Porte, D.J. Insulin binding to brain capillaries is reduced in genetically obese, hyperinsulinemic Zucker rats. Peptides 1990, 11, 467–472. [Google Scholar] [CrossRef]
- Fishel, M.A.; Watson, G.S.; Montine, T.J.; Wang, W.; Green, P.S.; Kulstad, J.J.; Cook, D.G.; Peskind, E.R.; Baker, L.D.; Goldgaber, D.; et al. Hyperinsulinemia provokes synchronous increases in central inflammation and β-amyloid in normal adults. Arch. Neurol. 2005, 62, 1539–1544. [Google Scholar] [CrossRef]
- Obici, S.; Zhang, B.B.; Karkanias, G.; Rossetti, L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nat. Med. 2002, 8, 1376–1382. [Google Scholar] [CrossRef]
- Heni, M.; Wagner, R.; Kullmann, S.; Gancheva, S.; Roden, M.; Peter, A.; Stefan, N.; Preissl, H.; Haring, H.-U.; Fritsche, A. Hypothalamic and striatal insulin action suppresses endogenous glucose production and may stimulate glucose uptake during hyperinsulinemia in lean but not in overweight men. Diabetes 2017, 66, 1797–1806. [Google Scholar] [CrossRef]
- Gancheva, S.; Koliaki, C.; Bierwagen, A.; Nowotny, P.; Heni, M.; Fritsche, A.; Haring, H.-U.; Szendroedi, J.; Roden, M. Effects of intranasal insulin on hepatic fat accumulation and energy metabolism in humans. Diabetes 2015, 64, 1966–1975. [Google Scholar] [CrossRef]
- Chen, M.; Woods, S.C.; Porte, D.J. Effect of cerebral intraventricular insulin on pancreatic insulin secretion in the dog. Diabetes 1975, 24, 910–914. [Google Scholar] [CrossRef]
- Rebelos, E.; Mari, A.; Bucci, M.; Honka, M.-J.; Hannukainen, J.C.; Virtanen, K.A.; Hirvonen, J.; Nummenmaa, L.; Heni, M.; Iozzo, P.; et al. Brain substrate metabolism and ß-cell function in humans: A positron emission tomography study. Endocrinol. Diabetes Metab. 2020, 3, e00136. [Google Scholar] [CrossRef]
- Mari, A.; Tura, A.; Gastaldelli, A.; Ferrannini, E. Assessing insulin secretion by modeling in multiple-meal tests: Role of potentiation. Diabetes 2002, 51, S221–S226. [Google Scholar] [CrossRef] [PubMed]
- Rebelos, E.; Hirvonen, J.; Bucci, M.; Pekkarinen, L.; Nyman, M.; Hannukainen, J.C.; Iozzo, P.; Salminen, P.; Nummenmaa, L.; Ferrannini, E.; et al. Brain free fatty acid uptake is elevated in morbid obesity, and is irreversible 6 months after bariatric surgery: A positron emission tomography study. Diabetes Obes. Metab. 2020, 22, 1074–1082. [Google Scholar] [CrossRef] [PubMed]
- Benedict, C.; Brede, S.; Schiöth, H.B.; Lehnert, H.; Schultes, B.; Born, J.; Hallschmid, M. Intranasal insulin enhances postprandial thermogenesis and lowers postprandial serum insulin levels in healthy men. Diabetes 2011, 60, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Kellar, D.; Craft, S. Brain insulin resistance in Alzheimer’s disease and related disorders: Mechanisms and therapeutic approaches. Lancet Neurol. 2020, 19, 758–766. [Google Scholar] [CrossRef]
- de la Monte, S.M. Type 3 diabetes is sporadic Alzheimer’s disease: Mini-review. Eur. Neuropsychopharmacol. 2014, 24, 1954–1960. [Google Scholar] [CrossRef]
- Craft, S.; Cholerton, B.; Baker, L.D. Insulin and Alzheimer’s disease: Untangling the web. J. Alzheimers Dis. 2013, 33, S263–S275. [Google Scholar] [CrossRef]
- Moreira, P.I.; Duarte, A.I.; Santos, M.S.; Rego, A.C.; Oliveira, C.R. An integrative view of the role of oxidative stress, mitochondria and insulin in Alzheimer’s disease. J. Alzheimers Dis. 2009, 16, 741–761. [Google Scholar] [CrossRef]
- Lee, C.-C.; Kuo, Y.-M.; Huang, C.-C.; Hsu, K.-S. Insulin rescues amyloid beta-induced impairment of hippocampal long-term potentiation. Neurobiol. Aging 2009, 30, 377–387. [Google Scholar] [CrossRef]
- Figlewicz, D.P.; Szot, P.; Israel, P.A.; Payne, C.; Dorsa, D.M. Insulin reduces norepinephrine transporter mRNA in vivo in rat locus coeruleus. Brain Res. 1993, 602, 161–164. [Google Scholar] [CrossRef]
- Kopf, S.R.; Baratti, C.M. Effects of posttraining administration of insulin on retention of a habituation response in mice: Participation of a central cholinergic mechanism. Neurobiol. Learn. Mem. 1999, 71, 50–61. [Google Scholar] [CrossRef]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the cholinergic system. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef]
- Gasparini, L.; Gouras, G.K.; Wang, R.; Gross, R.S.; Beal, M.F.; Greengard, P.; Xu, H. Stimulation of beta-amyloid precursor protein trafficking by insulin reduces intraneuronal beta-amyloid and requires mitogen-activated protein kinase signaling. J. Neurosci. 2001, 21, 2561–2570. [Google Scholar] [CrossRef]
- Schubert, M.; Brazil, D.P.; Burks, D.J.; Kushner, J.A.; Ye, J.; Flint, C.L.; Farhand-Fallah, J.; Dikkes, P.; Warot, X.M.; Rio, C.; et al. Insulin receptor substrate-2 deficiency impairs brain growth and promotes tau phosphorylation. J. Neurosci. 2003, 23, 7084–7092. [Google Scholar] [CrossRef]
- Claxton, A.; Baker, L.D.; Hanson, A.; Trittschuh, E.H.; Cholerton, B.; Morgan, A.; Callaghan, M.; Arbuckle, M.; Behl, C.; Craft, S. Long-acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer’s disease dementia. J. Alzheimers Dis. 2015, 44, 897–906. [Google Scholar] [CrossRef]
- Craft, S.; Baker, L.D.; Montine, T.J.; Minoshima, S.; Watson, G.S.; Claxton, A.; Arbuckle, M.; Callaghan, M.; Tsai, E.; Plymate, S.R.; et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: A pilot clinical trial. Arch. Neurol. 2012, 69, 29–38. [Google Scholar] [CrossRef]
- Reger, M.A.; Watson, G.S.; Green, P.S.; Wilkinson, C.W.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Breitner, J.C.S.; DeGroodt, W.; et al. Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology 2008, 70, 440–448. [Google Scholar] [CrossRef]
- Claxton, A.; Baker, L.D.; Wilkinson, C.W.; Trittschuh, E.H.; Chapmana, D.; Watson, G.S.; Cholerton, B.; Plymate, S.R.; Arbuckle, M.; Craft, S. Sex and ApoE genotype differences in treatment response to two doses of intranasal insulin in adults with mild cognitive impairment or Alzheimer’s disease. J. Alzheimers Dis. 2013, 35, 789–797. [Google Scholar] [CrossRef]
- Reivich, M.; Kuhl, D.; Wolf, A.; Greenberg, J.; Phelps, M.; Casella, V.; Ido, T.; Fowler, J.; Hoffman, E.; Alavi, A.; et al. The [18F] fluorodeoxyglucose method for the measurement of local cerebral glucose utilization in man. Circ. Res. 1979, 44, 127–137. [Google Scholar] [CrossRef]
- Garibotto, V.; Herholz, K.; Boccardi, M.; Picco, A.; Varrone, A.; Nordberg, A.; Nobili, F.; Ratib, O.; Geneva Task Force for the Roadmap of Alzheimer’s Biomarkers. Clinical validity of brain fluorodeoxyglucose positron emission tomography as a biomarker for Alzheimer’s disease in the context of a structured 5-phase development framework. Neurobiol. Aging 2017, 52, 183–195. [Google Scholar] [CrossRef]
- Nobili, F.; Arbizu, J.; Bouwman, F.; Drzezga, A.; Agosta, F.; Nestor, P.; Walker, Z.; Boccardi, M.; EANM-EAN Task Force for the Prescription of FDG-PET for Dementing Neurodegenerative Disorders. European Association of Nuclear Medicine and European Academy of Neurology recommendations for the use of brain (18) F-fluorodeoxyglucose positron emission tomography in neurodegenerative cognitive impairment and dementia: Delphi consensus. Eur. J. Neurol. 2018, 25, 1201–1217. [Google Scholar] [CrossRef]
- Varrone, A.; Asenbaum, S.; Vander Borght, T.; Booji, J.; Nobili, F.; Nagren, K.; Darcourt, J.; Kapucu, O.L.; Tatsch, K.; Bartenstein, P.; et al. EANM procedure guidelines for PET brain imaging using [18F] FDG, version 2. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 2103–2110. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, L.; Tsui, W.H.; Rusinek, H.; De Santi, S.; Li, Y.; Wang, G.-J.; Pupi, A.; Fowler, J.; de Leon, M.J. Quantitation, regional vulnerability, and kinetic modeling of brain glucose metabolism in mild Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2007, 34, 1467–1479. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, L.; McHugh, P.F. FDG- and amyloid-PET in Alzheimer’s disease: Is the whole greater than the sum of the parts? Q. J. Nucl. Med. Mol. Imaging 2011, 55, 250–264. [Google Scholar] [PubMed]
- Jack, C.R.J.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Feldman, H.H.; Frisoni, G.B.; Hampel, H.; Jagust, W.J.; Johnson, K.A.; Knopman, D.S.; et al. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016, 87, 539–547. [Google Scholar] [CrossRef]
- Zimmer, E.R.; Parent, M.J.; Souza, D.G.; Leuzy, A.; Lecrux, C.; Kim, H.-I.; Gauthier, S.; Pellerin, L.; Hamel, E.; Rosa-Neto, P. [(18)F] FDG PET signal is driven by astroglial glutamate transport. Nat. Neurosci. 2017, 20, 393–395. [Google Scholar] [CrossRef]
- Mächler, P.; Wyss, M.T.; Elsayed, M.; Stobart, J.; Gutierrez, R.; von Faber-Castell, A.; Kaelin, V.; Zuend, M.; San Martin, A.; Romero-Gomez, I.; et al. In vivo evidence for a lactate gradient from astrocytes to neurons. Cell Metab. 2016, 23, 94–102. [Google Scholar] [CrossRef]
- Simpson, I.A.; Chundu, K.R.; Davies-Hill, T.; Honer, W.G.; Davies, P. Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with Alzheimer’s disease. Ann. Neurol. 1994, 35, 546–551. [Google Scholar] [CrossRef]
- Ashraf, A.; Fan, Z.; Brooks, D.J.; Edison, P. Cortical hypermetabolism in MCI subjects: A compensatory mechanism? Eur. J. Nucl. Med. Mol. Imaging 2015, 42, 447–458. [Google Scholar] [CrossRef]
- Croteau, E.; Castellano, C.A.; Fortier, M.; Bocti, C.; Fulop, T.; Paquet, N.; Cunnane, S.C. A cross-sectional comparison of brain glucose and ketone metabolism in cognitively healthy older adults, mild cognitive impairment and early Alzheimer’s disease. Exp. Gerontol. 2018, 107, 18–26. [Google Scholar] [CrossRef]
- Mosconi, L.; Tsui, W.-H.; De Santi, S.; Rusinek, H.; Convit, A.; Li, Y.; Boppana, M.; de Leon, M.J. Reduced hippocampal metabolism in MCI and AD: Automated FDG-PET image analysis. Neurology 2005, 64, 1860–1867. [Google Scholar] [CrossRef]
- Minoshima, S.; Giordani, B.; Berent, S.; Frey, K.A.; Foster, N.L.; Kuhl, D.E. Metabolic reduction in the posterior cingulate cortex in very early Alzheimer’s disease. Ann. Neurol. 1997, 42, 85–94. [Google Scholar] [CrossRef]
- Gauthier, S.; Reisberg, B.; Zaudig, M.; Petersen, R.C.; Ritchie, K.; Broich, K.; Belleville, S.; Brodaty, H.; Bennett, D.; Chertkow, H.; et al. Mild cognitive impairment. Lancet 2006, 367, 1262–1270. [Google Scholar] [CrossRef]
- Petersen, R.C. Mild cognitive impairment. Continuum 2016, 22, 404–418. [Google Scholar] [CrossRef]
- Bailly, M.; Destrieux, C.; Hommet, C.; Mondon, K.; Cottier, J.P.; Beaufils, E.; Vierron, E.; Vercouillie, J.; Ibazizene, M.; Voisin, T.; et al. Precuneus and cingulate cortex atrophy and hypometabolism in patients with Alzheimer’s Disease and mild cognitive impairment: MRI and (18)F-FDG PET quantitative analysis using FreeSurfer. Biomed. Res. Int. 2015, 2015, 583931. [Google Scholar] [CrossRef]
- Del Sole, A.; Clerici, F.; Chiti, A.; Lecchi, M.; Mariani, C.; Maggiore, L.; Mosconi, L.; Lucignani, G. Individual cerebral metabolic deficits in Alzheimer’s disease and amnestic mild cognitive impairment: An FDG PET study. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 1357–1366. [Google Scholar] [CrossRef]
- Edison, P.; Archer, H.A.; Gerhard, A.; Hinz, R.; Pavese, N.; Turkheimer, F.E.; Hammers, A.; Tai, Y.F.; Fox, N.; Kennedy, A.; et al. Microglia, amyloid, and cognition in Alzheimer’s disease: An [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol. Dis. 2008, 32, 412–419. [Google Scholar] [CrossRef]
- Fan, Z.; Dani, M.; Femminella, G.D.; Wood, M.; Calsolaro, V.; Veronese, M.; Turkheimer, F.; Gentleman, S.; Brooks, D.J.; Hinz, R.; et al. Parametric mapping using spectral analysis for (11)C-PBR28 PET reveals neuroinflammation in mild cognitive impairment subjects. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 1432–1441. [Google Scholar] [CrossRef]
- Fan, Z.; Brooks, D.J.; Okello, A.; Edison, P. An early and late peak in microglial activation in Alzheimer’s disease trajectory. Brain 2017, 140, 792–803. [Google Scholar] [CrossRef]
- Matthews, D.R.; Hosker, J.P.; Rudenski, A.S.; Naylor, B.A.; Treacher, D.F.; Turner, R.C. Homeostasis model assessment: Insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985, 28, 412–419. [Google Scholar] [CrossRef]
- Willette, A.A.; Bendlin, B.B.; Starks, E.J.; Birdsill, A.C.; Johnson, S.C.; Christian, B.T.; Okonkwo, O.C.; La Rue, A.; Hermann, B.P.; Koscik, R.L.; et al. Association of insulin resistance with cerebral glucose uptake in late middle-aged adults at risk for Alzheimer disease. JAMA Neurol. 2015, 72, 1013–1020. [Google Scholar] [CrossRef]
- Roberts, R.O.; Knopman, D.S.; Cha, R.H.; Miekle, M.M.; Pankratz, V.S.; Boeve, B.F.; Kantarci, K.; Geda, Y.E.; Jack, C.R., Jr.; Petersen, R.C.; et al. Diabetes and elevated hemoglobin A1c levels are associated with brain hypometabolism but not amyloid accumulation. J. Nucl. Med. 2014, 55, 759–764. [Google Scholar] [CrossRef]
- Pegueroles, J.; Pané, A.; Vilaplana, E.; Montal, V.; Bejanin, A.; Videla, L.; Carmona-Iragui, M.; Barroeta, I.; Ibarzabal, A.; Casajoana, A.; et al. Obesity impacts brain metabolism and structure independently of amyloid and tau pathology in healthy elderly. Alzheimer’s Dement. 2020, 12, e12052. [Google Scholar] [CrossRef]
- Sala, A.; Malpetti, M.; Ferrulli, A.; Gianolli, L.; Luzi, L.; Perani, D. High body mass index, brain metabolism and connectivity: An unfavorable effect in elderly females. Aging 2019, 11, 8573–8586. [Google Scholar] [CrossRef]
- Jais, A.; Solas, M.; Backes, H.; Chaurasia, B.; Kleinridders, A.; Theurich, S.; Mauer, J.; Steculorum, S.M.; Hampel, B.; Goldau, J.; et al. Myeloid-cell-derived VEGF maintains brain glucose uptake and limits cognitive impairment in obesity. Cell 2016, 165, 882–895. [Google Scholar] [CrossRef]
- Iozzo, P.; Guzzardi, M.A. Imaging of brain glucose uptake by PET in obesity and cognitive dysfunction: Life-course perspective. Endocr. Connect. 2019, 8, R169–R183. [Google Scholar] [CrossRef]
- Goyal, M.S.; Vlassenko, A.G.; Blazey, T.M.; Su, Y.; Couture, L.E.; Durbin, T.J.; Bateman, R.J.; Benzinger, T.L.; Morris, J.C.; Raichle, M.E. Loss of brain aerobic glycolysis in normal human aging. Cell Metab. 2017, 26, 353–360.e3. [Google Scholar] [CrossRef]
- Hasselbalch, S.G.; Knudsen, G.M.; Videbaek, C.; Pinborg, L.H.; Schmidt, J.F.; Holm, S.; Paulson, O.B. No effect of insulin on glucose blood-brain barrier transport and cerebral metabolism in humans. Diabetes 1999, 48, 1915–1921. [Google Scholar] [CrossRef]
- Bingham, E.M.; Hopkins, D.; Smith, D.; Pernet, A.; Hallett, W.; Reed, L.; Marsden, P.K.; Amiel, S.A. The role of insulin in human brain glucose metabolism: An 18fluoro-deoxyglucose positron emission tomography study. Diabetes 2002, 51, 3384–3390. [Google Scholar] [CrossRef]
- Hirvonen, J.; Virtanen, K.A.; Nummenmaa, L.; Hannukainen, J.C.; Honka, M.-J.; Bucci, M.; Nesterov, S.V.; Parkkola, R.; Rinne, J.; Iozzo, P.; et al. Effects of insulin on brain glucose metabolism in impaired glucose tolerance. Diabetes 2011, 60, 443–447. [Google Scholar] [CrossRef]
- Bahri, S.; Horowitz, M.; Malbert, C.-H. Inward glucose transfer accounts for insulin-dependent increase in brain glucose metabolism associated with diet-induced obesity. Obesity 2018, 26, 1322–1331. [Google Scholar] [CrossRef]
- Button, K.S.; Ioannidis, J.P.A.; Mokrysz, C.; Nosek, B.A.; Flint, J.; Robinson, E.S.J.; Munafo, M.R. Power failure: Why small sample size undermines the reliability of neuroscience. Nat. Rev. Neurosci. 2013, 14, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Bonadonna, R.C.; Groop, L.; Kraemer, N.; Ferrannini, E.; Del Prato, S.; DeFronzo, R.A. Obesity and insulin resistance in humans: A dose-response study. Metabolism 1990, 39, 452–459. [Google Scholar] [CrossRef]
- Kapogiannis, D.; Mustapic, M.; Shardell, M.D.; Berkowitz, S.T.; Diehl, T.C.; Spangler, R.D.; Tran, J.; Lazaropoulos, M.P.; Chawla, S.; Gulyani, S.; et al. Association of extracellular vesicle biomarkers with Alzheimer disease in the Baltimore longitudinal study of aging. JAMA Neurol. 2019, 76, 1340–1351. [Google Scholar] [CrossRef]
- Mustapic, M.; Tran, J.; Craft, S.; Kapogiannis, D. Extracellular vesicle biomarkers track cognitive changes following intranasal insulin in Alzheimer’s disease. J. Alzheimers Dis. 2019, 69, 489–498. [Google Scholar] [CrossRef]
- Mansur, R.B.; Delgado-Peraza, F.; Subramaniapillai, M.; Lee, Y.; Iacobucci, M.; Nasri, F.; Rodrigues, N.; Rosenblat, J.D.; Britzke, E.; Cosgrove, V.E.; et al. Exploring brain insulin resistance in adults with bipolar depression using extracellular vesicles of neuronal origin. J. Psychiatr. Res. 2021, 133, 82–92. [Google Scholar] [CrossRef]
- Tschritter, O.; Preissl, H.; Hennige, A.M.; Stumvoll, M.; Porubska, K.; Frost, R.; Marx, H.; Klosel, B.; Lutzenberger, W.; Birbaumer, N.; et al. The cerebrocortical response to hyperinsulinemia is reduced in overweight humans: A magnetoencephalographic study. Proc. Natl. Acad. Sci. USA 2006, 103, 12103–12108. [Google Scholar] [CrossRef]
- Hwang, J.J.; Jiang, L.; Hamza, M.; Rangel, E.S.; Dai, F.; Belfort-DeAguiar, R.; Parikh, L.; Koo, B.B.; Rothman, D.L.; Mason, G.; et al. Blunted rise in brain glucose levels during hyperglycemia in adults with obesity and T2DM. JCI Insight 2017, 2, e95913. [Google Scholar] [CrossRef]
- Choi, I.-Y.; Seaquist, E.R.; Gruetter, R. Effect of hypoglycemia on brain glycogen metabolism in vivo. J. Neurosci. Res. 2003, 72, 25–32. [Google Scholar] [CrossRef]
- Kobayashi, M.; Nikami, H.; Morimatsu, M.; Saito, M. Expression and localization of insulin-regulatable glucose transporter (GLUT4) in rat brain. Neurosci. Lett. 1996, 213, 103–106. [Google Scholar] [CrossRef]
- Thaler, J.P.; Yi, C.-X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Investig. 2012, 122, 153–162. [Google Scholar] [CrossRef]
- Baron, A.D.; Steinberg, H.O.; Chaker, H.; Leaming, R.; Johnson, A.; Brechtel, G. Insulin-mediated skeletal muscle vasodilation contributes to both insulin sensitivity and responsiveness in lean humans. J. Clin. Investig. 1995, 96, 786–792. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rebelos, E.; Rinne, J.O.; Nuutila, P.; Ekblad, L.L. Brain Glucose Metabolism in Health, Obesity, and Cognitive Decline—Does Insulin Have Anything to Do with It? A Narrative Review. J. Clin. Med. 2021, 10, 1532. https://doi.org/10.3390/jcm10071532
Rebelos E, Rinne JO, Nuutila P, Ekblad LL. Brain Glucose Metabolism in Health, Obesity, and Cognitive Decline—Does Insulin Have Anything to Do with It? A Narrative Review. Journal of Clinical Medicine. 2021; 10(7):1532. https://doi.org/10.3390/jcm10071532
Chicago/Turabian StyleRebelos, Eleni, Juha O. Rinne, Pirjo Nuutila, and Laura L. Ekblad. 2021. "Brain Glucose Metabolism in Health, Obesity, and Cognitive Decline—Does Insulin Have Anything to Do with It? A Narrative Review" Journal of Clinical Medicine 10, no. 7: 1532. https://doi.org/10.3390/jcm10071532
APA StyleRebelos, E., Rinne, J. O., Nuutila, P., & Ekblad, L. L. (2021). Brain Glucose Metabolism in Health, Obesity, and Cognitive Decline—Does Insulin Have Anything to Do with It? A Narrative Review. Journal of Clinical Medicine, 10(7), 1532. https://doi.org/10.3390/jcm10071532