Bscl2 Deficiency Does Not Directly Impair the Innate Immune Response in a Murine Model of Generalized Lipodystrophy

 , ,
, ,  , ,
, ,  and
and 
Abstract
1. Introduction
2. Experimental Section
2.1. Animal Studies
2.2. Metabolic Studies
2.3. Gene Expression
2.4. Bone-Marrow-Derived Macrophages (BMDM)
2.5. Bacterial Infection
2.6. Immunofluorescence
2.7. Data Analysis
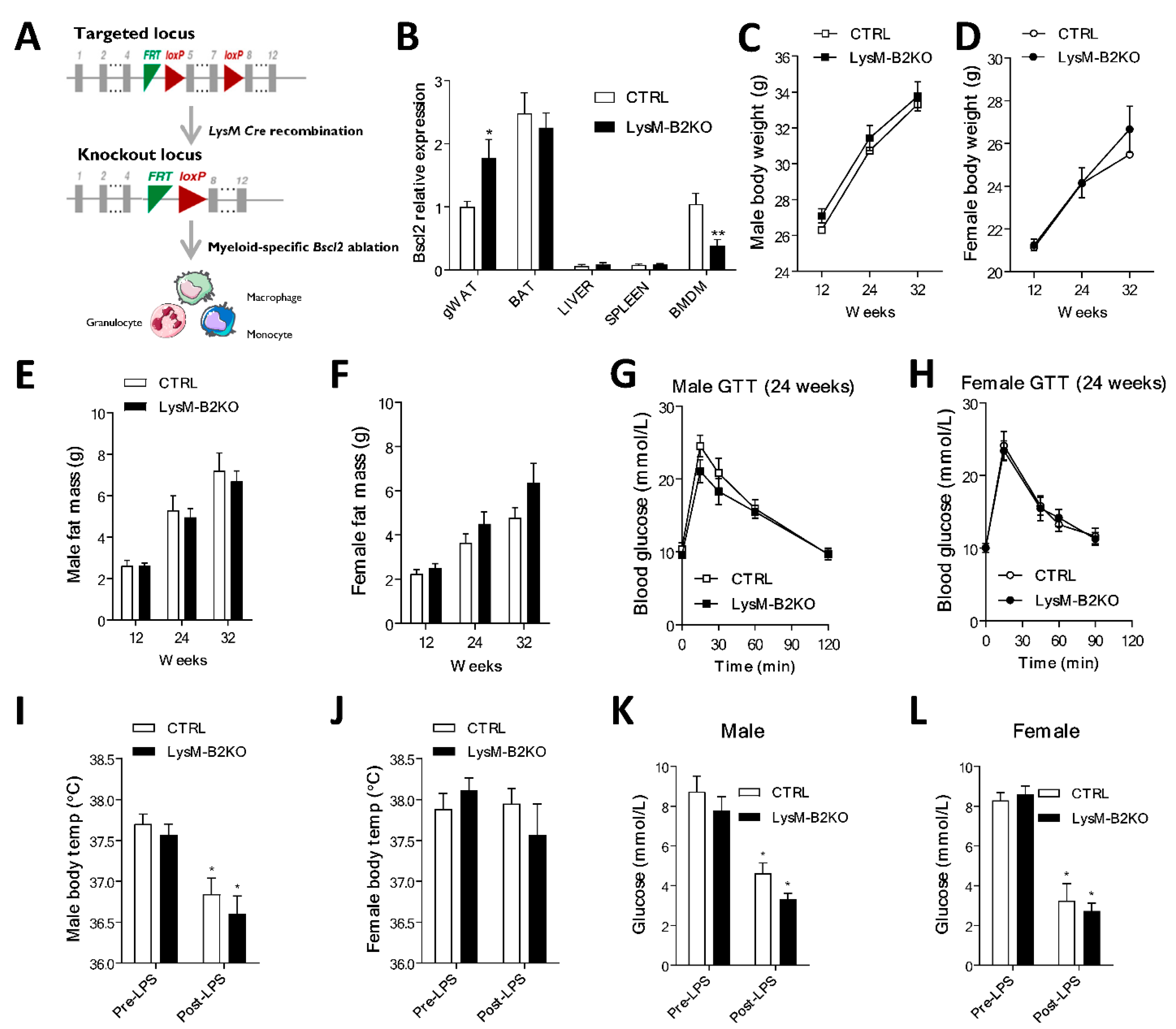
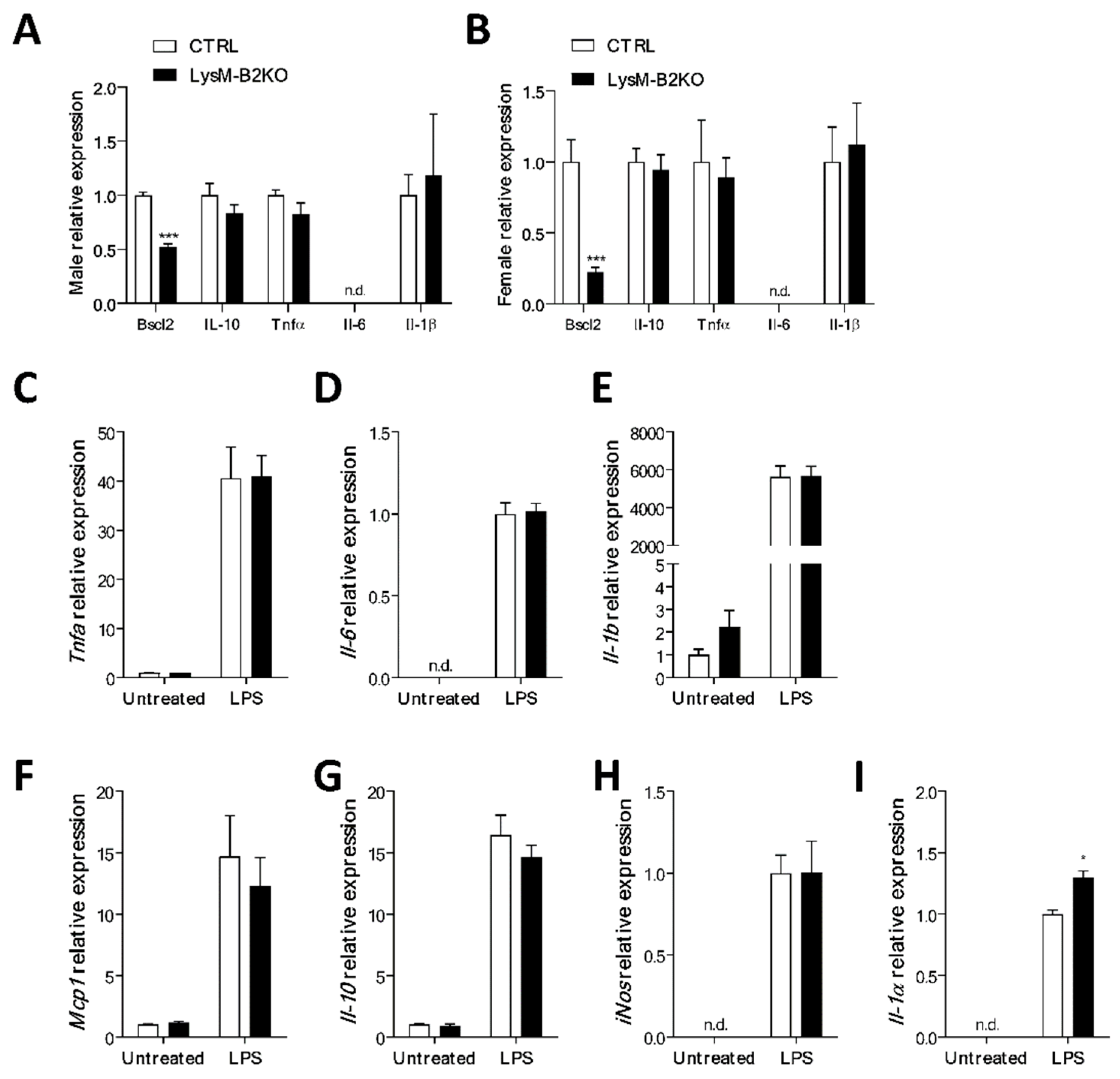
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Garg, A. Lipodystrophies: Genetic and acquired body fat disorders. J. Clin. Endocrinol. Metab. 2011, 96, 3313–3325. [Google Scholar] [CrossRef]
- Magre, J.; Delepine, M.; Khallouf, E.; Gedde-Dahl, T., Jr.; Van Maldergem, L.; Sobel, E.; Papp, J.; Meier, M.; Megarbane, A.; Bachy, A.; et al. Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13. Nat. Genet. 2001, 28, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Payne, V.A.; Grimsey, N.; Tuthill, A.; Virtue, S.; Gray, S.L.; Dalla Nora, E.; Semple, R.K.; O’Rahilly, S.; Rochford, J.J. The human lipodystrophy gene BSCL2/seipin may be essential for normal adipocyte differentiation. Diabetes 2008, 57, 2055–2060. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Yechoor, V.K.; Chang, B.H.; Li, M.V.; March, K.L.; Chan, L. The human lipodystrophy gene product Berardinelli-Seip congenital lipodystrophy 2/seipin plays a key role in adipocyte differentiation. Endocrinology 2009, 150, 4552–4561. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Wang, Y.; Tang, Y.; Liu, Y.; Zhao, L.; Deng, J.; Xu, G.; Peng, X.; Ju, S.; Liu, G.; et al. Seipin ablation in mice results in severe generalized lipodystrophy. Hum. Mol. Genet. 2011, 20, 3022–3030. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Chang, B.; Saha, P.; Hartig, S.M.; Li, L.; Reddy, V.T.; Yang, Y.; Yechoor, V.; Mancini, M.A.; Chan, L. Berardinelli-seip congenital lipodystrophy 2/seipin is a cell-autonomous regulator of lipolysis essential for adipocyte differentiation. Mol. Cell Biol. 2012, 32, 1099–1111. [Google Scholar] [CrossRef] [PubMed]
- Prieur, X.; Dollet, L.; Takahashi, M.; Nemani, M.; Pillot, B.; Le May, C.; Mounier, C.; Takigawa-Imamura, H.; Zelenika, D.; Matsuda, F.; et al. Thiazolidinediones partially reverse the metabolic disturbances observed in Bscl2/seipin-deficient mice. Diabetologia 2013, 56, 1813–1825. [Google Scholar] [CrossRef]
- Mcilroy, G.D.; Suchacki, K.; Roelofs, A.J.; Yang, W.; Fu, Y.; Bai, B.; Wallace, R.J.; De Bari, C.; Cawthorn, W.P.; Han, W.; et al. Adipose specific disruption of seipin causes early-onset generalised lipodystrophy and altered fuel utilisation without severe metabolic disease. Mol. Metab. 2018, 10, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Sim, M.F.; Dennis, R.J.; Aubry, E.M.; Ramanathan, N.; Sembongi, H.; Saudek, V.; Ito, D.; O’Rahilly, S.; Siniossoglou, S.; Rochford, J.J. The human lipodystrophy protein seipin is an ER membrane adaptor for the adipogenic PA phosphatase lipin 1. Mol. Metab. 2012, 2, 38–46. [Google Scholar] [CrossRef]
- Talukder, M.M.; Sim, M.F.; O’Rahilly, S.; Edwardson, J.M.; Rochford, J.J. Seipin oligomers can interact directly with AGPAT2 and lipin 1, physically scaffolding critical regulators of adipogenesis. Mol. Metab. 2015, 4, 199–209. [Google Scholar] [CrossRef]
- Castro, I.G.; Eisenberg-Bord, M.; Persiani, E.; Rochford, J.J.; Schuldiner, M.; Bohnert, M. Promethin Is a Conserved Seipin Partner Protein. Cells 2019, 8, 268. [Google Scholar] [CrossRef] [PubMed]
- Sim, M.F.M.; Persiani, E.; Talukder, M.M.U.; Mcilroy, G.D.; Roumane, A.; Edwardson, J.M.; Rochford, J.J. Oligomers of the lipodystrophy protein seipin may co-ordinate GPAT3 and AGPAT2 enzymes to facilitate adipocyte differentiation. Sci. Rep. 2020, 10, 3259. [Google Scholar] [CrossRef]
- Lima, J.G.; Nobrega, L.H.C.; Lima, N.N.; Dos Santos, M.C.F.; Silva, P.H.D.; Baracho, M.F.P.; Lima, D.N.; de Melo Campos, J.T.A.; Ferreira, L.C.; Freire Neto, F.P.; et al. Causes of death in patients with Berardinelli-Seip congenital generalized lipodystrophy. PLoS ONE 2018, 13, e0199052. [Google Scholar] [CrossRef] [PubMed]
- Bohnert, M. New friends for seipin—Implications of seipin partner proteins in the life cycle of lipid droplets. Semin. Cell Dev. Biol. 2020, 108, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Dichlberger, A.; Kovanen, P.T.; Schneider, W.J. Mast cells: From lipid droplets to lipid mediators. Clin. Sci. 2013, 125, 121–130. [Google Scholar] [CrossRef]
- Melo, R.C.; Dvorak, A.M. Lipid body-phagosome interaction in macrophages during infectious diseases: Host defense or pathogen survival strategy? PLoS Pathog. 2012, 8, e1002729. [Google Scholar] [CrossRef][Green Version]
- Kim, K.; Kim, H.; Lee, D. Site-specific modification of genome with cell-permeable Cre fusion protein in preimplantation mouse embryo. Biochem. Biophys. Res. Commun. 2009, 388, 122–126. [Google Scholar] [CrossRef]
- Katz, A.; Nambi, S.S.; Mather, K.; Baron, A.D.; Follmann, D.A.; Sullivan, G.; Quon, M.J. Quantitative insulin sensitivity check index: A simple, accurate method for assessing insulin sensitivity in humans. J. Clin. Endocrinol. Metab. 2000, 85, 2402–2410. [Google Scholar] [CrossRef]
- Owen, C.; Czopek, A.; Agouni, A.; Grant, L.; Judson, R.; Lees, E.K.; Mcilroy, G.D.; Goransson, O.; Welch, A.; Bence, K.K.; et al. Adipocyte-specific protein tyrosine phosphatase 1B deletion increases lipogenesis, adipocyte cell size and is a minor regulator of glucose homeostasis. PLoS ONE 2012, 7, e32700. [Google Scholar] [CrossRef]
- Cai, K.C.; van Mil, S.; Murray, E.; Mallet, J.F.; Matar, C.; Ismail, N. Age and sex differences in immune response following LPS treatment in mice. Brain Behav. Immun. 2016, 58, 327–337. [Google Scholar] [CrossRef]
- Raetzsch, C.F.; Brooks, N.L.; Alderman, J.M.; Moore, K.S.; Hosick, P.A.; Klebanov, S.; Akira, S.; Bear, J.E.; Baldwin, A.S.; Mackman, N.; et al. Lipopolysaccharide inhibition of glucose production through the Toll-like receptor-4, myeloid differentiation factor 88, and nuclear factor kappa b pathway. Hepatology 2009, 50, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Ciesielska, A.; Matyjek, M.; Kwiatkowska, K. TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell Mol. Life Sci. 2020. [Google Scholar] [CrossRef] [PubMed]
- Craveiro Sarmento, A.S.; de Azevedo Medeiros, L.B.; Agnez-Lima, L.F.; Lima, J.G.; de Melo Campos, J.T.A. Exploring Seipin: From Biochemistry to Bioinformatics Predictions. Int. J. Cell Biol. 2018, 2018, 5207608. [Google Scholar] [CrossRef]
- Remmerie, A.; Scott, C.L. Macrophages and lipid metabolism. Cell Immunol. 2018, 330, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Ozata, M.; Ozdemir, I.C.; Licinio, J. Human leptin deficiency caused by a missense mutation: Multiple endocrine defects, decreased sympathetic tone, and immune system dysfunction indicate new targets for leptin action, greater central than peripheral resistance to the effects of leptin, and spontaneous correction of leptin-mediated defects. J. Clin. Endocrinol. Metab. 1999, 84, 3686–3695. [Google Scholar] [PubMed]
- Maurya, R.; Bhattacharya, P.; Dey, R.; Nakhasi, H.L. Leptin Functions in Infectious Diseases. Front. Immunol. 2018, 9, 2741. [Google Scholar] [CrossRef]
- Ratter, J.M.; Tack, C.J.; Netea, M.G.; Stienstra, R. Environmental Signals Influencing Myeloid Cell Metabolism and Function in Diabetes. Trends Endocrinol. Metab. 2018, 29, 468–480. [Google Scholar] [CrossRef]
- Dollet, L.; Magre, J.; Cariou, B.; Prieur, X. Function of seipin: New insights from Bscl2/seipin knockout mouse models. Biochimie 2014, 96, 166–172. [Google Scholar] [CrossRef]
- Liao, J.; Liu, X.; Gao, M.; Wang, M.; Wang, Y.; Wang, F.; Huang, W.; Liu, G. Dyslipidemia, steatohepatitis and atherogenesis in lipodystrophic apoE deficient mice with Seipin deletion. Gene 2018, 648, 82–88. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Male | Female | ||||
|---|---|---|---|---|---|
| Genotype | Pre-LPS Treatment | Post-LPS Treatment | Pre-LPS Treatment | Post-LPS Treatment | |
| Insulin (µg/L) | CTRL | 0.28 ± 0.15 | 1.75 ± 1.51 | 0.16 ± 0.03 | 0.21 ± 0.07 |
| LysM-B2KO | 0.34 ± 0.13 | 0.99 ± 0.46 | 0.18 ± 0.11 | 0.39 ± 0.08 | |
| QUICKI | CTRL | 0.34 ± 0.03 | 0.30 ± 0.04 | 0.36 ± 0.02 | 0.39 ± 0.04 |
| LysM-B2KO | 0.33 ± 0.02 | 0.33 ± 0.02 | 0.37 ± 0.03 | 0.38 ± 0.02 | |
| IL-10 (pg/mL) | CTRL | - | 453.91 ± 104.65 | - | 561.13 ± 113.92 |
| LysM-B2KO | - | 404.87 ± 114.11 | - | 865.17 ± 408.16 | |
| TNF-α (pg/mL) | CTRL | - | 390.78 ± 68.26 | - | 394.17 ± 387.63 |
| LysM-B2KO | - | 313.94 ± 97.05 | - | 200.21 ± 107.88 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roumane, A.; Mcilroy, G.D.; Balci, A.; Han, W.; Delibegović, M.; Baldassarre, M.; Newsholme, P.; Rochford, J.J. Bscl2 Deficiency Does Not Directly Impair the Innate Immune Response in a Murine Model of Generalized Lipodystrophy. J. Clin. Med. 2021, 10, 441. https://doi.org/10.3390/jcm10030441
Roumane A, Mcilroy GD, Balci A, Han W, Delibegović M, Baldassarre M, Newsholme P, Rochford JJ. Bscl2 Deficiency Does Not Directly Impair the Innate Immune Response in a Murine Model of Generalized Lipodystrophy. Journal of Clinical Medicine. 2021; 10(3):441. https://doi.org/10.3390/jcm10030441
Chicago/Turabian StyleRoumane, Ahlima, George D. Mcilroy, Arda Balci, Weiping Han, Mirela Delibegović, Massimiliano Baldassarre, Philip Newsholme, and Justin J. Rochford. 2021. "Bscl2 Deficiency Does Not Directly Impair the Innate Immune Response in a Murine Model of Generalized Lipodystrophy" Journal of Clinical Medicine 10, no. 3: 441. https://doi.org/10.3390/jcm10030441
APA StyleRoumane, A., Mcilroy, G. D., Balci, A., Han, W., Delibegović, M., Baldassarre, M., Newsholme, P., & Rochford, J. J. (2021). Bscl2 Deficiency Does Not Directly Impair the Innate Immune Response in a Murine Model of Generalized Lipodystrophy. Journal of Clinical Medicine, 10(3), 441. https://doi.org/10.3390/jcm10030441