
Inflammatory Biomarkers in Postural Orthostatic Tachycardia Syndrome with Elevated G-Protein-Coupled Receptor Autoantibodies

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
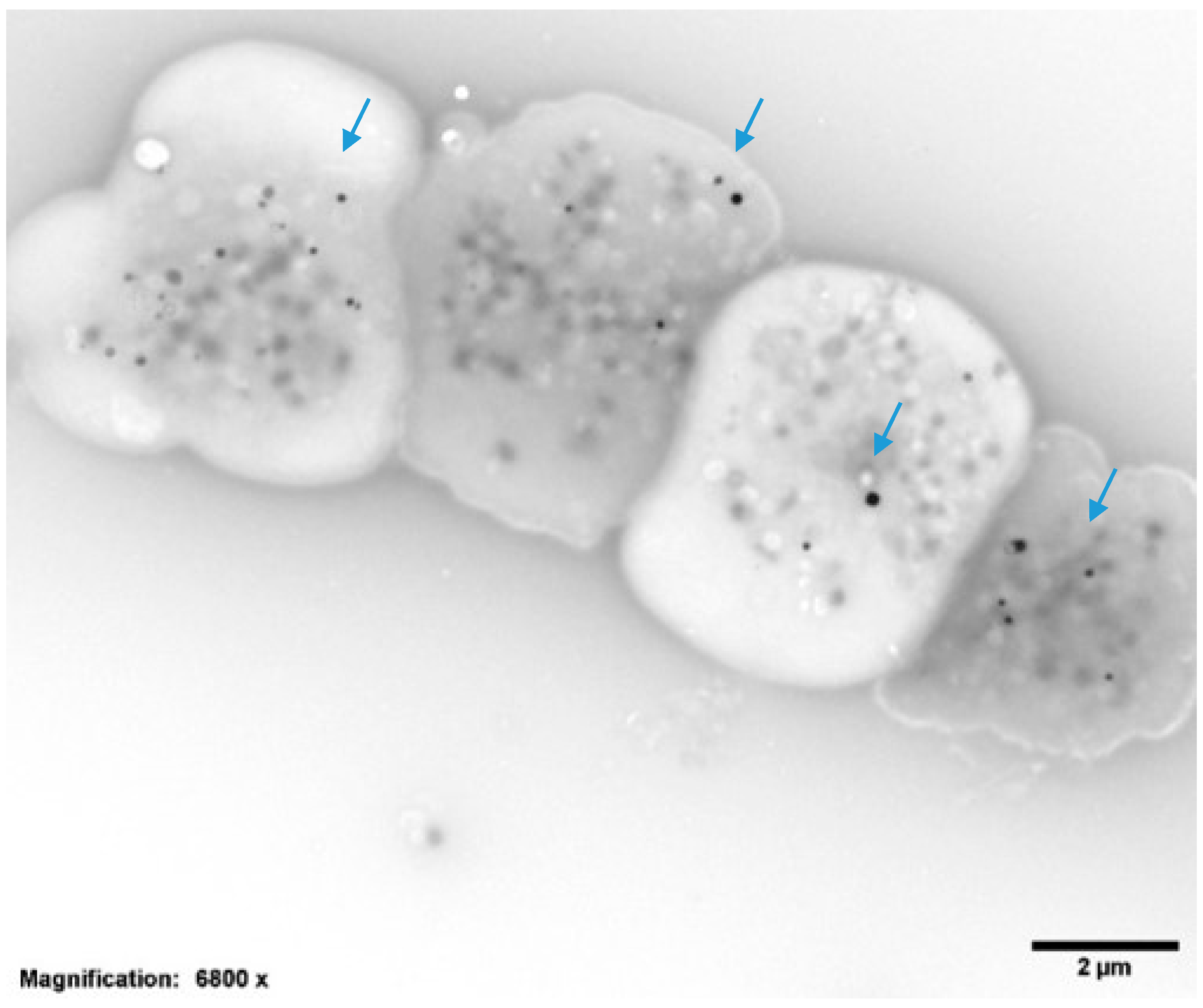
2.2. Platelet Preparations for Electron Microscopy
2.3. ELISA Sample Preparation
2.4. Inflammation Biomarker Preparations
2.5. Statistical Methods
3. Results
3.1. Bleeding Assessments
3.2. Complete Blood Cell Count and Platelet Storage Pool
3.3. Biomarkers of Inflammation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Low, P.A.; Opfer-Gehrking, T.L.; Textor, S.C.; Benarroch, E.E.; Shen, W.K.; Schondorf, R.; Suarez, G.A.; Rummans, T.A. Pos-tural tachycardia syndrome (POTS). Neurology 1995, 45, S19–S25. [Google Scholar]
- Raj, S.R. The Postural Tachycardia Syndrome (POTS): Pathophysiology, Diagnosis & Management. Indian Pacing Electrophysiol. J. 2006, 6, 84–99. [Google Scholar] [PubMed]
- Mar, P.L.; Raj, S.R. Neuronal and hormonal perturbations in postural tachycardia syndrome. Front. Physiol. 2014, 5, 220. [Google Scholar] [CrossRef] [PubMed]
- Garland, E.M.; Raj, S.R.; Black, B.K.; Harris, P.A.; Robertson, D. The hemodynamic and neurohumoral phenotype of postural tachycardia syndrome. Neurology 2007, 69, 790–798. [Google Scholar] [CrossRef] [PubMed]
- Peggs, K.J.; Nguyen, H.; Enayat, D.; Keller, N.R.; Al-Hendy, A.; Raj, S.R. Gynecologic disorders and menstrual cycle lightheadedness in postural tachycardia syndrome. Int. J. Gynecol. Obstet. 2012, 118, 242–246. [Google Scholar] [CrossRef]
- Boris, J.R.; Bernadzikowski, T. Demographics of a large paediatric Postural Orthostatic Tachycardia Syndrome Program. Cardiol. Young 2018, 28, 668–674. [Google Scholar] [CrossRef]
- Gunning, W.T., III; Karabin, B.L.; Blomquist, T.M.; Grubb, B.P. Postural orthostatic tachycardia syndrome is associated with platelet storage pool deficiency. Medicine 2016, 95, e4849. [Google Scholar] [CrossRef]
- Guo, Y.; Walsh, A.M.; Fearon, U.; Smith, M.D.; Wechalekar, M.D.; Yin, X.; Cole, S.; Orr, C.; McGarry, T.; Canavan, M.; et al. CD40L-Dependent Pathway Is Active at Various Stages of Rheumatoid Arthritis Disease Progression. J. Immunol. 2017, 198, 4490–4501. [Google Scholar] [CrossRef]
- Benarroch, E.E. Postural Tachycardia Syndrome: A Heterogeneous and Multifactorial Disorder. Mayo Clin. Proc. 2012, 87, 1214–1225. [Google Scholar] [CrossRef]
- Sheldon, R.S.; Grubb, B.P.; Olshansky, B.; Shen, W.-K.; Calkins, H.; Brignole, M.; Raj, S.R.; Krahn, A.D.; Morillo, C.A.; Stewart, J.M.; et al. 2015 Heart Rhythm Society Expert Consensus Statement on the Diagnosis and Treatment of Postural Tachycardia Syndrome, Inappropriate Sinus Tachycardia, and Vasovagal Syncope. Heart. Rhythm. 2015, 12, e41–e63. [Google Scholar] [CrossRef]
- Shaw, B.H.; Stiles, L.E.; Bourne, K.; Green, E.A.; Shibao, C.A.; Okamoto, L.E.; Garland, E.M.; Gamboa, A.; Diedrich, A.; Raj, V.; et al. The face of postural tachycardia syndrome—Insights from a large cross-sectional online community-based survey. J. Intern. Med. 2019, 286, 438–448. [Google Scholar] [CrossRef]
- Vernino, S.; Adamski, J.; Kryzer, T.J.; Fealey, R.D.; Lennon, V.A. Neuronal nicotinic ACh receptor antibody in subacute autonomic neuropathy and cancer-related syndromes. Neurology 1998, 50, 1806–1813. [Google Scholar] [CrossRef]
- Thieben, M.J.; Sandroni, P.; Sletten, D.M.; Benrud-Larson, L.M.; Fealey, R.D.; Vernino, S.; Low, P.A.; Lennon, V.A.; Shen, W.-K. Postural Orthostatic Tachycardia Syndrome: The Mayo Clinic Experience. Mayo Clin. Proc. 2007, 82, 308–313. [Google Scholar] [CrossRef]
- Sandroni, P.; Vernino, S.; Klein, C.M.; Lennon, V.A.; Benrud-Larson, L.; Sletten, D.; Low, P. Idiopathic Autonomic Neuropathy. Arch. Neurol. 2004, 61, 44–48. [Google Scholar] [CrossRef]
- Vernino, S.; Lennon, V.A. Neuronal ganglionic acetylcholine receptor autoimmunity. Ann. N. Y. Acad. Sci. 2003, 998, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.M.; Vernino, S.; Lennon, V.A.; Sandroni, P.; Fealey, R.D.; Benrud-Larson, L.; Sletten, D.; Low, P. The spectrum of autoimmune autonomic neuropathies. Ann. Neurol. 2003, 53, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, G.; Zhou, L.; Nuss, Z.; Beel, M.; Hines, B.; Murphy, T.; Liles, J.; Zhang, L.; Kem, D.C.; et al. Adrenergic Autoantibody-Induced Postural Tachycardia Syndrome in Rabbits. J. Am. Heart Assoc. 2019, 8, e013006. [Google Scholar] [CrossRef] [PubMed]
- Gunning, W.T., III; Kvale, H.; Kramer, P.M.; Karabin, B.L.; Grubb, B.P. Postural Orthostatic Tachycardia Syndrome Is Associated with Elevated G-Protein Coupled Receptor Autoantibodies. J. Am. Heart Assoc. 2019, 8, e013602. [Google Scholar] [CrossRef] [PubMed]
- Fedorowski, A.; Hongliang, L.; Xichun, Y.; Kristi, A.K.; Valerie, M.H.; Campbell, L.; Taylor, A.M.; Syed, M.S.Q.; Robert, H.S.; Richard, S.; et al. Postural orthostatic tachycardia syndrome is associated with platelet storage pool deficiency. Europace 2017, 7, 1211–1219. [Google Scholar] [CrossRef]
- Yu, X.; Li, H.; Murphy, T.A.; Nuss, Z.; Liles, J.; Liles, C.; Aston, C.E.; Raj, S.R.; Fedorowski, A.; Kem, D.C. Angiotensin II Type 1 Receptor Autoantibodies in Postural Tachycardia Syndrome. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef]
- Li, H.; Yu, X.; Liles, C.; Khan, M.; Vanderlinde-Wood, M.; Galloway, A.; Zillner, C.; Benbrook, A.; Reim, S.; Collier, D.; et al. Autoimmune Basis for Postural Tachycardia Syndrome. J. Am. Heart Assoc. 2014, 3, e000755. [Google Scholar] [CrossRef]
- Blitshteyn, S.; Brinth, L.; Hendrickson, J.E.; Martinez-Lavin, M. Autonomic dysfunction and HPV immunization: An over-view. Immunol. Res. 2018. [Google Scholar] [CrossRef]
- Barboi, A.; Gibbons, C.H.; Axelrod, F.; Benarroch, E.E.; Biaggioni, I.; Chapleau, M.W.; Chelimsky, G.; Chelimsky, T.; Cheshire, W.P.; Claydon, V.E.; et al. Human papillomavirus (HPV) vaccine and autonomic disorders: A position statement from the American Autonomic Society. Auton. Neurosci. 2020, 223, 13–18. [Google Scholar] [CrossRef]
- Ward, D.; Thorsen, N.M.; Frisch, M.; Valentiner-Branth, P.; Mølbak, K.; Hviid, A. A cluster analysis of serious adverse event reports after human papillomavirus (HPV) vaccination in Danish girls and young women, September 2009 to August 2017. Eurosurveillance 2019, 24, 1800380. [Google Scholar] [CrossRef]
- Bonamichi-Santos, R.; Yoshimi-Kanamori, K.; Giavina-Bianchi, P.; Aun, M.V. Association of Postural Tachycardia Syndrome and Ehlers-Danlos Syndrome with Mast Cell Activation Disorders. Immunol. Allergy Clin. N. Am. 2018, 38, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Arnold, A.C.; Okamoto, L.E.; Diedrich, A.; Paranjape, S.Y.; Raj, S.R.; Biaggioni, I.; Gamboa, A. Low-dose propranolol and exercise capacity in postural tachycardia syndrome: A randomized study. Neurology 2013, 80, 1927–1933. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, K.; Nair, S.; Kulkarni, B.; Khare, A.; Shetty, S.; Mohanty, D. Platelet function tests using platelet aggregometry: Need for repetition of the test for diagnosis of defective platelet function. Platelets 2003, 14, 351–354. [Google Scholar] [CrossRef] [PubMed]
- Lewandowski, K.; Zozulińska, M.; Samborski, W.; Zawilska, K. Acquired platelet storage pool deficiency in rheumatoid arthritis. Pol. Arch. Intern. Med. 1991, 86, 46–52. [Google Scholar]
- Łukasik, Z.M.; Makowski, M.A.; Makowska, J.S. From blood coagulation to innate and adaptive immunity: The role of platelets in the physiology and pathology of autoimmune disorders. Rheumatol. Int. 2018, 38, 959–974. [Google Scholar] [CrossRef] [PubMed]
- Tunjungputri, R.N.; Li, Y.; De Groot, P.G.; Dinarello, C.A.; Smeekens, S.P.; Jaeger, M.; Doppenberg-Oosting, M.; Cruijsen, M.; Lemmers, H.; Toenhake-Dijkstra, H.; et al. The Inter-Relationship of Platelets with Interleukin-1β-Mediated Inflammation in Humans. Thromb. Haemost. 2018, 118, 2112–2125. [Google Scholar] [CrossRef]
- Janssen, C.A.; Scholten, P.C.; Heintz, A.P. A simple visual assessment technique to discriminate between menorrhagia and normal menstrual blood loss. Obstet. Gynecol. 1995, 85, 977–982. [Google Scholar] [CrossRef]
- Tosetto, A.; Castaman, G.; Rodeghiero, F. Assessing bleeding in von Willebrand disease with bleeding score. Blood Rev. 2007, 21, 89–97. [Google Scholar] [CrossRef]
- Jain, S.; Zhang, S.; Acosta, M.; Malone, K.; Kouides, P.; Zia, A. Prospective evaluation of ISTH-BAT as a predictor of bleeding disorder in adolescents presenting with heavy menstrual bleeding in a multidisciplinary hematology clinic. J. Thromb. Haemost. 2020, 18, 2542–2550. [Google Scholar] [CrossRef] [PubMed]
- Gunning, W.T., III; Raghavan, M.; Calomeni, E.P.; Turner, J.; Roysam, B.; Roysam, S.; Smith, M.R.; Kouides, P.A.; Lachant, N.A.; Gunning, I.W.T. A Morphometric Analysis of Platelet Dense Granules of Patients with Unexplained Bleeding: A New Entity of Delta-Microgranular Storage Pool Deficiency. J. Clin. Med. 2020, 9, 1734. [Google Scholar] [CrossRef] [PubMed]
- Brunet, J.G.; Iyer, J.K.; Badin, M.S.; Graf, L.; Moffat, K.A.; Timleck, M.; Spitzer, E.; Hayward, C.P.M. Electron microscopy ex-amination of platelet whole mount preparations to quantitate platelet dense granule numbers: Implications for diagnosing suspected platelet function disorders due to dense granule deficiency. Int. J. Lab. Hematol. 2018, 40, 400–407. [Google Scholar] [CrossRef]
- Giil, L.M.; Aarsland, D.; Hellton, K.; Lund, A.; Heidecke, H.; Schulze-Forster, K.; Riemekasten, G.; Vik-Mo, A.O.; Kristoffersen, E.K.; Vedeler, C.A.; et al. Antibodies to Multiple Receptors are Associated with Neuropsychiatric Symptoms and Mortality in Alzheimer’s Disease: A Longitudinal Study. J. Alzheimer’s Dis. 2018, 64, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Loebel, M.; Grabowski, P.; Heidecke, H.; Bauer, S.; Hanitsch, L.G.; Wittke, K.; Meisel, C.; Reinke, P.; Volk, H.-D.; Fluge, Ø.; et al. Antibodies to β adrenergic and muscarinic cholinergic receptors in patients with Chronic Fatigue Syndrome. Brain Behav. Immun. 2016, 52, 32–39. [Google Scholar] [CrossRef]
- Ponomarev, E.D. Fresh Evidence for Platelets as Neuronal and Innate Immune Cells: Their Role in the Activation, Differentiation, and Deactivation of Th1, Th17, and Tregs during Tissue Inflammation. Front. Immunol. 2018, 9, 406. [Google Scholar] [CrossRef]
- Selle, F.; James, C.; Tuffigo, M.; Pillois, X.; Viallard, J.-F.; Alessi, M.-C.; Fiore, M. Clinical and Laboratory Findings in Patients with δ-Storage Pool Disease: A Case Series. Semin. Thromb. Hemost. 2016, 43, 48–58. [Google Scholar] [CrossRef]
- Yun, S.-H.; Sim, E.-H.; Goh, R.-Y.; Park, J.-I.; Han, J. Platelet Activation: The Mechanisms and Potential Biomarkers. BioMed. Res. Int. 2016, 2016, 1–5. [Google Scholar] [CrossRef]
- Mantovani, A.; Dinarello, C.A.; Molgora, M.; Garlanda, C. Interleukin-1 and Related Cytokines in the Regulation of Inflammation and Immunity. Immunity 2019, 50, 778–795. [Google Scholar] [CrossRef]
- Akdis, M.; Aab, A.; Altunbulakli, C.; Azkur, K.; Costa, R.A.; Crameri, R.; Duan, S.; Eiwegger, T.; Eljaszewicz, A.; Ferstl, R.; et al. Interleukins (from IL-1 to IL-38), interferons, transforming growth factor β, and TNF-α: Receptors, functions, and roles in diseases. J. Allergy Clin. Immunol. 2016, 138, 984–1010. [Google Scholar] [CrossRef]
- Ferreira, V.L.; Borba, H.H.; Bonetti, A.D.F.; Leonart, L.P.; Pontarolo, R. Cytokines and Interferons: Types and Functions. In Autoantibodies and Cytokines; IntechOpen: London, UK, 2019; ISBN 978-1-83962-130-7. [Google Scholar]
- Cantarini, L.; Lopalco, G.; Cattalini, M.; Vitale, A.; Galeazzi, M.; Rigante, D. Interleukin-1: Ariadne’s Thread in Autoinflam-matory and Autoimmune Disorders. Isr. Med. Assoc. J. 2015, 17, 93–97. [Google Scholar] [PubMed]
- Van Kempen, T.S.; Wenink, M.H.; Leijten, E.F.A.; Radstake, T.R.D.J.; Boes, M. Perception of self: Distinguishing autoimmunity from autoinflammation. Nat. Rev. Rheumatol. 2015, 11, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Havnaer, A.; Han, G. Autoinflammatory Disorders: A Review and Update on Pathogenesis and Treatment. Am. J. Clin. Dermatol. 2019, 20, 539–564. [Google Scholar] [CrossRef]
- Rolfes, V.; Ribeiro, L.S.; Hawwari, I.; Böttcher, L.; Rosero, N.; Maasewerd, S.; Santos, M.L.S.; Próchnicki, T.; Silva, C.M.S.; Wanderley, C.W.S.; et al. Platelets Fuel the Inflammasome Activation of Innate Immune Cells. Cell Rep. 2020, 6, 107615. [Google Scholar] [CrossRef]
- Papa, R.; Picco, P.; Gattorno, M. The expanding pathways of autoinflammation: A lesson from the first 100 genes related to autoinflammatory manifestations. Adv. Protein Chem. Struct. Biol. 2020, 120, 1–44. [Google Scholar] [CrossRef]
- Leonard, W.J.; Wan, C.-K. IL-21 Signaling in Immunity. F1000Research 2016, 5, 224. [Google Scholar] [CrossRef] [PubMed]
- Gensous, N.; Schmitt, N.; Richez, C.; Ueno, H.; Blanco, P. T follicular helper cells, interleukin-21 and systemic lupus erythematosus. Rheumatology 2016, 56, kew297. [Google Scholar] [CrossRef]
- Iervasi, E.; Auricchio, R.; Strangio, A.; Greco, L.; Saverino, D. Serum IL-21 levels from celiac disease patients correlates with anti-tTG IgA autoantibodies and mucosal damage. Autoimmunity 2020, 53, 225–230. [Google Scholar] [CrossRef]
- Zubchenko, S.; Potemkina, G.; Havrylyuk, A.; Lomikovska, M.; Sharikadze, O. Analysis of the level of cytokines with anti-viral activity in patients with allergopathology in active and latent phases of chronic persistent Epstein-Barr infection. Georgian Med. News 2019, 289, 158–162. [Google Scholar]
- Barnes, J.; Mondelli, V.; Pariante, C.M. Genetic Contributions of Inflammation to Depression. Neuropsychopharmacology 2017, 42, 81–98. [Google Scholar] [CrossRef]
- Meyer, O. Interferons and autoimmune disorders. Jt. Bone Spine 2009, 76, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Borden, E.C.; Sen, G.C.; Uzé, G.; Silverman, R.H.; Ransohoff, R.M.; Foster, G.R.; Stark, G.R. Interferons at age 50: Past, current and future impact on biomedicine. Nat. Rev. Drug Discov. 2007, 6, 975–990. [Google Scholar] [CrossRef]
- Crow, M.K.; Olferiev, M.; Kirou, K.A. Type I Interferons in Autoimmune Disease. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 369–393. [Google Scholar] [CrossRef] [PubMed]
- Psarras, A.; Emery, P.; Vital, E.M. Type I interferon–mediated autoimmune diseases: Pathogenesis, diagnosis and targeted therapy. Rheumatology 2017, 56, 1662–1675. [Google Scholar] [CrossRef] [PubMed]
- Van Der Weyden, C.A.; Pileri, S.A.; Feldman, A.L.; Whisstock, J.; Prince, H.M. Understanding CD30 biology and therapeutic targeting: A historical perspective providing insight into future directions. Blood Cancer J. 2017, 7, e603. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, A.; Dolcino, M.; Tinazzi, E.; Rigo, A.; Argentino, G.; Patuzzo, G.; Ottria, A.; Beri, R.; Puccetti, A.; Lunardi, C. Characterization of CD30/CD30L+Cells in Peripheral Blood and Synovial Fluid of Patients with Rheumatoid Arthritis. J. Immunol. Res. 2015, 2015, 1–10. [Google Scholar] [CrossRef]
- Opat, S.; Gaston, J. CD30: CD30 Ligand Interactions in the Immune Response. Autoimmunity 2001, 33, 45–60. [Google Scholar] [CrossRef]
- Aloui, C.; Prigent, A.; Sut, C.; Tariket, S.; Hamzeh-Cognasse, H.; Pozzetto, B.; Richard, Y.; Cognasse, F.; Laradi, S.; Garraud, O. The Signaling Role of CD40 Ligand in Platelet Biology and in Platelet Component Transfusion. Int. J. Mol. Sci. 2014, 15, 22342–22364. [Google Scholar] [CrossRef]
- Wang, H.; Liu, C.; Chen, W.; Ding, G. The skewed frequency of B-cell subpopulation CD19 + CD24 hi CD38 hi cells in peripheral blood mononuclear cells is correlated with the elevated serum sCD40L in patients with active systemic lupus erythematosus. J. Cell. Biochem. 2019, 120, 11490–11497. [Google Scholar] [CrossRef] [PubMed]
- Karnell, J.L.; Albulescu, M.; Drabic, S.; Wang, L.; Moate, R.; Baca, M.; Oganesyan, V.; Gunsior, M.; Thisted, T.; Yan, L.; et al. A CD40L-targeting protein reduces autoantibodies and improves disease activity in patients with autoimmunity. Sci. Transl. Med. 2019, 11, eaar6584. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q.; Gao, X.; Wang, J.; Sun, Y.; Shekhar, S. Cytokines in Autoimmune Disease. Mediat. Inflamm. 2017, 2017, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Nézondet, A.C.; Poubelle, P.E.; Pelletier, M. The evaluation of cytokines to help establish diagnosis and guide treatment of autoinflammatory and autoimmune diseases. J. Leukoc. Biol. 2020, 108, 647–657. [Google Scholar] [CrossRef]
- Thomsen, R.W.; Öztürk, B.; Pedersen, L.; Nicolaisen, S.K.; Petersen, I.; Olsen, J.; Sørensen, H.T. Hospital Records of Pain, Fatigue, or Circulatory Symptoms in Girls Exposed to Human Papillomavirus Vaccination: Cohort, Self-Controlled Case Series, and Population Time Trend Studies. Am. J. Epidemiol. 2020, 189, 277–285. [Google Scholar] [CrossRef]
- Blitshteyn, S.; Brook, J. Postural tachycardia syndrome (POTS) with anti-NMDA receptor antibodies after human papillomavirus vaccination. Immunol. Res. 2016, 65, 282–284. [Google Scholar] [CrossRef]
- Fedorowski, A. Postural orthostatic tachycardia syndrome: Clinical presentation, aetiology and management. J. Intern. Med. 2019, 285, 352–366. [Google Scholar] [CrossRef]
- Waisbren, B.A. Acquired autoimmunity after viral vaccination is caused by molecular mimicry and antigen complimentarity in the presence of an immunologic adjuvant and specific HLA patterns. Med. Hypotheses 2008, 70, 346–348. [Google Scholar] [CrossRef]
- Miglis, M.G.; Prieto, T.; Shaik, R.; Muppidi, S.; Sinn, D.-I.; Jaradeh, S. A case report of postural tachycardia syndrome after COVID-19. Clin. Auton. Res. 2020, 30, 449–451. [Google Scholar] [CrossRef]
- Kanjwal, K.; Jamal, S.; Kichloo, A.; Grubb, B.P. New-onset Postural Orthostatic Tachycardia Syndrome Following Coronavirus Disease 2019 Infection. J. Innov. Card. Rhythm. Manag. 2020, 11, 4302–4304. [Google Scholar] [CrossRef]
- Eshak, N.; Abdelnabi, M.; Ball, S.; Elgwairi, E.; Creed, K.; Test, V.; Nugent, K. Dysautonomia: An Overlooked Neurological Manifestation in a Critically Ill COVID-19 Patient. Am. J. Med. Sci. 2020, 360, 427–429. [Google Scholar] [CrossRef]
- Chey, W.D.; Kurlander, J.; Eswaran, S. Irritable Bowel Syndrome. JAMA 2015, 313, 949–958. [Google Scholar] [CrossRef] [PubMed]
- El-Salhy, M. Irritable bowel syndrome: Diagnosis and pathogenesis. World J. Gastroenterol. 2012, 18, 5151–5163. [Google Scholar] [CrossRef] [PubMed]
- Long, D.; Chen, Y.; Wu, H.; Zhao, M.; Lu, Q. Clinical significance and immunobiology of IL-21 in autoimmunity. J. Autoimmun. 2019, 99, 1–14. [Google Scholar] [CrossRef]
- Kunz, M.; Ibrahim, S.M. Cytokines and Cytokine Profiles in Human Autoimmune Diseases and Animal Models of Autoimmunity. Mediat. Inflamm. 2009, 2009, 1–20. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Demographic/Symptom | Mean (STD)/Percentage/(N) |
|---|---|
| N | 34 |
| Age | 31.1 ± 11.5 |
| Females | 94.1% (32/34) |
| Menses score | 202.6 ± 145.3 (8/32), Normal: <185 |
| Bleeding score | 6.9 ± 7.9 (21), Normal: <5 for women |
| Easy bruising | 67.6% (23) |
| Dense Granules per Platelet (DG/PL) | 3.04 ± 0.9, Normal = 4–6 DG/PL |
| Cytokine/Chemokine | POTS Patients (n = 34) (pg/mL) | Normal (pg/mL) | Major Function |
|---|---|---|---|
| IL 1β | 332 ± 100 | <10 | Regulates cell proliferation |
| IL 10 | 16 ± 3.6 | <6 | Inhibitory to T helper cells |
| IL 21 | 1918 ± 410 | <200 | Controls NK and T cells |
| TNFα | 342 ± 78 | <3 | Regulates inflammation |
| INFγ | 226 ± 62 | <5 | Antiviral |
| CD30 | 193 ± 59 | <10 | Regulates cell proliferation |
| CD40 L | 119 ± 11 | 350–90 | Recruits leukocytes |
| RANTES (CCL5) | 995 ± 123 | 5000–6100 | Chemotactic for T cells |
| P-Selectin | 12,540 ± 1094 | 10,000–130,000 | Recruits leukocytes |
| MCP-1 | 78 ± 5 | 65–1025 | Recruits monocytes |
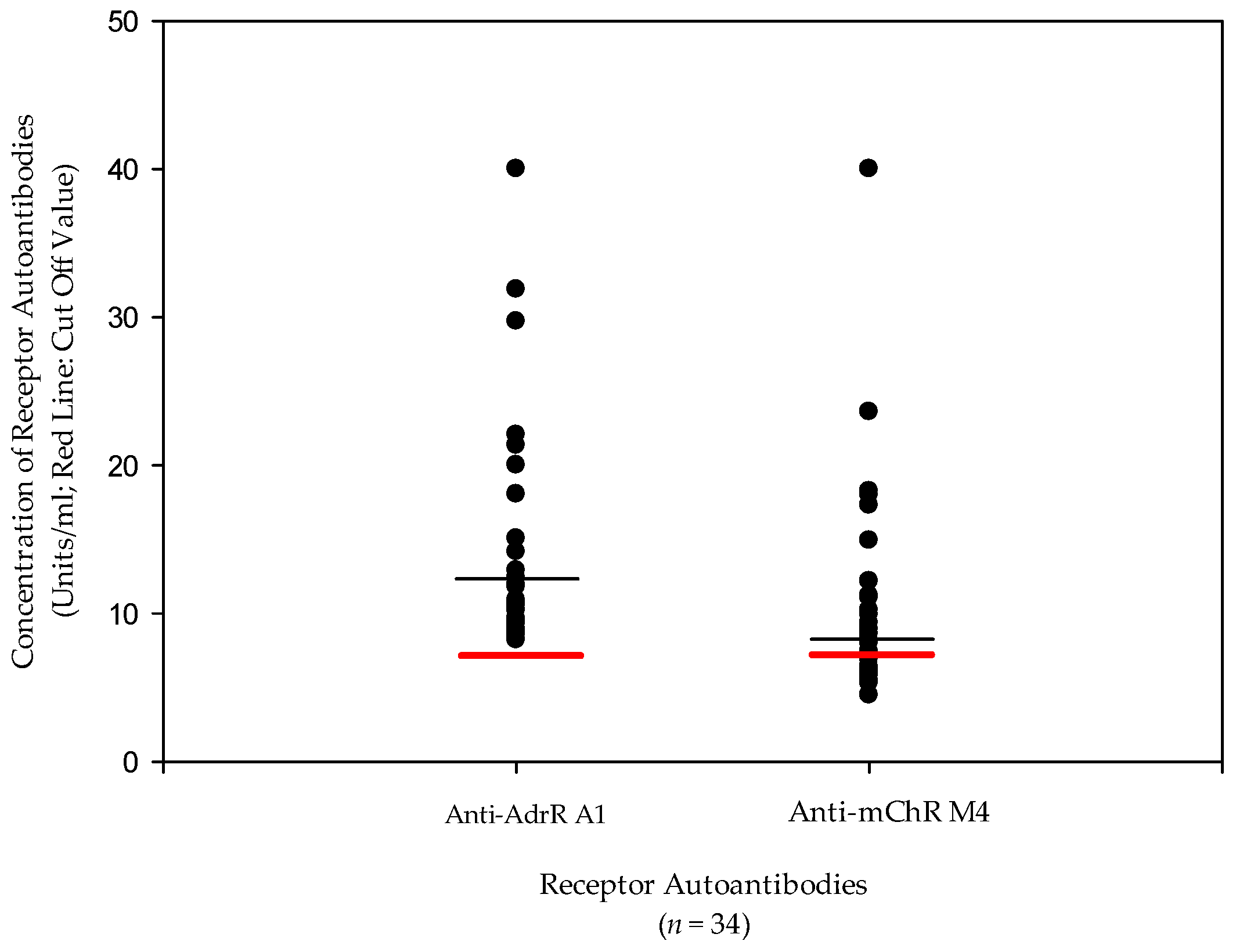
| AdR A1 antibodies | 16.6 U/mL | <7 U/mL | Autoantibody |
| AChR M4 Abs | 11.2 U/mL | <7 U/mL | Autoantibody |
| Autoantibodies | IL-1β | IL-10 | IL-21 | TNFα | INFγ | CD30 | CD40L | RANTES | P-Selectin | MCP-1 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| AdrR A1 | r | −0.067 | 0.194 | −0.007 | 0.093 | 0.086 | 0.100 | −0.170 | −0.029 | −0.198 | −0.132 |
| p | 0.705 | 0.272 | 0.966 | 0.602 | 0.627 | 0.573 | 0.338 | 0.236 | 0.261 | 0.458 | |
| AChR M4 | r | 0.018 | 0.281 | −0.061 | 0.254 | 0.210 | 0.211 | −0.264 | −0.139 | 0.203 | 0.156 |
| p | 0.919 | 0.107 | 0.731 | 0.147 | 0.238 | 0.231 | 0.231 | 0.131 | 0.433 | 0.379 | |
| DG | r | −0.107 | 0.101 | 0.445 | −0.025 | 0.196 | 0.338 | −0.182 | −0.634 | −0.357 | −0.077 |
| p | 0.100 | 0.871 | 0.883 | 0.968 | 0.752 | 0.578 | 0.769 | 0.250 | 0.555 | 0.902 | |
| IL-1β | IL-10 | IL-21 | TNFα | INFγ | CD30 | CD40L | RANTES | P-Selectin | MCP-1 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| IL-1β | r | 0.653 | 0.520 | 0.606 | 0.565 | <0.87 | 0.106 | 0.866 | 0.196 | 0.379 | |
| p | <0.001 | 0.002 | <0.001 | <0.001 | <0.001 | 0.552 | <0.001 | 0.266 | 0.027 | ||
| IL-10 | r | 0.555 | 0.731 | 0.694 | 0.83 | 0.011 | 0.218 | −0.141 | 0.093 | ||
| p | <0.001 | <0.001 | <0.001 | <0.001 | 0.950 | <0.001 | 0.427 | 0.601 | |||
| IL-21 | r | 0.74 | 0.76 | 0.626 | 0.133 | −0.019 | −0.121 | 0.242 | |||
| p | <0.001 | <0.001 | <0.001 | 0.453 | 0.912 | 0.497 | 0.168 | ||||
| TNFα | r | 0.93 | 0.81 | −0.041 | 0.105 | −0.150 | 0.447 | ||||
| p | <0.001 | <0.001 | 0.817 | 0.553 | 0.398 | 0.008 | |||||
| INFγ | r | 0.73 | 0.043 | 0.073 | −0.075 | 0.388 | |||||
| p | <0.001 | 0.808 | 0.680 | 0.674 | 0.024 | ||||||
| CD30 | r | 0.049 | 0.215 | 0.007 | 0.338 | ||||||
| p | 0.784 | 0.222 | 0.970 | 0.051 | |||||||
| CD40L | r | 0.367 | 0.134 | −0.011 | |||||||
| p | 0.033 | 0.449 | 0.95 | ||||||||
| RANTES | r | 0.231 | −0.008 | ||||||||
| p | 0.188 | 0.962 | |||||||||
| P-Selectin | r | 0.076 | |||||||||
| p | 0.671 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gunning, W.T., III; Stepkowski, S.M.; Kramer, P.M.; Karabin, B.L.; Grubb, B.P. Inflammatory Biomarkers in Postural Orthostatic Tachycardia Syndrome with Elevated G-Protein-Coupled Receptor Autoantibodies. J. Clin. Med. 2021, 10, 623. https://doi.org/10.3390/jcm10040623
Gunning WT III, Stepkowski SM, Kramer PM, Karabin BL, Grubb BP. Inflammatory Biomarkers in Postural Orthostatic Tachycardia Syndrome with Elevated G-Protein-Coupled Receptor Autoantibodies. Journal of Clinical Medicine. 2021; 10(4):623. https://doi.org/10.3390/jcm10040623
Chicago/Turabian StyleGunning, William T., III, Stanislaw M. Stepkowski, Paula M. Kramer, Beverly L. Karabin, and Blair P. Grubb. 2021. "Inflammatory Biomarkers in Postural Orthostatic Tachycardia Syndrome with Elevated G-Protein-Coupled Receptor Autoantibodies" Journal of Clinical Medicine 10, no. 4: 623. https://doi.org/10.3390/jcm10040623
APA StyleGunning, W. T., III, Stepkowski, S. M., Kramer, P. M., Karabin, B. L., & Grubb, B. P. (2021). Inflammatory Biomarkers in Postural Orthostatic Tachycardia Syndrome with Elevated G-Protein-Coupled Receptor Autoantibodies. Journal of Clinical Medicine, 10(4), 623. https://doi.org/10.3390/jcm10040623