
Circulating AQP4 Levels in Patients with Cerebral Amyloid Angiopathy-Associated Intracerebral Hemorrhage

 , , , ,
, , , ,
Abstract
1. Introduction
2. Methods
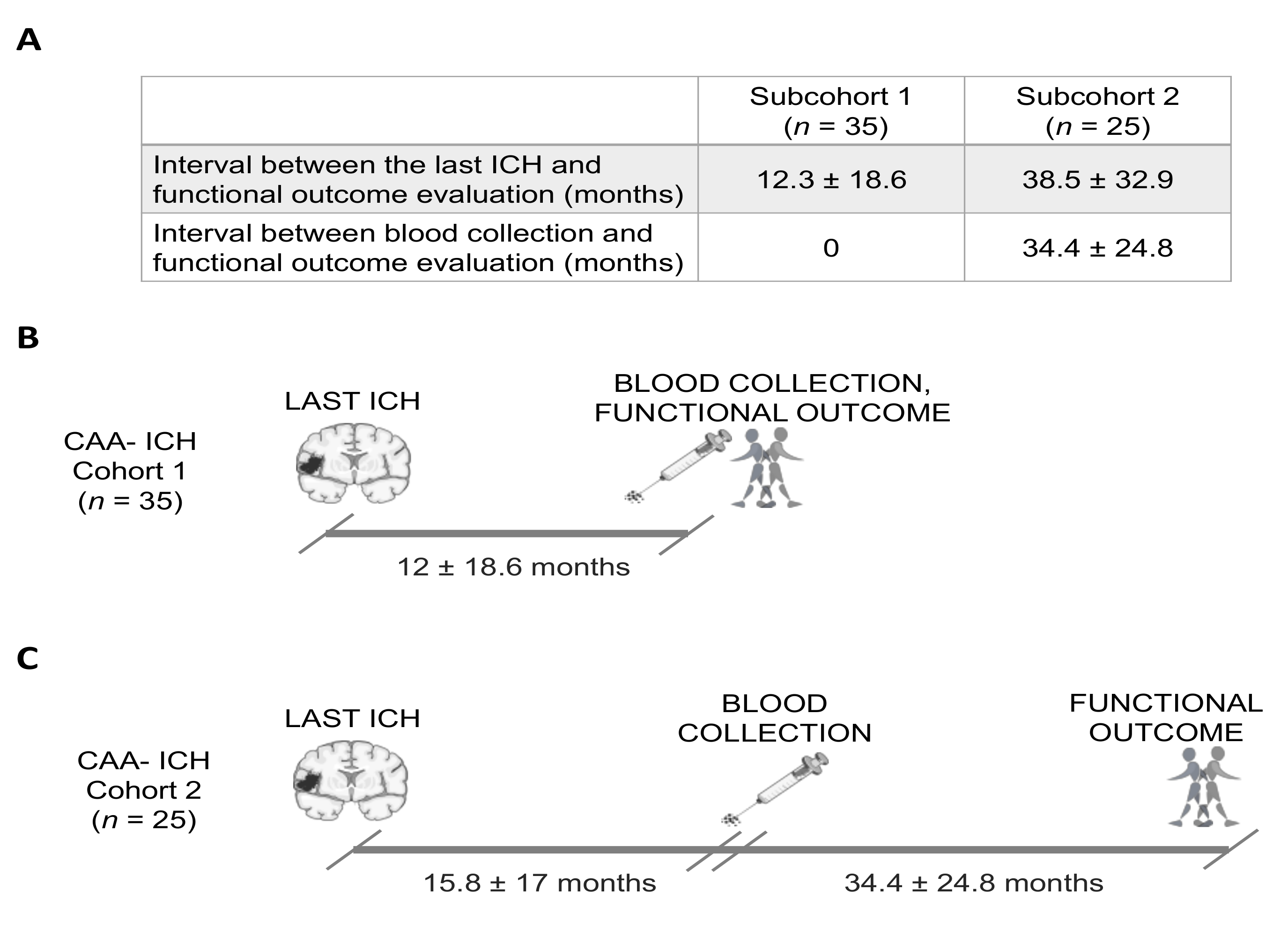
2.1. Study Population
2.2. MRI Protocol and Radiological Data
2.3. Serum AQP4 Determination
2.4. Statistical Analyses
3. Results
3.1. Baseline Characteristics
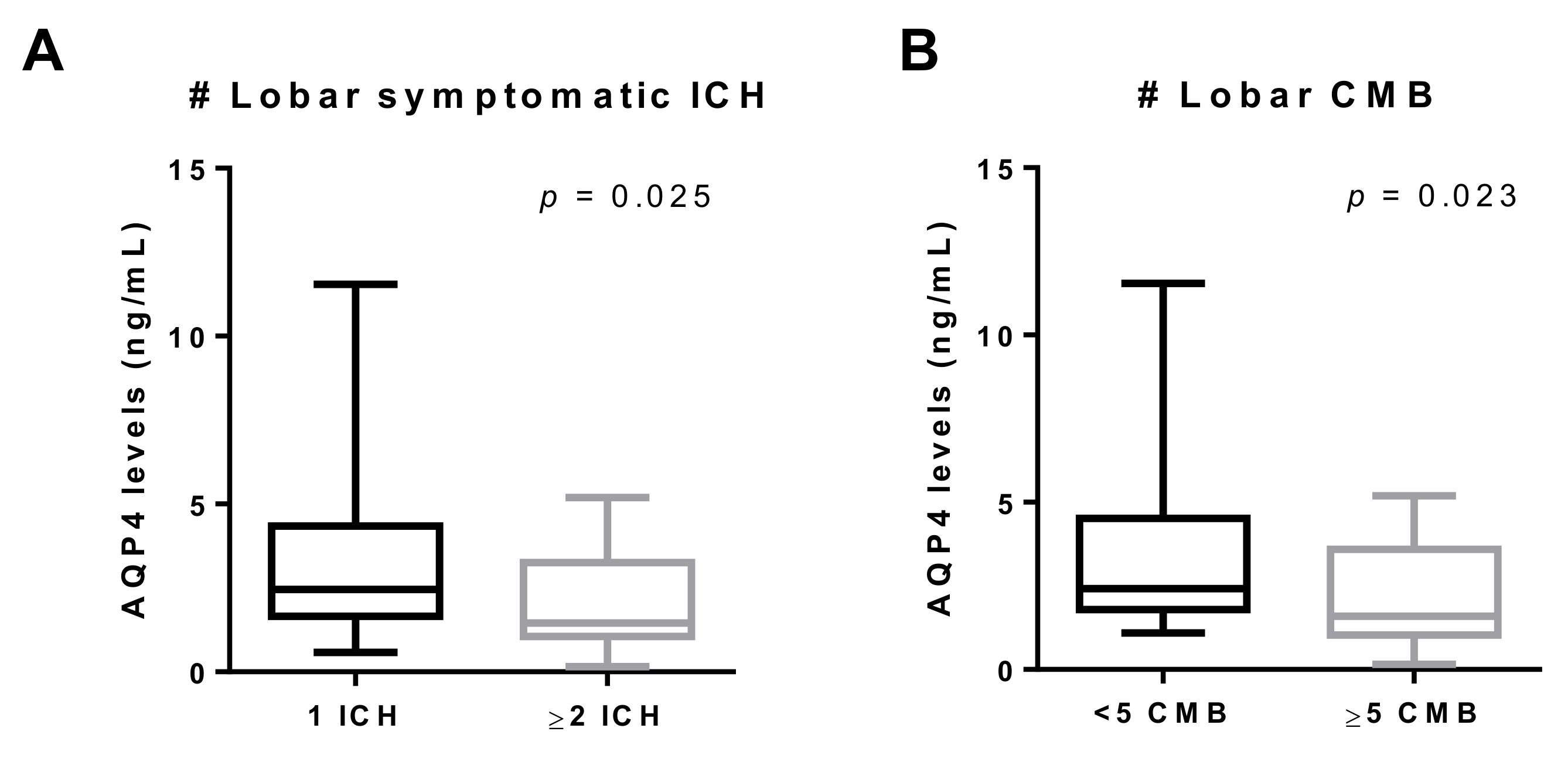
3.2. AQP4 Levels According to Clinical and Radiological Characteristics
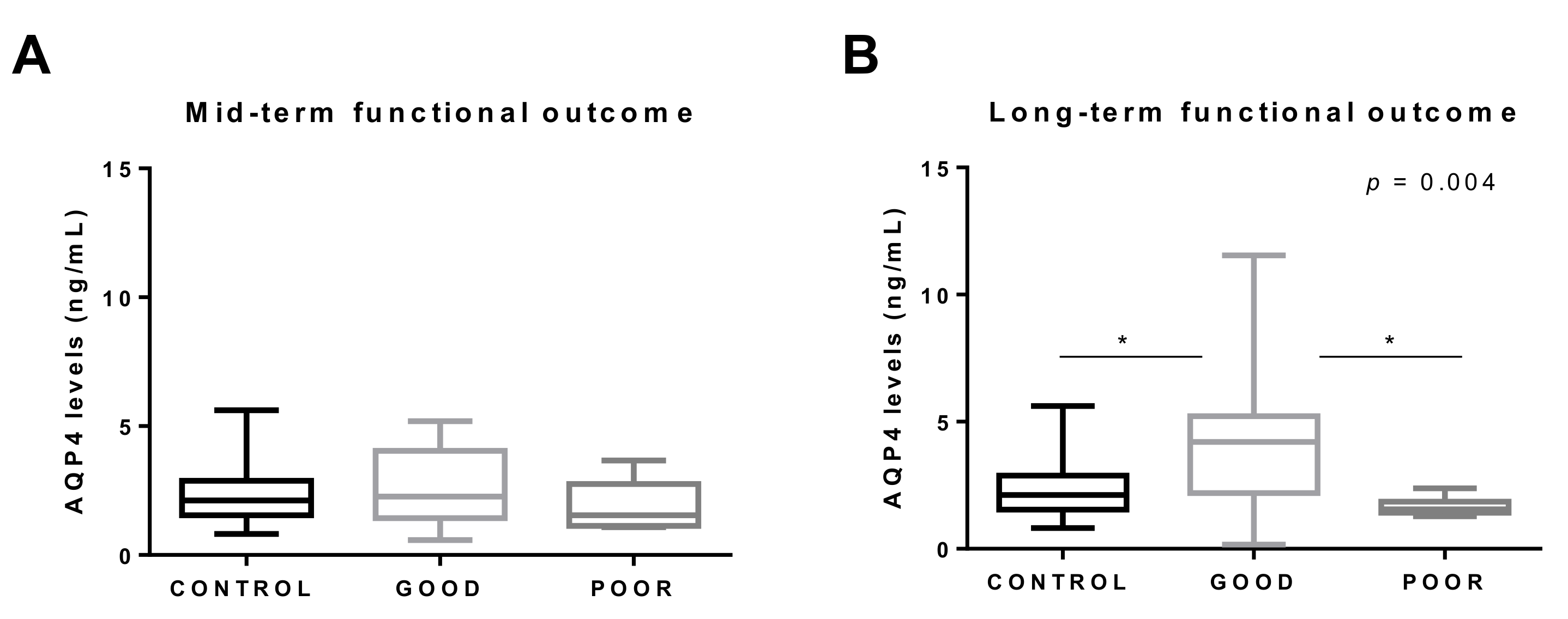
3.3. AQP4 and Functional Outcome
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A


{kind=link}
{kind=link}
{kind=link}
| Cognitive impairment | ≥2 ICHs | |||||
|---|---|---|---|---|---|---|
| Variable | YES | NO | p-value | YES | NO | p-Value |
| AQP4 | 1.69 (1.27–2.76) n = 30 | 3.09 (1.81–4.38) n = 30 | 0.030 | 1.45 (1.08–3.22) n = 17 | 2.45 (1.69–4.27) n = 43 | 0.026 |
| Age | 77 (73–79) | 74 (69–79) | 0.406 | 77 (72–78) | 76 (71.5–79) | 0.941 |
| Sex, female | 18 (60.0%) | 12 (40.0%) | 0.196 | 10 (58.8%) | 20 (46.5%) | 0.567 |
| Hypertension | 15 (53.6%) | 14 (48.3%) | 0.793 | 10 (66.7%) | 19 (45.2%) | 0.230 |
| Diabetes | 4 (14.3%) | 3 (11.1%) | 1 | 2 (14.3%) | 5 (12.2%) | 1 |
| Dyslipidemia | 8 (29.6%) | 9 (34.6%) | 0.773 | 8 (53.3%) | 9 (23.7%) | 0.053 |
| APOE genotype, ε2 carriers | 4 (13.3%) | 4 (13.3%) | 1 | 5 (29.4%) | 3 (7.0%) | 0.035 |
| APOE genotype, ε4 carriers | 10 (33.3%) | 4 (13.3%) | 0.125 | 6 (35.3%) | 8 (18.6%) | 0.190 |
| Cognitive impairment | - | - | - | 9 (52.9%) | 21 (48.8%) | 1 |
| ≥2 lobar ICHs | 9 (30.0%) | 8 (26.7%) | 1 | - | - | - |
| WMH | 29 (96.7%) | 28 (93.3%) | 1 | 17 (100%) | 40 (93.0%) | 0.551 |
| Periventricular | 29 (96.7%) | 22 (73.3%) | 0.026 | 14 (82.4%) | 37 (86.05%) | 0.704 |
| Deep subcortical WMH | 28 (93.3%) | 22 (73.3%) | 0.080 | 15 (88.2%) | 35 (81.4) | 0.709 |
| Lobar CMB | 21 (70.0%) | 19 (63.3%) | 0.785 | 13 (76.5%) | 27 (62.8%) | 0.375 |
| EPVS | 25 (83.3%) | 28 (93.3%) | 0.424 | 13 (76.5%) | 40 (93.0%) | 0.092 |
| EPVS basal ganglia | 25 (83.3%) | 27 (90.0%) | 0.706 | 12 (70.6%) | 40 (93.0%) | 0.035 |
| EPVS CSO | 19 (63.3%) | 22 (73.3%) | 0.580 | 11 (64.7%) | 30 (69.8%) | 0.763 |
| cSS | 18 (60.0%) | 12 (40.0%) | 0.196 | 13 (76.5%) | 17 (39.5%) | 0.020 |
| Chronic infarct | 10 (33.3%) | 3 (11.1%) | 0.061 | 5 (33.3%) | 8 (19.0%) | 0.294 |
| Atrophy | 13 (43.3%) | 10 (33.3%) | 0.596 | 12 (70.6%) | 11 (25.6%) | 0.003 |
| High SVD burden (score 4–6) | 21 (55.53%) | 17 (44.7%) | 0.422 | 15 (88.2%) | 23 (53.5%) | 0.017 |
| CAA–ICH subcohort 2 (n = 25) | |||
|---|---|---|---|
| Variable | Poor outcome | Good outcome | p-Value |
| AQP4 | 1.56 (1.42–1.81) n = 8 | 4.20 (2.33–4.89) n = 17 | 0.002 |
| Age | 77 (74.5–81) | 77 (72–79) | 0.549 |
| Sex, female | 6 (75.0%) | 9 (52.9%) | 0.294 |
| Hypertension | 3 (42.9%) | 11 (64.7%) | 0.324 |
| Diabetes | 1 (14.3%) | 4 (25.7%) | 0.519 |
| Dyslipidemia | 2 (25.0%) | 1 (7.7%) | 0.271 |
| APOE genotype, ε2 carriers | 1 (12.5%) | 2 (11.8%) | 0.958 |
| APOE genotype, ε4 carriers | 2 (25.0%) | 3 (17.5%) | 0.668 |
| Cognitive impairment | 6 (75%) | 6 (35.3%) | 0.064 |
| ≥2 lobar ICHs | 3 (37.5%) | 3 (17.6%) | 0.278 |
| WMH | 8 (100%) | 16 (94.1%) | 0.484 |
| Periventricular | 8 (100%) | 13 (76.5%) | 0.134 |
| Deep subcortical WMH | 8 (100%) | 12 (70.6%) | 0.086 |
| Lobar CMB | 7 (87.5%) | 10 (58.8%) | 0.152 |
| EPVS | 8 (100%) | 17 (100%) | - |
| EPVS basal ganglia | 7 (87.5%) | 17 (100%) | 0.137 |
| EPVS CSO | 8 (100%) | 15 (88.2%) | 0.312 |
| cSS | 3 (37.5%) | 9 (52.9%) | 0.471 |
| Chronic Infarct | 3 (42.9%) | 2 (11.8%) | 0.088 |
| Atrophy | 3 (37.5%) | 6 (35.3%) | 0.915 |
| High SVD burden (score 4–6) | 4 (50.0%) | 10 (58.8%) | 0.678 |
References
- Vinters, H.V. Cerebral amyloid angiopathy a critical review. Stroke 1987. [Google Scholar] [CrossRef]
- Weller, R.O.; Preston, S.D.; Subash, M.; Carare, R.O. Cerebral amyloid angiopathy in the aetiology and immunotherapy of Alzheimer disease. Alzheimers. Res. Ther. 2009, 1, 6. [Google Scholar] [CrossRef]
- Viswanathan, A.; Greenberg, S.M. Cerebral amyloid angiopathy in the elderly. Ann. Neurol. 2011, 70, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Charidimou, A.; Boulouis, G.; Gurol, M.E.; Ayata, C.; Bacskai, B.J.; Frosch, M.P.; Viswanathan, A.; Greenberg, S.M. Emerging concepts in sporadic cerebral amyloid angiopathy. Brain 2017, 140, 1829–1850. [Google Scholar] [CrossRef] [PubMed]
- Vinters, H.V. Emerging Concepts in Alzheimer’s Disease. Annu. Rev. Pathol. Mech. Dis. 2015. [Google Scholar] [CrossRef] [PubMed]
- Hanger, H.C.; Wilkinson, T.J.; Fayez-Iskander, N.; Sainsbury, R. The risk of recurrent stroke after intracerebral haemorrhage. J. Neurol. Neurosurg. Psychiatry 2007, 78, 836–840. [Google Scholar] [CrossRef] [PubMed]
- Charidimou, A.; Peeters, A.; Fox, Z.; Gregoire, S.M.; Vandermeeren, Y.; Laloux, P.; Jäger, H.R.; Baron, J.-C.; Werring, D.J. Spectrum of Transient Focal Neurological Episodes in Cerebral Amyloid Angiopathy. Stroke 2012, 43, 2324–2330. [Google Scholar] [CrossRef]
- Arvanitakis, Z.; Leurgans, S.E.; Wang, Z.; Wilson, R.S.; Bennett, D.A.; Schneider, J.A. Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Ann. Neurol. 2011. [Google Scholar] [CrossRef]
- Banerjee, G.; Carare, R.; Cordonnier, C.; Greenberg, S.M.; Schneider, J.A.; Smith, E.E.; van Buchem, M.; van der Grond, J.; Verbeek, M.M.; Werring, D.J. The increasing impact of cerebral amyloid angiopathy: Essential new insights for clinical practice. J. Neurol. Neurosurg. Psychiatry 2017, 88, 982–994. [Google Scholar] [CrossRef]
- Greenberg, S.M.; Charidimou, A. Diagnosis of Cerebral Amyloid Angiopathy. Stroke 2018, 49, 491–497. [Google Scholar] [CrossRef]
- Linn, J.; Halpin, A.; Demaerel, P.; Ruhland, J.; Giese, A.D.; Dichgans, M.; Van Buchem, M.A.; Bruckmann, H.; Greenberg, S.M. Prevalence of superficial siderosis in patients with cerebral amyloid angiopathy. Neurology 2010, 74, 1346–1350. [Google Scholar] [CrossRef] [PubMed]
- Charidimou, A.; Shakeshaft, C.; Werring, D.J. Cerebral microbleeds on magnetic resonance imaging and anticoagulant-associated intracerebral hemorrhage risk. Front. Neurol. 2012, 3, 133. [Google Scholar] [CrossRef] [PubMed]
- Doubal, F.N.; MacLullich, A.M.J.; Ferguson, K.J.; Dennis, M.S.; Wardlaw, J.M. Enlarged Perivascular Spaces on MRI Are a Feature of Cerebral Small Vessel Disease. Stroke 2010. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-C.; Chabriat, H.; Godin, O.; Dufouil, C.; Rosand, J.; Greenberg, S.M.; Smith, E.E.; Tzourio, C.; Viswanathan, A. Distribution of white matter hyperintensity in cerebral hemorrhage and healthy aging. J. Neurol. 2012, 259, 530–536. [Google Scholar] [CrossRef]
- Charidimou, A.; Meegahage, R.; Fox, Z.; Peeters, A.; Vandermeeren, Y.; Laloux, P.; Baron, J.C.; Jäger, H.R.; Werring, D.J. Enlarged perivascular spaces as a marker of underlying arteriopathy in intracerebral haemorrhage: A multicentre MRI cohort study. J. Neurol. Neurosurg. Psychiatry 2013. [Google Scholar] [CrossRef]
- Greenberg, S.M.; Bacskai, B.J.; Hernandez-Guillamon, M.; Pruzin, J.; Sperling, R.; van Veluw, S.J. Cerebral amyloid angiopathy and Alzheimer disease—One peptide, two pathways. Nat. Rev. Neurol. 2020, 16, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Gatti, L.; Tinelli, F.; Scelzo, E.; Arioli, F.; Di Fede, G.; Obici, L.; Pantoni, L.; Giaccone, G.; Caroppo, P.; Parati, E.A.; et al. Understanding the pathophysiology of cerebral amyloid angiopathy. Int. J. Mol. Sci. 2020, 10, 3435. [Google Scholar] [CrossRef] [PubMed]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain—implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef]
- Bakker, E.N.T.P.; Bacskai, B.J.; Arbel-Ornath, M.; Aldea, R.; Bedussi, B.; Morris, A.W.J.; Weller, R.O.; Carare, R.O. Lymphatic Clearance of the Brain: Perivascular, Paravascular and Significance for Neurodegenerative Diseases. Cell. Mol. Neurobiol. 2016, 36, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Ihara, M. New therapeutic approaches for Alzheimer’s disease and cerebral amyloid angiopathy. Front. Aging Neurosci. 2014, 6, 1–11. [Google Scholar] [CrossRef]
- Weller, R.O.; Subash, M.; Preston, S.D.; Mazanti, I.; Carare, R.O. SYMPOSIUM: Clearance of Aβ from the Brain in Alzheimer’s Disease: Perivascular Drainage of Amyloid-β Peptides from the Brain and Its Failure in Cerebral Amyloid Angiopathy and Alzheimer’s Disease. Brain Pathol. 2007, 18, 253–266. [Google Scholar] [CrossRef]
- Hawkes, C.A.; Jayakody, N.; Johnston, D.A.; Bechmann, I.; Carare, R.O. Failure of Perivascular Drainage of β-amyloid in Cerebral Amyloid Angiopathy. Brain Pathol. 2014, 24, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Day, R.E.; Kitchen, P.; Owen, D.S.; Bland, C.; Marshall, L.; Conner, A.C.; Bill, R.M.; Conner, M.T. Human aquaporins: Regulators of transcellular water flow. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 1492–1506. [Google Scholar] [CrossRef]
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin water channels in the nervous system. Nat. Rev. Neurosci. 2013, 14, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Mader, S.; Brimberg, L. Aquaporin-4 Water Channel in the Brain and Its Implication for Health and Disease. Cells 2019, 2, 90. [Google Scholar] [CrossRef]
- Abbott, N.J.; Pizzo, M.E.; Preston, J.E.; Janigro, D.; Thorne, R.G. The role of brain barriers in fluid movement in the CNS: Is there a ‘ glymphatic ’ system? Acta Neuropathol. 2018, 135, 387–407. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.K.; Mestre, H.; Nedergaard, M. The glymphatic pathway in neurological disorders. Lancet Neurol. 2018, 17, 1016–1024. [Google Scholar] [CrossRef]
- Lan, Y.-L.; Zhao, J.; Ma, T.; Li, S. The Potential Roles of Aquaporin 4 in Alzheimer’s Disease. Mol. Neurobiol. 2016, 53, 5300–5309. [Google Scholar] [CrossRef]
- Chu, H.; Huang, C.; Ding, H.; Dong, J.; Gao, Z.; Yang, X.; Tang, Y.; Dong, Q. Aquaporin-4 and cerebrovascular diseases. Int. J. Mol. Sci. 2016, 8, 1249. [Google Scholar] [CrossRef]
- Yang, C.; Huang, X.; Huang, X.; Mai, H.; Li, J.; Jiang, T.; Wang, X.; Lü, T. Aquaporin-4 and Alzheimer’s Disease. J. Alzheimer’s Dis. 2016, 52, 391–402. [Google Scholar] [CrossRef]
- Riba-Llena, I.; Jarca, C.I.; Mundet, X.; Tovar, J.L.; Orfila, F.; López-Rueda, A.; Nafría, C.; Fernández, J.L.; Castañé, X.; Domingo, M.; et al. Investigating silent strokes in hypertensives: A magnetic resonance imaging study (ISSYS): Rationale and protocol design. BMC Neurol. 2013, 13, 130. [Google Scholar] [CrossRef] [PubMed]
- Cordonnier, C.; Potter, G.M.; Jackson, C.A.; Doubal, F.; Keir, S.; Sudlow, C.L.M.; Wardlaw, J.M.; Salman, R.A.-S. Improving Interrater Agreement About Brain Microbleeds. Stroke 2009, 40, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Fazekas, F.; Chawluk, J.B.; Alavi, A.; Hurtig, H.I.; Zimmerman, R.A. MR signal abnormalities at 1.5 T in Alzheimer’s dementia and normal aging. Am. J. Roentgenol. 1987. [Google Scholar] [CrossRef]
- Wardlaw, J.M.; Smith, E.E.; Biessels, G.J.; Cordonnier, C.; Fazekas, F.; Frayne, R.; Lindley, R.I.; O’Brien, J.T.; Barkhof, F.; Benavente, O.R.; et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. 2013, 12, 822–838. [Google Scholar] [CrossRef]
- Charidimou, A.; Martinez-Ramirez, S.; Reijmer, Y.D.; Oliveira-filho, J.; Lauer, A.; Roongpiboonsopit, D.; Frosch, M.; Vashkevich, A.; Ayres, A.; Rosand, J.; et al. Total MRI small vessel disease burden in cerebral amyloid angiopathy: A concept validation imaging-pathological study. JAMA Neurol. 2016, 73, 994–1001. [Google Scholar] [CrossRef]
- Rosu, G.C.; Catalin, B.; Balseanu, T.A.; Laurentiu, M.; Claudiu, M.; Kumar-Singh, S.; Daniel, P. Inhibition of Aquaporin 4 Decreases Amyloid Aβ40 Drainage Around Cerebral Vessels. Mol. Neurobiol. 2020. [Google Scholar] [CrossRef]
- Wilcock, D.M.; Vitek, M.P.; Colton, C.A. Vascular amyloid alters astrocytic water and potassium channels in mouse models and humans with Alzheimer’s disease. Neuroscience 2009. [Google Scholar] [CrossRef] [PubMed]
- Moftakhar, P.; Lynch, M.D.; Pomakian, J.L.; Vinters, H.V. Aquaporin expression in the brains of patients with or without cerebral amyloid angiopathy. J. Neuropathol. Exp. Neurol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Hoshi, A.; Tsunoda, A.; Yamamoto, T.; Tada, M.; Kakita, A.; Ugawa, Y. Altered expression of glutamate transporter-1 and water channel protein aquaporin-4 in human temporal cortex with Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Boespflug, E.L.; Simon, M.J.; Leonard, E.; Grafe, M.; Woltjer, R.; Silbert, L.C.; Kaye, J.A.; Iliff, J.J. Targeted Assessment of Enlargement of the Perivascular Space in Alzheimer’s Disease and Vascular Dementia Subtypes Implicates Astroglial Involvement Specific to Alzheimer’s Disease. J. Alzheimer’s Dis. 2018. [Google Scholar] [CrossRef]
- Hoshi, A.; Yamamoto, T.; Shimizu, K.; Ugawa, Y.; Nishizawa, M.; Takahashi, H.; Kakita, A. Characteristics of Aquaporin Expression Surrounding Senile Plaques and Cerebral Amyloid Angiopathy in Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2012, 71, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Owasil, R.; O’neill, R.; Keable, A.; Nimmo, J.; Sharp, M.M.; Kelly, L.; Saito, S.; Simpson, J.E.; Weller, R.O.; Smith, C.; et al. The pattern of AQP4 expression in the ageing human brain and in cerebral amyloid angiopathy. Int. J. Mol. Sci. 2020, 4, 1225. [Google Scholar] [CrossRef]
- Debette, S.; Markus, H.S. The clinical importance of white matter hyperintensities on brain magnetic resonance imaging: Systematic review and meta-analysis. BMJ 2010, 341, c3666. [Google Scholar] [CrossRef] [PubMed]
- Mortamais, M.; Artero, S.; Ritchie, K. White Matter Hyperintensities as Early and Independent Predictors of Alzheimer’s Disease Risk. J. Alzheimer’s Dis. 2014, 42, S393–S400. [Google Scholar] [CrossRef]
- Fan, Y.; Liu, M.; Wu, X.; Wang, F.; Ding, J.; Chen, J.; Hu, G. Aquaporin-4 promotes memory consolidation in Morris water maze. Brain Struct. Funct. 2013. [Google Scholar] [CrossRef] [PubMed]
- Skucas, V.A.; Mathews, I.B.; Yang, J.; Cheng, Q.; Treister, A.; Duffy, A.M.; Verkman, A.S.; Hempstead, B.L.; Wood, M.A.; Binder, D.K.; et al. Impairment of select forms of spatial memory and neurotrophin-dependent synaptic plasticity by deletion of glial aquaporin-4. J. Neurosci. 2011. [Google Scholar] [CrossRef]
- Zhang, J.; Li, Y.; Chen, Z.G.; Dang, H.; Ding, J.H.; Fan, Y.; Hu, G. Glia protein aquaporin-4 regulates aversive motivation of spatial memory in morris water maze. CNS Neurosci. Ther. 2013. [Google Scholar] [CrossRef]
- Xu, Z.; Xiao, N.; Chen, Y.; Huang, H.; Marshall, C.; Gao, J.; Cai, Z.; Wu, T.; Hu, G.; Xiao, M. Deletion of aquaporin-4 in APP/PS1 mice exacerbates brain Aβ accumulation and memory deficits. Mol. Neurodegener. 2015. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Ikegawa, N.; Yoshida, K.; Muramatsu, K.; Hattori, S.; Kawai, K.; Murakami, M.; Tanaka, T.; Goda, W.; Goto, M.; et al. Behavioral and electrophysiological evidence for a neuroprotective role of aquaporin-4 in the 5xFAD transgenic mice model. Acta Neuropathol. Commun. 2020. [Google Scholar] [CrossRef] [PubMed]
- Boulouis, G.; Charidimou, A.; Pasi, M.; Roongpiboonsopit, D.; Xiong, L.; Auriel, E.; van Etten, E.S.; Martinez-Ramirez, S.; Ayres, A.; Vashkevich, A.; et al. Hemorrhage recurrence risk factors in cerebral amyloid angiopathy: Comparative analysis of the overall small vessel disease severity score versus individual neuroimaging markers. J. Neurol. Sci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wu, P.; Su, J.; Xiang, J.; Cai, D.; Dong, Q. Effects of Aquaporin-4 on edema formation following intracerebral hemorrhage. Exp. Neurol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Xiang, J.; Wu, P.; Su, J.; Ding, H.; Tang, Y.; Dong, Q. The role of aquaporin 4 in apoptosis after intracerebral hemorrhage. J. Neuroinflammation 2014. [Google Scholar] [CrossRef] [PubMed]
- Appelboom, G.; Bruce, S.; Duren, A.; Piazza, M.; Monahan, A.; Christophe, B.; Zoller, S.; LoPresti, M.; Connolly, E.S. Aquaporin-4 gene variant independently associated with oedema after intracerebral haemorrhage. Neurol. Res. 2015. [Google Scholar] [CrossRef] [PubMed]
- Dardiotis, E.; Siokas, V.; Marogianni, C.; Aloizou, A.M.; Sokratous, M.; Paterakis, K.; Dardioti, M.; Grigoriadis, S.; Brotis, A.; Kapsalaki, E.; et al. AQP4 tag SNPs in patients with intracerebral hemorrhage in Greek and Polish population. Neurosci. Lett. 2019. [Google Scholar] [CrossRef]
- O’Donnell, H.C.; Rosand, J.; Knudsen, K.A.; Furie, K.L.; Segal, A.Z.; Chiu, R.I.; Ikeda, D.; Greenberg, S.M. Apolipoprotein E Genotype and the Risk of Recurrent Lobar Intracerebral Hemorrhage. N. Engl. J. Med. 2000. [Google Scholar] [CrossRef]
- Brouwers, H.B.; Biffi, A.; McNamara, K.A.; Ayres, A.M.; Valant, V.; Schwab, K.; Romero, J.M.; Viswanathan, A.; Greenberg, S.M.; Rosand, J.; et al. Apolipoprotein e genotype is associated with ct angiography spot sign in lobar intracerebral hemorrhage. Stroke 2012. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, J.A.R.; McCarron, M.O. APOE gene polymorphism as a risk factor for cerebral amyloid angiopathy-related hemorrhage. Amyloid 2001, 8, 51–55. [Google Scholar] [PubMed]
- Nicchia, G.P.; Nico, B.; Camassa, L.M.A.; Mola, M.G.; Loh, N.; Dermietzel, R.; Spray, D.C.; Svelto, M.; Frigeri, A. The role of aquaporin-4 in the blood-brain barrier development and integrity: Studies in animal and cell culture models. Neuroscience 2004. [Google Scholar] [CrossRef]
- Saadoun, S.; Tait, M.J.; Reza, A.; Davies, D.C.; Bell, B.A.; Verkman, A.S.; Papadopoulos, M.C. AQP4 gene deletion in mice does not alter blood-brain barrier integrity or brain morphology. Neuroscience 2009. [Google Scholar] [CrossRef]
- Chu, H.; Tang, Y.; Dong, Q. Protection of granulocyte-colony stimulating factor to hemorrhagic brain injuries and its involved mechanisms: Effects of vascular endothelial growth factor and aquaporin-4. Neuroscience 2014. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Kong, H.; Hua, X.; Xiao, M.; Ding, J.; Hu, G. Altered blood-brain barrier integrity in adult aquaporin-4 knockout mice. Neuroreport 2008. [Google Scholar] [CrossRef] [PubMed]
- Nico, B.; Frigeri, A.; Nicchia, G.P.; Quondamatteo, F.; Herken, R.; Errede, M.; Ribatti, D.; Svelto, M.; Roncali, L. Role of aquaporin-4 water channel in the development and integrity of the blood-brain barrier. J. Cell Sci. 2001, 129, 935–945. [Google Scholar]
- Ramiro, L.; Simats, A.; Penalba, A.; Garcia-Tornel, A.; Rovira, A.; Mancha, F.; Bustamante, A.; Montaner, J. Circulating Aquaporin-4 as A biomarker of early neurological improvement in stroke patients: A pilot study. Neurosci. Lett. 2020. [Google Scholar] [CrossRef] [PubMed]


| Variable | Control (n = 19) | CAA–ICH (n = 60) | p-Value |
|---|---|---|---|
| Age, years, median (IQR) | 74 (73.5–74) | 76.5 (71.5–70) | 0.130 |
| Sex, female, n (%) | 10 (52.6%) | 30 (50%) | 1 |
| Hypertension | 19 (100%) | 29 (48.3%) | 0.000 |
| Diabetes | 6 (31.3%) | 7 (11.7%) | 0.063 |
| Dyslipidemia | 16 (78.9%) | 17 (28.3%) | 0.000 |
| APOE genotype, ε2 carriers | 1 (5.3%) | 8 (13.3%) | 0.679 |
| APOE genotype, ε4 carriers | 7 (36.8%) | 14 (23.3%) | 0.251 |
| Lobar ICH | 0 (0.0%) | 60 (100%) | 0.000 |
| WMH, n (%) | 9 (47.4%) | 57 (95.0%) | 0.000 |
| CMB | 0 (0.0%) | 40 (66.7%) | 0.000 |
| Serum AQP4, ng/mL, median (IQR) | 2.12 (1.63–2.67) | 2.15 (1.44–4.12) | 0.626 |
| CAA–ICH (n = 60) | |
|---|---|
| Boston Criteria | |
| Possible | 12 (20.0%) |
| Probable | 45 (75.0%) |
| Probable with supporting pathology | 3 (5.0%) |
| WMH | 57 (95.0%) |
| Periventricular | 51 (85.0%) |
| Moderate (1–2 Fazekas) | 10 (16.7%) |
| Severe (3–4 Fazekas) | 41 (68.3%) |
| Deep subcortical WMH | 52 (86.7%) |
| Moderate (1–2 Fazekas) | 21 (35.0%) |
| Severe (3–4 Fazekas) | 31 (51.6%) |
| CMB | 40 (66.7%) |
| Lobar CMB | 40 (66.7%) |
| 1–5 | 14 (23.3%) |
| 6–10 | 9 (15.0%) |
| 10–20 | 3 (5.0%) |
| >20 | 14 (23.3%) |
| Deep CMB | 0 (0.0%) |
| Cerebellar CMB | 4 (6.7%) |
| EPVS | 53 (88.3%) |
| EPVS basal ganglia | 52 (86.7%) |
| Moderate (1–20) | 43 (71.7%) |
| Severe (21 to >40) | 9 (15.0%) |
| EPVS CSO | 41 (68.3%) |
| Moderate (1–20) | 19 (31.7%) |
| Severe (21 to >40) | 22 (36.7%) |
| cSS | 30 (50.0%) |
| Focal | 9 (15.0%) |
| Disseminated | 21 (35.0%) |
| Atrophy | 23 (38.3%) |
| Small vessel disease burden | |
| Low (0–3) | 22 (36.7%) |
| High (4–6) | 38 (63.3%) |
| Variable | YES | NO | p-Value |
|---|---|---|---|
| Age | r = 0.147 | 0.264 | |
| Sex, female | 2.19 (1.46–4.20) n = 30 | 2.08 (1.41–4.04) n = 30 | 0.684 |
| Hypertension | 1.85 (1.45–3.29) n = 29 | 2.89 (1.77–4.27) n = 28 | 0.102 |
| Diabetes | 2.60 (2.11–3.58) n = 7 | 2.14 (1.44–4.27) n = 48 | 0.435 |
| Dyslipidemia | 1.81 (1.41–2.89) n = 17 | 2.11 (1.44–4.41) n = 36 | 0.331 |
| APOE genotype, ε2 carriers | 1.75 (1.18–3.00) n = 8 | 2.30 (1.45–4.27) n = 52 | 0.317 |
| APOE genotype, ε4 carriers | 1.46 (1.03–2.60) n = 14 | 2.41 (1.65–4.20) n = 46 | 0.028 |
| Cognitive impairment | 1.69 (1.27–2.76) n = 30 | 3.09 (1.81–4.38) n = 30 | 0.030 |
| Previous stroke | 1.28 (0.99–1.67) n = 12 | 2.68 (1.69–4.35) n = 48 | 0.002 |
| Previous ischemic stroke | 1.53 (1.26–3.27) n = 4 | 2.68 (1.69–4.35) n = 48 | 0.261 |
| Previous hemorrhagic stroke | 1.12 (0.79–1.61) n = 8 | 2.68 (1.69–4.35) n = 48 | 0.001 |
| Variable | YES | NO | p-Value |
|---|---|---|---|
| Interval between the last ICH and the date of blood collection | r = –0.052 | 0.703 | |
| Number of lobar ICHs | r = –0.307 | 0.017 | |
| WMH | 2.04 (1.43–3.71) n = 57 | 4.46 (3.15–5.7) n = 3 | 0.163 |
| Periventricular | 2.02 (1.42–3.69) n = 51 | 3.29 (1.84–5.02) n = 9 | 0.092 |
| Deep subcortical WMH | 1.88 (1.42–3.67) n = 50 | 3.41 (2.04–5.19) n = 10 | 0.045 |
| Lobar CMB | 1.83 (1.41–3.79) n = 40 | 2.84 (1.93–4.25) n = 20 | 0.052 |
| EPVS | 2.26 (1.46–4.04) n = 53 | 1.81 (1.27–3.78) n = 7 | 0.718 |
| EPVS basal ganglia | 2.30 (1.47–4.12) n = 52 | 1.63 (1.27–3.78) n = 8 | 0.521 |
| EPVS CSO | 2.45 (1.50–4.36) n = 41 | 1.84 (1.10–3.00) n = 19 | 0.144 |
| cSS | 2.11 (1.16–4.38) n = 30 | 2.15 (1.65–2.90) n = 30 | 0.988 |
| Chronic Infarct | 1.53 (1.27–2.02) n = 13 | 2.52 (1.55–4.19) n = 44 | 0.146 |
| Atrophy | 2.26 (1.43–4.00) n = 23 | 2.04 (1.50–4.04) n = 37 | 0.715 |
| High SVD burden (score 4–6) | 2.35 (1.27–4.34) n = 23 | 2.03 (1.81–2.90) n = 37 | 0.570 |
| Regression Cognitive Impairment | Regression ≥ 2 ICHs | |
|---|---|---|
| Variable | OR (95% CI) p-value | OR (95% CI) p-value |
| AQP4 | - | 0.520 (0.286–0.976) p = 0.042 |
| APOE genotype, ε2 carriers | - | 22.536 (1.989–255.296) p = 0.034 |
| WMH periventricular | 10.545 (1.227–90.662) p = 0.032 | - |
| Atrophy | - | 6.167 (1.080–35.213) p = 0.004 |
| High SVD burden (score 4–6) | - | 11.280 (1.109–114.739) p = 0.025 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marazuela, P.; Bonaterra-Pastra, A.; Faura, J.; Penalba, A.; Pizarro, J.; Pancorbo, O.; Rodríguez-Luna, D.; Vert, C.; Rovira, A.; Pujadas, F.; et al. Circulating AQP4 Levels in Patients with Cerebral Amyloid Angiopathy-Associated Intracerebral Hemorrhage. J. Clin. Med. 2021, 10, 989. https://doi.org/10.3390/jcm10050989
Marazuela P, Bonaterra-Pastra A, Faura J, Penalba A, Pizarro J, Pancorbo O, Rodríguez-Luna D, Vert C, Rovira A, Pujadas F, et al. Circulating AQP4 Levels in Patients with Cerebral Amyloid Angiopathy-Associated Intracerebral Hemorrhage. Journal of Clinical Medicine. 2021; 10(5):989. https://doi.org/10.3390/jcm10050989
Chicago/Turabian StyleMarazuela, Paula, Anna Bonaterra-Pastra, Júlia Faura, Anna Penalba, Jesús Pizarro, Olalla Pancorbo, David Rodríguez-Luna, Carla Vert, Alex Rovira, Francesc Pujadas, and et al. 2021. "Circulating AQP4 Levels in Patients with Cerebral Amyloid Angiopathy-Associated Intracerebral Hemorrhage" Journal of Clinical Medicine 10, no. 5: 989. https://doi.org/10.3390/jcm10050989
APA StyleMarazuela, P., Bonaterra-Pastra, A., Faura, J., Penalba, A., Pizarro, J., Pancorbo, O., Rodríguez-Luna, D., Vert, C., Rovira, A., Pujadas, F., Freijo, M. M., Tur, S., Martínez-Zabaleta, M., Cardona Portela, P., Vera, R., Lebrato-Hernández, L., Arenillas, J. F., Pérez-Sánchez, S., Montaner, J., ... Hernández-Guillamon, M. (2021). Circulating AQP4 Levels in Patients with Cerebral Amyloid Angiopathy-Associated Intracerebral Hemorrhage. Journal of Clinical Medicine, 10(5), 989. https://doi.org/10.3390/jcm10050989