
Valproic Acid-Induced Liver Injury: A Case-Control Study from a Prospective Pharmacovigilance Program in a Tertiary Hospital




Abstract
1. Introduction
2. Material and Methods
2.1. Setting
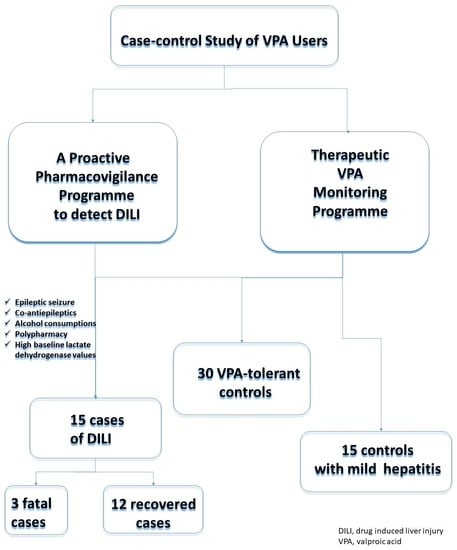
2.2. Definition of Cases and Controls
2.3. Case Definition
2.4. Control Definition
2.5. Data Collection
2.6. Data Analysis
2.6.1. Sample Size Calculation
2.6.2. Statistical Analysis
2.7. Ethical Statement
3. Results
4. Discussion
5. Conclusions
5.1. What Is Already Known about This Subject
- -
- Valproic acid (VPA) therapy is known to cause liver injury in a small percentage of patients, with some fatal outcomes.
- -
- Mechanisms associated with an altered mitochondrial β-oxidation pathway and excessive oxidative stress have been postulated.
- -
- Certain risk factors (such as polytherapy and younger age) could determine the chance of developing hepatotoxicity.
5.2. What This Study Adds
- -
- A greater characterisation of VPA hepatotoxicity (e.g., predominance of hepatocellular vs. cholestatic hepatitis).
- -
- A number of other risk factors appear to be related to VPA-induced liver injury, such as alcohol consumption, seizures and increased baseline LDH levels.
- -
- Considering these factors, patients at higher risk of liver injury should be identified and closely monitored.
- -
- Future research is needed to confirm these findings.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Peterson, G.M.; Naunton, M. Valproate: A simple chemical with so much to offer. J. Clin. Pharm. Ther. 2005, 30, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Sztajnkrycer, M.D. Valproic Acid Poisoning; UpToDate: Wellesley, MA, USA, 2020; Available online: www.uptodate.com/contents/valproic-acid-poisoning?search=valproic-acid&source=search_result&selectedTitle=1~150&usage_type=default&display_rank=1 (accessed on 13 June 2020).
- Romoli, M.; Mazzocchetti, P.; D’Alonzo, R.; Siliquini, S.; Rinaldi, V.E.; Verrotti, A.; Calabresi, P.; Costa, C. Valproic Acid and Epilepsy: From Molecular Mechanisms to Clinical Evidences. Curr. Neuropharmacol. 2019, 17, 926–946. [Google Scholar] [CrossRef]
- López-Muñoz, F.; Shen, W.W.; D’Ocon, P.; Romero, A.; Álamo, C. A History of the Pharmacological Treatment of Bipolar Disorder. Int. J. Mol. Sci. 2018, 19, 2143. [Google Scholar] [CrossRef] [PubMed]
- CIMA. DEPAKINE Crono 300 mg. Summary Product Characteristics; CIMA: Madrid, Spain, 1995; Available online: https://cima.aemps.es/cima/dochtml/ft/60351/FT_60351.html (accessed on 13 June 2020).
- Löscher, W. Basic Pharmacology of Valproate. CNS Drugs 2002, 16, 669–694. [Google Scholar] [CrossRef]
- Perucca, E. Pharmacological and Therapeutic Properties of Valproate. CNS Drugs 2002, 16, 695–714. [Google Scholar] [CrossRef] [PubMed]
- Gobbi, G.; Janiri, L. Sodium- and magnesium-valproate in vivo modulate glutamatergic and GABAergic synapses in the medial prefrontal cortex. Psychopharmacology 2006, 185, 255–262. [Google Scholar] [CrossRef]
- Zeise, M.; Kasparow, S.; Zieglgänsberger, W. Valproate suppresses N-methyl-d-aspartate-evoked, transient depolarizations in the rat neocortex in vitro. Brain Res. 1991, 544, 345–348. [Google Scholar] [CrossRef]
- Ichikawa, J.; Meltzer, H.Y. Valproate and carbamazepine increase prefrontal dopamine release by 5-HT1A receptor activation. Eur. J. Pharmacol. 1999, 380, R1–R3. [Google Scholar] [CrossRef]
- Zhu, M.-M.; Li, H.-L.; Shi, L.-H.; Chen, X.-P.; Luo, J.; Zhang, Z.-L. The pharmacogenomics of valproic acid. J. Hum. Genet. 2017, 62, 1009–1014. [Google Scholar] [CrossRef]
- Ponchaut, S.; Veitch, K. Valproate and mitochondria. Biochem. Pharmacol. 1993, 46, 199–204. [Google Scholar] [CrossRef]
- Nanau, R.M.; Neuman, M.G. Adverse drug reactions induced by valproic acid. Clin. Biochem. 2013, 46, 1323–1338. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.-L.; Jing, X.; Sun, J.-Y.; Hu, Y.-H.; Xu, Z.-J.; Ni, M.-M.; Chen, F.; Lu, X.-P.; Qiu, J.-C.; Wang, T. Valproic Acid and the Liver Injury in Patients with Epilepsy: An Update. Curr. Pharm. Des. 2019, 25, 343–351. [Google Scholar] [CrossRef]
- Ramirez, E.; Carcas, A.J.; Borobia, A.M.; Lei, S.H.; Piñana, E.; Fudio, S.; Frias, J. A Pharmacovigilance Program from Laboratory Signals for the Detection and Reporting of Serious Adverse Drug Reactions in Hospitalized Patients. Clin. Pharmacol. Ther. 2009, 87, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.-C.; Drug-induced Liver Injury (DILI) Study Group; Mao, Y.-M.; Chen, C.-W.; Chen, J.-J.; Chen, J.; Cong, W.-M.; Ding, Y.; Duan, Z.-P.; Fu, Q.-C.; et al. CSH guidelines for the diagnosis and treatment of drug-induced liver injury. Hepatol. Int. 2017, 11, 221–241. [Google Scholar] [CrossRef] [PubMed]
- Aithal, G.P.; Watkins, P.B.; Andrade, R.J.; Larrey, D.; Molokhia, M.; Takikawa, H.; Hunt, C.M.; A Wilke, R.; Avigan, M.; Kaplowitz, N.; et al. Case Definition and Phenotype Standardization in Drug-Induced Liver Injury. Clin. Pharmacol. Ther. 2011, 89, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Danan, G.; Benichou, C. Causality assessment of adverse reactions to drugs—I. A novel method based on the conclusions of international consensus meetings: Application to drug-induced liver injuries. J. Clin. Epidemiol. 1993, 46, 1323–1330. [Google Scholar] [CrossRef]
- Danan, G.; Teschke, R. Drug-Induced Liver Injury: Why is the Roussel Uclaf Causality Assessment Method (RUCAM) Still Used 25 Years After Its Launch? Drug Saf. 2018, 41, 735–743. [Google Scholar] [CrossRef] [PubMed]
- IMIM. Sample Size Calculator; IMIM: Barcelona, Spain, 2012; Available online: www.imim.es/ofertadeserveis/software-public/granmo/ (accessed on 13 June 2020).
- ECIJA. Organic law 3/2018, December 5, 2018, Protection of Personal Data and Guarantee of Digital Rights; ECIJA: Madrid, Spain, 2018; pp. 119788–119857. Available online: https://apdcat.gencat.cat/web/.content/01-autoritat/normativa/documentos/Llei-organica-pd-2018.pdf (accessed on 1 June 2020).
- Schmid, M.M.; Freudenmann, R.; Keller, F.; Connemann, B.J.; Hiemke, C.; Gahr, M.; Kratzer, W.; Fuchs, M.; Schönfeldt-Lecuona, C. Non-Fatal and Fatal Liver Failure Associated with Valproic Acid. Pharmacopsychiatry 2012, 46, 63–68. [Google Scholar] [CrossRef]
- Star, K.; Edwards, I.R.; Choonara, I. Valproic Acid and Fatalities in Children: A Review of Individual Case Safety Reports in VigiBase. PLoS ONE 2014, 9, e108970. [Google Scholar] [CrossRef]
- Xiong, H.; Liu, C.-T.; Zhang, Y.-H.; Bao, X.-H.; Jiang, Y.-W.; Zhao, H.; Wu, X.-P.; Qin, J. Valproic acid-induced idiosyncratic liver injury in 4 cases. Chin. J. Pediatr. 2012, 50, 890–894. [Google Scholar]
- Pessayre, D.; Larrey, D. 8 Acute and chronic drug-induced hepatitis. Baillière Clin. Gastroenterol. 1988, 2, 385–422. [Google Scholar] [CrossRef]
- Devarbhavi, H.; Patil, M.; Reddy, V.V.; Singh, R.; Joseph, T.; Ganga, D. Drug-induced acute liver failure in children and adults: Results of a single-centre study of 128 patients. Liver Int. 2017, 38, 1322–1329. [Google Scholar] [CrossRef]
- Walker, C.P.; Deb, S. Rhabdomyolysis and Hepatotoxicity from Valproic Acid: Case Reports. J. Pharm. Pract. 2019, 0897190019882880. [Google Scholar] [CrossRef]
- Rocco, A.; Compare, D.; Angrisani, D.; Zamparelli, M.S.; Nardone, G. Alcoholic disease: Liver and beyond. World J. Gastroenterol. 2014, 20, 14652–14659. [Google Scholar] [CrossRef] [PubMed]
- Ishak, K.G.; Zimmerman, H.J.; Ray, M.B. Alcoholic Liver Disease: Pathologic, Pathogenetic and Clinical Aspects. Alcohol. Clin. Exp. Res. 1991, 15, 45–66. [Google Scholar] [CrossRef]
- Beier, J.I.; McClain, C.J. Mechanisms and cell signaling in alcoholic liver disease. Biol. Chem. 2010, 391, 1249–1264. [Google Scholar] [CrossRef] [PubMed]
- Pourahmad, J.; Eskandari, M.R.; Kaghazi, A.; Shaki, F.; Shahraki, J.; Fard, J.K. A new approach on valproic acid induced hepatotoxicity: Involvement of lysosomal membrane leakiness and cellular proteolysis. Toxicol. Vitr. 2012, 26, 545–551. [Google Scholar] [CrossRef]
- Hassan, H.M.; Guo, H.; Yousef, B.A.; Guerram, M.; Hamdi, A.M.; Zhang, L.; Jiang, Z. Role of Inflammatory and Oxidative Stress, Cytochrome P450 2E1, and Bile Acid Disturbance in Rat Liver Injury Induced by Isoniazid and Lipopolysaccharide Cotreatment. Antimicrob. Agents Chemother. 2016, 60, 5285–5293. [Google Scholar] [CrossRef]
- Neuman, M.G.; Shear, N.H.; Jacobson-Brown, P.M.; Katz, G.G.; Neilson, H.K.; Malkiewicz, I.M.; Cameron, R.G.; Abbott, F. CYP2E1-mediated modulation of valproic acid-induced hepatocytotoxicity. Clin. Biochem. 2001, 34, 211–218. [Google Scholar] [CrossRef]
- Turnbull, D.M.; Rawlins, M.D.; Weightman, D.; Chadwick, D.W. Plasma concentrations of sodium valproate: Their clinical value. Ann. Neurol. 1983, 14, 38–42. [Google Scholar] [CrossRef]
- Chadwick, D.W. Concentration-Effect Relationships of Valproic Acid. Clin. Pharmacokinet. 1985, 10, 155–163. [Google Scholar] [CrossRef]
- Patsalos, P.N.; Berry, D.J.; Bourgeois, B.F.D.; Cloyd, J.C.; Glauser, T.A.; Johannessen, S.I.; Leppik, I.E.; Tomson, T.; Perucca, E. Antiepileptic drugsbest practice guidelines for therapeutic drug monitoring: A position paper by the subcommission on therapeutic drug monitoring, ILAE Commission on Therapeutic Strategies. Epilepsia 2008, 49, 1239–1276. [Google Scholar] [CrossRef]
- Legrand, C.; Bour, J.; Jacob, C.; Capiaumont, J.; Martial, A.; Marc, A.; Wudtke, M.; Kretzmer, G.; Demangel, C.; Duval, D.; et al. Lactate dehydrogenase (LDH) activity of the number of dead cells in the medium of cultured eukaryotic cells as marker. J. Biotechnol. 1992, 25, 231–243. [Google Scholar] [CrossRef]
- Chan, F.K.-M.; Moriwaki, K.; De Rosa, M.J. Detection of Necrosis by Release of Lactate Dehydrogenase Activity. Method Mol. Biol. 2013, 979, 65–70. [Google Scholar] [CrossRef]
- Chu, X.-M.; Zhang, L.-F.; Wang, G.-J.; Zhang, S.-N.; Zhou, J.-H.; Hao, H.-P. Influence of UDP-glucuronosyltransferase polymorphisms on valproic acid pharmacokinetics in Chinese epilepsy patients. Eur. J. Clin. Pharmacol. 2012, 68, 1395–1401. [Google Scholar] [CrossRef]
- Kiang, T.K.L.; Ho, P.C.; Anari, M.R.; Tong, V.; Abbott, F.S.; Chang, T.K.H. Contribution of CYP2C9, CYP2A6, and CYP2B6 to Valproic Acid Metabolism in Hepatic Microsomes from Individuals with the CYP2C9*1/*1 Genotype. Toxicol. Sci. 2006, 94, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Amini-Shirazi, N.; Ghahremani, M.H.; Ahmadkhaniha, R.; Mandegary, A.; Dadgar, A.; Abdollahi, M.; Shadnia, S.; Pakdaman, H.; Kebriaeezadeh, A. Influence of CYP2C9 polymorphism on metabolism of valproate and its hepatotoxin metabolite in Iranian patients. Toxicol. Mech. Methods 2010, 20, 452–457. [Google Scholar] [CrossRef]
- Saneto, R.P.; Lee, I.-C.; Koenig, M.K.; Bao, X.; Weng, S.-W.; Naviaux, R.K.; Wong, L.-J.C. POLG DNA testing as an emerging standard of care before instituting valproic acid therapy for pediatric seizure disorders. Seizure 2010, 19, 140–146. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Cases DILI | Mild Liver Injury Controls | Cases vs. Mild Liver Injury Controls | VPA-Tolerant Controls | Cases vs. VPA-Tolerant Controls | |
|---|---|---|---|---|---|
| (n = 15) | (n = 15) | p Value | (n = 30) | p Value | |
| Age, years * | 45.73 (30.8) | 46.2 (31.1) | 0.967 | 44.70 (29.8) | 0.840 |
| Adults, n (%) | 10 (66.7) | 10 (66.7) | 1.00 | 20 (66.7) | 1.00 |
| Median age, years (range) | 68 (27–85) | 69 (19–80) | 0.977 | 68.5 (32–77) | 0.890 |
| Children, n (%) | 5 (33.39) | 5 (33.39) | 1.00 | 10 (33.3) | 1.00 |
| Median age, years (range) | 9 (2–17) | 9 (3–17) | 0.977 | 9 (2–16) | 0.89 |
| Women, n (%) * | 4 (26.7) | 4 (26.7) | 1.00 | 8 (26.7) | 1.00 |
| Weight, kg | 58.1 (25.4) | 54 (25) | 0.662 | 58.0 (23.3) | 0.580 |
| Height, cm | 153 (32) | 143.7 (24.2) | 0.647 | 155.5 (11.2) | 0.18 |
| BMI, kg/m2 | 23.1 (6.8) | 22.7 (5.6) | 0.918 | 24.7 (4.2) | 0.16 |
| Family history | |||||
| Neurodegenerative disorder without diagnosis | 1 (6.7) | 0 | 0.012 | 0 | 0.012 |
| Medical history, n (%) | |||||
| CNS mass | 4 (26.7) | 5 (33.3) | 0.28 | 7 (23.3) | 0.623 |
| Hydrocephalus | 1 (6.7) | 0 | 0.012 | 0 | 0.012 |
| Dravet syndrome | 1 (6.7) | 1 (6.7) | 1 | 0 | 0.012 |
| Generalised epilepsy | 4 (26.7) | 6 (40.0) | 0.035 | 8 (26.7) | 1 |
| Focal motor epilepsy | 3 (20) | 1 (6.7) | 0.003 | 3 (10) | 0.047 |
| Bipolar disorder | 1 (6.7) | 2 (13.3) | 0.091 | 6 (20) | 0.003 |
| Lennox Gastaut syndrome | 1 (6.7) | 0 | 0.012 | 1 (3.3) | 0.31 |
| Myoclonic epilepsy | 0 | 0 | −1 | 1 (3.3) | 0.081 |
| Schizoaffective disorder/schizophrenia | 0 | 0 | −1 | 3 (10) | 0.001 |
| Temporal seizures | 0 | 0 | −1 | 1 (3.3) | 0.081 |
| Previous drug allergies | 1 (6.7) | 4 (26.6) | <0.001 | 3 (10) | 0.300 |
| Azithromycin | Amoxicillin clavulanic Itraconazole Propyphenazone Unfinished study | Metamizole (2) Amoxicillin clavulanic | |||
| Indication of VPA therapy | |||||
| Seizures | 13 (86.7) | 14 (93.3) | 0.111 | 20 (66.7) | <0.001 |
| Bipolar disorder | 2 (13.3) | 1 (6.7) | 0.091 | 10 (33.3) | <0.001 |
| VPA dosage, mg/kg/day | 26.4 (13.5) | 24.5 (24.0) | 0.795 | 21.7 (11.1) | 0.213 |
| Treatment time to ADR, days | 61.6 (73.8) | 304 (309) | <0.001 | NA | - |
| Toxics, n (%) | |||||
| Alcohol | 8 (53.3) | 2 (13.3) | <0.001 | 1 (3.3) | <0.001 |
| Tobacco | 2 (13.3) | 2 (13.3) | 1 | 6 (20) | 0.180 |
| Alcohol and Tobacco | 1 (6.7) | 1 (6.7) | 1 | 1 (3.3) | 0.310 |
| Concomitant medication | 4.27 (3.1) | 4.2 (3.6) | 0.957 | 3.6 (2.8) | 0.759 |
| Concomitant antiepileptics, median (range) | 2 (0–3) | 1 (0–2) | 0.047 | 2 (0–3) | 0.049 |
| Levetiracetam | 4 | 5 | - | 2 | - |
| Clobazam | 2 | 2 | - | 1 | - |
| Clonazepam | 2 | 0 | - | 4 | - |
| Lamotrigine | 1 | 0 | - | 1 | - |
| Gabapentin | 2 | 0 | - | 0 | - |
| Diazepam | 1 | 0 | - | 1 | - |
| Rufinamide | 1 | 1 | - | 0 | - |
| Phenytoin | 0 | 0 | - | 2 | - |
| Lacosamide | 0 | 0 | - | 1 | - |
| Oxcarbazepine | 0 | 0 | - | 2 | - |
| Tiagabine | 0 | 1 | - | 0 | - |
| Topiramate | 0 | 2 | - | 0 | - |
| Brivaracetam | 0 | 1 | - | 0 | - |
| Interactions # | |||||
| Red | 7 (46.7) | 2 (13.3) | <0.001 | 4 (13.3) | <0.001 |
| Yellow | 9 (60) | 2 (13.3) | <0.001 | 4 (13.3) | <0.001 |
| Green | 3 (20) | 0 | <0.001 | 0 | <0.001 |
| Liver Injury | |||||
| Hepatocellular | 11 (73.3) | 1 (6.7) | <0.001 | NA | - |
| Mixed | 2 (13.3) | 2 (13.3) | 1 | NA | - |
| Cholestatic | 1 (6.7) | 8 (53.3) | <0.001 | NA | - |
| Severity | |||||
| Acute liver injury | 7 (46.6) | 15 (100) | <0.001 | NA | - |
| Severe liver dysfunction (INR > 1.5) | 1 (6.7) | - | NA | - | |
| Acute liver failure, INR > 1.5 and any degree of encephalopathy: | - | ||||
| Encephalopathy G1 | 6 (40) | - | NA | - | |
| Encephalopathy G2 | 0 | - | NA | - | |
| Encephalopathy G3 | 1 (6.7) | - | NA | - | |
| Encephalopathy G4 | 0 | - | NA | - | |
| Liver Injury duration, days | 18.7 (13.9) | 204.4 (241.5) | 0.02 | NA | - |
| Outcome, n (%) | |||||
| Recovered | 12 (80) | 12 (80) | 1 | NA | - |
| Death | 3 (20) | 1 (6.7) | 0.003 | NA | - |
| Not recovered | 0 | 2 (13.3) | 0.012 | NA | - |
| Laboratory Data | |||||
| Baseline | |||||
| ALT, UI/L | 24 (12) | 23 (18) | 0.877 | 17 (10) | 0.706 |
| AST, UI/L | 23 (8) | 29 (16) | 0.28 | 21 (10) | 0.637 |
| GGT, UI/L | 55 (57) | 42 (33) | 0.528 | 32 (35) | 0.041 |
| ALP, UI/L | 85 (54) | 94 (38) | 0.7 | 91 (58) | 0.737 |
| TB, mg/dL | 0.6 (0.3) | 0.5 (0.3) | 0.537 | 1.7 (5.9) | 0.214 |
| PA, % | 82.5 (17.5) | 76.8 (33.9) | 0.766 | 84.7 (26.3) | 0.43 |
| LDH, UI/L | 333 (55) | 227 (26) | 0.033 | 202 (67) | 0.039 |
| Cr, mg/dL | 0.79 (0.79) | 1.0 (0.62) | 0.21 | 0.71 (0.30) | 0.108 |
| Albumin, g/dL | 4.4 (1.4) | 9.5 (17.0) | 0.409 | 3.7 (0.7) | 0.444 |
| Protein, g/dL | 5.7 (2.0) | 6.8 (0.9) | 0.922 | 11.2 (16.1) | 0.121 |
| Ammonium, µmol/L | 58 (-) | - | - | - | - |
| Onset | |||||
| ALT, UI/L | 435 (347) | 200 (387) | 0.025 | NA | - |
| AST, UI/L | 446 (333) | 163 (405) | 0.009 | NA | - |
| GGT, UI/L | 207 (95) | 77 (87) | 0.042 | NA | - |
| ALP, UI/L | 166 (193) | 147 (103) | 0.824 | NA | - |
| TB, mg/dL | 1.6 (1.8) | 0.6 (0.2) | 0.036 | NA | - |
| PA, % | 74.4 (28.7) | 85.3 (16.0) | 0.37 | NA | - |
| LDH, UI/L | 580 (36) | 428 (35) | 0.009 | NA | - |
| Cr, mg/dL | 0.82 (0.48) | 0.75 (0.54) | 0.668 | NA | - |
| Albumin, g/dL | 3.4 (0.6) | 2.8 (1.5) | 0.374 | NA | - |
| Protein, g/dL | 5.8 (0.5) | 5.7 (2.1) | 0.336 | NA | - |
| Ammonium, µmol/L | 62 (20–92) ** | 201 (-) | 0.013 | NA | - |
| Peak | |||||
| ALT, UI/L | 732 (335) | 232 (303) | 0.005 | NA | - |
| AST, UI/L | 811 (223) | 163 (386) | 0.023 | NA | - |
| GGT, UI/L | 1084 (259) | 140 (139) | <0.001 | NA | - |
| ALP, UI/L | 195 (146) | 146 (104) | 0.385 | NA | - |
| TB, mg/dL | 4.5 (7.7) | 0.5 (0.5) | 0.002 | NA | - |
| PA, % | 64.88 (32.8) | 81.3 (8.7) | 0.147 | NA | - |
| LDH, UI/L | 652 (50) | 513 (32) | 0.005 | NA | - |
| Cr, mg/dL | 1.04 (0.67) | 0.82 (0.55) | 0.668 | NA | - |
| Albumin, g/dL | 3.1 (0.6) | 2.7 (1.3) | 0.362 | NA | - |
| Protein, g/dL | 5.2 (0.8) | 6.0 (2.1) | 0.245 | NA | - |
| Ammonium, µmol/L | 142.5 (75–432) ** | - | - | NA | - |
| Recovery | |||||
| ALT, UI/L | 77 (106) | 58 (98) | 0.64 | NA | - |
| AST, UI/L | 56 (63) | 30 (14) | 0.219 | NA | - |
| GGT, UI/L | 90 (89) | 76 (127) | 0.342 | NA | - |
| ALP, UI/L | 110 (66) | 72 (39) | 0.222 | NA | - |
| TB, mg/dL | 7.1 (17.1) | 1.2 (2.2) | 0.242 | NA | - |
| PA, % | 84.8 (20.0) | 95.3 (8.7) | 0.154 | NA | - |
| LDH, UI/L | 246 (106.7) | 255 (76.7) | 0.861 | NA | - |
| Cr, mg/dL | 0.84 (0.45) | 0.79 (0.30) | 0.411 | NA | - |
| Albumin, g/dL | 3.6 (0.6) | 3.8 (0.5) | 0.401 | NA | - |
| Protein, g/dL | 6.4 (0.5) | 6.2 (2.1) | 0.893 | NA | - |
| Ammonium, µmol/L | 101 (66–116) ** | - | - | NA | - |
| VPA concentration | |||||
| VPA 1, ug/mL | 59.4 (28.3) | 59.6 (20.6) | 0.985 | 57.9 (20.8) | 0.003 |
| Time since onset, days | −38 (−12) | −49 (19) | 0.122 | −31 (12) | 0.437 |
| VPA 2, ug/mL | 56.3 (22.2) | 58.3 (27.5) | 0.92 | 57.7 (26.5) | 0.152 |
| Time since onset, days | −1.17 (3.7) | 10 (13) | 0.097 | 9 (9) | 0.897 |
| VPA 3, ug/mL | 51.0 (16.8) | 76.3 (19.4) | 0.018 | 61.6 (16.2) | 0.014 |
| Time since onset, days | 5 (9.8) | 44.8 (57.3) | 0.004 | 34.6 (67.1) | 0.090 |
| Pseudo-OR * | 95% BCa CI | p-Value | |
|---|---|---|---|
| Toxics (alcohol) | 7.30 | 6.17–8.44 | <0.001 |
| Co-antiepileptics, n | 4.33 | 2.57–6.09 | <0.001 |
| Indication (Epilepsy) | 2.34 | 1.94–2.74 | 0.002 |
| Co-medication, n | 1.58 | 1.39–1.77 | 0.001 |
| Lactate dehydrogenase, IU/L | 1.41 | 1.18–1.64 | 0.02 |
| Pseudo R2 = 0.908 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meseguer, E.S.; Elizalde, M.U.; Borobia, A.M.; Ramírez, E. Valproic Acid-Induced Liver Injury: A Case-Control Study from a Prospective Pharmacovigilance Program in a Tertiary Hospital. J. Clin. Med. 2021, 10, 1153. https://doi.org/10.3390/jcm10061153
Meseguer ES, Elizalde MU, Borobia AM, Ramírez E. Valproic Acid-Induced Liver Injury: A Case-Control Study from a Prospective Pharmacovigilance Program in a Tertiary Hospital. Journal of Clinical Medicine. 2021; 10(6):1153. https://doi.org/10.3390/jcm10061153
Chicago/Turabian StyleMeseguer, Enrique S., Mikel U. Elizalde, Alberto M. Borobia, and Elena Ramírez. 2021. "Valproic Acid-Induced Liver Injury: A Case-Control Study from a Prospective Pharmacovigilance Program in a Tertiary Hospital" Journal of Clinical Medicine 10, no. 6: 1153. https://doi.org/10.3390/jcm10061153
APA StyleMeseguer, E. S., Elizalde, M. U., Borobia, A. M., & Ramírez, E. (2021). Valproic Acid-Induced Liver Injury: A Case-Control Study from a Prospective Pharmacovigilance Program in a Tertiary Hospital. Journal of Clinical Medicine, 10(6), 1153. https://doi.org/10.3390/jcm10061153