
Amyloid Beta Dynamics in Biological Fluids—Therapeutic Impact



Abstract
:1. Introduction
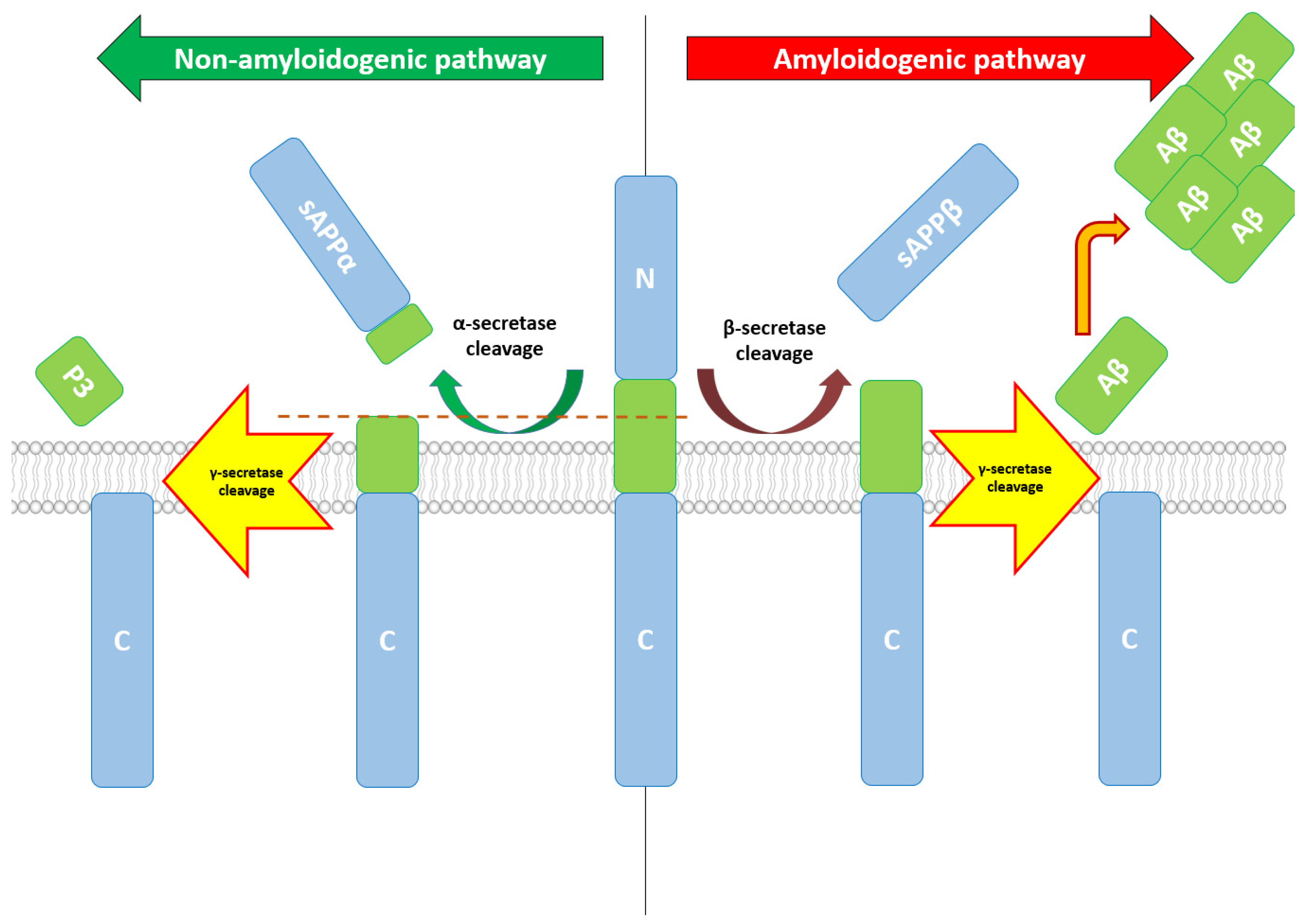
2. Aβ—Production, Structure, Functions, and Therapeutic Correlations
3. Aβ Dynamics between ISF, CSF, and Blood
4. Current and Future Therapeutic Directions for Aβ Reduction at CNS Level
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Briggs, R.; Kennelly, S.P.; O’Neill, D. Drug treatments in Alzheimer’s disease. Clin. Med. 2016, 16, 247–253. [Google Scholar] [CrossRef]
- Alzheimer’s Association 2016 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2016, 12, 459–509. [CrossRef]
- Sandoval, J.D.J.; Turra, C.M.; Loschi, R.H. Adjusted Mortality Rates Attributable to Alzheimer’s Disease Dementia, Brazil, 2009–2013. Cad. Saude Publica 2019, 35, e00091918. [Google Scholar] [CrossRef] [Green Version]
- Gresenz, C.R.; Mitchell, J.M.; Marrone, J.; Federoff, H.J. Effect of early-stage Alzheimer’s disease on household financial outcomes. Health Econ. 2020, 29, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, R.A. Risk factors for Alzheimer’s disease. Folia Neuropathol. 2019, 57, 87–105. [Google Scholar] [CrossRef] [Green Version]
- Abeysinghe, A.A.D.T.; Deshapriya, R.D.U.S.; Udawatte, C. Alzheimer’s disease; a review of the pathophysiological basis and therapeutic interventions. Life Sci. 2020, 256, 117996. [Google Scholar] [CrossRef]
- Hampel, H.; Mesulam, M.-M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimer’s Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef] [PubMed]
- Bakota, L.; Brandt, R. Tau Biology and Tau-Directed Therapies for Alzheimer’s Disease. Drugs 2016, 76, 301–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonnen, J.A.; Larson, E.B.; Gray, S.L.; Wilson, A.; Kohama, S.G.; Crane, P.K.; Breitner, J.C.S.; Montine, T.J. Free radical damage to cerebral cortex in Alzheimer’s disease, microvascular brain injury, and smoking. Ann. Neurol. 2009, 65, 226–229. [Google Scholar] [CrossRef] [Green Version]
- Beers, W.H.; Reich, E. Structure and Activity of Acetylcholine. Nature 1970, 228, 917–922. [Google Scholar] [CrossRef] [PubMed]
- Mautner, H.G.; Nachmansohn, D. Choline Acetyltransferas. CRC Crit. Rev. Biochem. 1977, 4, 341–370. [Google Scholar] [CrossRef] [PubMed]
- Prado, V.F.; Roy, A.; Kolisnyk, B.; Gros, R.; Prado, M.A.M. Regulation of cholinergic activity by the vesicular acetylcholine transporter. Biochem. J. 2013, 450, 265–274. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.A. Acetylcholine and cholinergic receptors. Brain Neurosci. Adv. 2019, 3, 2398212818820506. [Google Scholar] [CrossRef]
- Schliebs, R.; Arendt, T. The cholinergic system in aging and neuronal degeneration. Behav. Brain Res. 2011, 221, 555–563. [Google Scholar] [CrossRef]
- Lester, D.; Rogers, T.D.; Blaha, C.D. Acetylcholine-Dopamine Interactions in the Pathophysiology and Treatment of CNS Disorders. CNS Neurosci. Ther. 2010, 16, 137–162. [Google Scholar] [CrossRef] [PubMed]
- Bagri, K.; Kumar, A.; Manisha; Kumar, P. Computational Studies on Acetylcholinesterase Inhibitors: From Biochemistry to Chemistry. Mini-Rev. Med. Chem. 2020, 20, 1403–1435. [Google Scholar] [CrossRef]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef] [Green Version]
- Okuda, T.; Nomura, Y.; Konishi, A.; Misawa, H. Competitive inhibition of the high-affinity choline transporter by tetrahydropyrimidine anthelmintics. Eur. J. Pharmacol. 2021, 898, 173986. [Google Scholar] [CrossRef]
- Haga, T. Molecular properties of the high-affinity choline transporter CHT1. J. Biochem. 2014, 156, 181–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, L.; Duan, J.; Ran, D.; Fan, Z.; Yan, Y.; Huang, N.; Gu, H.; Zhu, Y. Amyloid-β depresses excitatory cholinergic synaptic transmission in Drosophila. Neurosci. Bull. 2012, 28, 585–594. [Google Scholar] [CrossRef] [Green Version]
- Majdi, A.; Sadigh-Eteghad, S.; Aghsan, S.R.; Farajdokht, F.; Vatandoust, S.M.; Namvaran, A.; Mahmoudi, J. Amyloid-β, tau, and the cholinergic system in Alzheimer’s disease: Seeking direction in a tangle of clues. Rev. Neurosci. 2020, 31, 391–413. [Google Scholar] [CrossRef]
- Murphy, M.P.; LeVine, H., 3rd. Alzheimer’s Disease and the Amyloid-β Peptide. J. Alzheimer’s Dis. 2010, 19, 311–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fjell, A.M.; McEvoy, L.; Holland, D.; Dale, A.M.; Walhovd, K.B.; Alzheimer’s Disease Neuroimaging Initiative. What is normal in normal aging? Effects of aging, amyloid and Alzheimer’s disease on the cerebral cortex and the hippocampus. Prog. Neurobiol. 2014, 117, 20–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampel, H.; Kötter, H.U.; Möller, H.-J. Blood-Cerebrospinal Fluid Barrier Dysfunction for High Molecular Weight Proteins in Alzheimer Disease and Major Depression: Indication for Disease Subsets. Alzheimer Dis. Assoc. Disord. 1997, 11, 78–87. [Google Scholar] [CrossRef]
- Cacace, R.; Sleegers, K.; Van Broeckhoven, C. Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimer’s Dement. 2016, 12, 733–748. [Google Scholar] [CrossRef] [Green Version]
- Pierce, A.L.; Bullain, S.S.; Kawas, C.H. Late-Onset Alzheimer Disease. Neurol. Clin. 2017, 35, 283–293. [Google Scholar] [CrossRef] [Green Version]
- Tellechea, P.; Pujol, N.; Esteve-Belloch, P.; Echeveste, B.; García-Eulate, M.; Arbizu, J.; Riverol, M. Early- and Late-Onset Alzheimer Disease: Are They the Same Entity? Neurologia 2018, 33, 244–253. [Google Scholar] [CrossRef]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased Clearance of CNS β-Amyloid in Alzheimer’s Disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-W.; Thompson, R.; Zhang, H.; Xu, H. APP processing in Alzheimer’s disease. Mol. Brain 2011, 4, 3. [Google Scholar] [CrossRef] [Green Version]
- Van Giau, V.; Bagyinszky, E.; Youn, Y.C.; An, S.S.A.; Kim, S. APP, PSEN1, and PSEN2 Mutations in Asian Patients with Early-Onset Alzheimer Disease. Int. J. Mol. Sci. 2019, 20, 4757. [Google Scholar] [CrossRef] [Green Version]
- Müller, U.C.; Zheng, H. Physiological Functions of APP Family Proteins. Cold Spring Harb. Perspect. Med. 2012, 2, a006288. [Google Scholar] [CrossRef] [Green Version]
- Lorent, K.; Overbergh, L.; Moechars, D.; de Strooper, B.; van Leuven, F.; van den Berghe, H. Expression in mouse embryos and in adult mouse brain of three members of the amyloid precursor protein family, of the alpha-2-macroglobulin receptor/low density lipoprotein receptor-related protein and of its ligands apolipoprotein E, lipoprotein lipase, alpha-2-macroglobulin and the 40,000 molecular weight receptor-associated protein. Neuroscience 1995, 65, 1009–1025. [Google Scholar] [CrossRef]
- Kojro, E.; Fahrenholz, F. The Non-Amyloidogenic Pathway: Structure and Function of α-Secretases. Subcell. Biochem. 2005, 38, 105–127. [Google Scholar] [CrossRef] [PubMed]
- Sammel, M.; Peters, F.; Lokau, J.; Scharfenberg, F.; Werny, L.; Linder, S.; Garbers, C.; Rose-John, S.; Becker-Pauly, C.; John, R.; et al. Differences in Shedding of the Interleukin-11 Receptor by the Proteases ADAM9, ADAM10, ADAM17, Meprin α, Meprin β and MT1-MMP. Int. J. Mol. Sci. 2019, 20, 3677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buxbaum, J.D.; Liu, K.-N.; Luo, Y.; Slack, J.L.; Stocking, K.L.; Peschon, J.J.; Johnson, R.S.; Castner, B.J.; Cerretti, D.P.; Black, R.A. Evidence That Tumor Necrosis Factor α Converting Enzyme Is Involved in Regulated α-Secretase Cleavage of the Alzheimer Amyloid Protein Precursor. J. Biol. Chem. 1998, 273, 27765–27767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tachida, Y.; Nakagawa, K.; Saito, T.; Saido, T.C.; Honda, T.; Saito, Y.; Murayama, S.; Endo, T.; Sakaguchi, G.; Kato, A.; et al. Interleukin-1β up-regulates TACE to enhance α-cleavage of APP in neurons: Resulting decrease in Aβ production. J. Neurochem. 2007, 104, 1387–1393. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, P.-H.; Wang, H.; Dislich, B.; Colombo, A.; Zeitschel, U.; Ellwart, J.W.; Kremmer, E.; Roßner, S.; Lichtenthaler, S.F. ADAM10 is the physiologically relevant, constitutive α-secretase of the amyloid precursor protein in primary neurons. EMBO J. 2010, 29, 3020–3032. [Google Scholar] [CrossRef] [Green Version]
- Jorissen, E.; Prox, J.; Bernreuther, C.; Weber, S.; Schwanbeck, R.; Serneels, L.; Snellinx, A.; Craessaerts, K.; Thathiah, A.; Tesseur, I.; et al. The Disintegrin/Metalloproteinase ADAM10 Is Essential for the Establishment of the Brain Cortex. J. Neurosci. 2010, 30, 4833–4844. [Google Scholar] [CrossRef]
- Livingstone, R.W.; Elder, M.K.; Barrett, M.C.; Westlake, C.M.; Peppercorn, K.; Tate, W.P.; Abraham, W.C.; Williams, J.M. Secreted Amyloid Precursor Protein-Alpha Promotes Arc Protein Synthesis in Hippocampal Neurons. Front. Mol. Neurosci. 2019, 12, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coronel, R.; Bernabeu-Zornoza, A.; Palmer, C.; Muñiz-Moreno, M.; Zambrano, A.; Cano, E.; Liste, I. Role of Amyloid Precursor Protein (APP) and Its Derivatives in the Biology and Cell Fate Specification of Neural Stem Cells. Mol. Neurobiol. 2018, 55, 7107–7117. [Google Scholar] [CrossRef]
- Koelsch, G. BACE1 Function and Inhibition: Implications of Intervention in the Amyloid Pathway of Alzheimer’s Disease Pathology. Molecules 2017, 22, 1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Das, B.; Hou, H.; He, W.; Yan, R. BACE1 deletion in the adult mouse reverses preformed amyloid deposition and improves cognitive functions. J. Exp. Med. 2018, 215, 927–940. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xu, Q.; Cai, F.; Liu, X.; Wu, Y.; Song, W. BACE2, a conditional β-secretase, contributes to Alzheimer’s disease pathogenesis. JCI Insight 2019, 4, e123431. [Google Scholar] [CrossRef] [Green Version]
- Voytyuk, I.; Mueller, S.A.; Herber, J.; Snellinx, A.; Moechars, D.; Van Loo, G.; Lichtenthaler, S.F.; De Strooper, B. BACE2 distribution in major brain cell types and identification of novel substrates. Life Sci. Alliance 2018, 1, e201800026. [Google Scholar] [CrossRef]
- Hook, V.; Hook, G.; Kindy, M. Pharmacogenetic features of cathepsin B inhibitors that improve memory deficit and reduce β-amyloid related to Alzheimer’s disease. Biol. Chem. 2010, 391, 861–872. [Google Scholar] [CrossRef]
- Wolfe, M.S. Structure and Function of the γ-Secretase Complex. Biochemistry 2019, 58, 2953–2966. [Google Scholar] [CrossRef] [PubMed]
- Kanyenda, L.J.; Verdile, G.; Boulos, S.; Krishnaswamy, S.; Taddei, K.; Meloni, B.P.; Mastaglia, F.L.; Martins, R.N. The Dynamics of CD147 in Alzheimer’s Disease Development and Pathology. J. Alzheimer’s Dis. 2011, 26, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Park, S.A.; Shaked, G.M.; Bredesen, D.E.; Koo, E.H. Mechanism of cytotoxicity mediated by the C31 fragment of the amyloid precursor protein. Biochem. Biophys. Res. Commun. 2009, 388, 450–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampel, H.; Vassar, R.; De Strooper, B.; Hardy, J.; Willem, M.; Singh, N.; Zhou, J.; Yan, R.; Vanmechelen, E.; De Vos, A.; et al. The β-Secretase BACE1 in Alzheimer’s Disease. Biol. Psychiatry 2021, 89, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Rowan, M.J.; Selkoe, D.J. Amyloid-β oligomers: Their production, toxicity and therapeutic inhibition. Biochem. Soc. Trans. 2002, 30, 552–557. [Google Scholar] [CrossRef] [Green Version]
- Glabe, C.C. Amyloid Accumulation and Pathogensis of Alzheimer’s Disease: Significance of Monomeric, Oligomeric and Fibrillar Abeta. Subcell. Biochem. 2005, 38, 167–177. [Google Scholar] [CrossRef]
- Katzmarski, N.; Ziegler-Waldkirch, S.; Scheffler, N.; Witt, C.; Abou-Ajram, C.; Nuscher, B.; Prinz, M.; Haass, C.; Meyer-Luehmann, M. Aβ oligomers trigger and accelerate Aβ seeding. Brain Pathol. 2019, 30, 36–45. [Google Scholar] [CrossRef] [Green Version]
- Ayala, S.; Genevaux, P.; Hureau, C.; Faller, P. (Bio)chemical Strategies To Modulate Amyloid-β Self-Assembly. ACS Chem. Neurosci. 2019, 10, 3366–3374. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Canto, I.; Breydo, L.; Rasool, S.; Lukacsovich, T.; Wu, J.; Albay, R., 3rd; Pensalfini, A.; Yeung, S.; Head, E.; et al. Conformation dependent monoclonal antibodies distinguish different replicating strains or conformers of prefibrillar Aβ oligomers. Mol. Neurodegener. 2010, 5, 57. [Google Scholar] [CrossRef] [Green Version]
- Sherman, M.A.; Lacroix, M.; Amar, F.; Larson, M.E.; Forster, C.; Aguzzi, A.; Bennett, D.A.; Ramsden, M.; Lesné, S.E. Soluble Conformers of A and Tau Alter Selective Proteins Governing Axonal Transport. J. Neurosci. 2016, 36, 9647–9658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wildsmith, K.R.; Holley, M.; Savage, J.C.; Skerrett, R.; Landreth, G.E. Evidence for impaired amyloid β clearance in Alzheimer’s disease. Alzheimer’s Res. Ther. 2013, 5, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Mader, S.; Brimberg, L. Aquaporin-4 Water Channel in the Brain and Its Implication for Health and Disease. Cells 2019, 8, 90. [Google Scholar] [CrossRef] [Green Version]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef] [Green Version]
- Harris, L.D.; Jasem, S.; Licchesi, J.D.F. The Ubiquitin System in Alzheimer’s Disease. Adv. Exp. Med. Biol. 2020, 1233, 195–221. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, I.; Jha, S. Mitochondrial Dysfunction and Alzheimer’s Disease: Role of Microglia. Front. Aging Neurosci. 2020, 12, 252. [Google Scholar] [CrossRef] [PubMed]
- Gosselet, F.; Saint-Pol, J.; Candela, P.; Fenart, L. Amyloid-β peptides, Alzheimer’s disease and the blood-brain barrier. Curr. Alzheimer Res. 2013, 10, 1015–1033. [Google Scholar] [CrossRef]
- Solár, P.; Zamani, A.; Kubíčková, L.; Dubový, P.; Joukal, M. Choroid plexus and the blood–cerebrospinal fluid barrier in disease. Fluids Barriers CNS 2020, 17, 1–29. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Pereira, C.D.; Martins, F.; Wiltfang, J.; da Cruz E Silva, O.A.B.; Rebelo, S. ABC Transporters Are Key Players in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 61, 463–485. [Google Scholar] [CrossRef] [PubMed]
- Mora Lagares, L.; Minovski, N.; Caballero Alfonso, A.Y.; Benfenati, E.; Wellens, S.; Culot, M.; Gosselet, F.; Novič, M. Homology Modeling of the Human P-glycoprotein (ABCB1) and Insights into Ligand Binding through Molecular Docking Studies. Int. J. Mol. Sci. 2020, 21, 4058. [Google Scholar] [CrossRef] [PubMed]
- Erickson, M.A.; Hartvigson, P.E.; Morofuji, Y.; Owen, J.B.; Butterfield, D.A.; Banks, W.A. Lipopolysaccharide impairs amyloid beta efflux from brain: Altered vascular sequestration, cerebrospinal fluid reabsorption, peripheral clearance and transporter function at the blood–brain barrier. J. Neuroinflammation 2012, 9, 150. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Liang, B.; Wang, Z.; Cheng, X.; Huang, Y.; Liu, Y.; Huang, Z. Influence of four polymorphisms in ABCA1 and PTGS2 genes on risk of Alzheimer’s disease: A meta-analysis. Neurol. Sci. 2016, 37, 1209–1220. [Google Scholar] [CrossRef]
- Rawat, V.; Wang, S.; Sima, J.; Bar, R.; Liraz, O.; Gundimeda, U.; Parekh, T.; Chan, J.; Johansson, J.O.; Tang, C.; et al. ApoE4 Alters ABCA1 Membrane Trafficking in Astrocytes. J. Neurosci. 2019, 39, 9611–9622. [Google Scholar] [CrossRef] [PubMed]
- Ulery, P.G.; Beers, J.; Mikhailenko, I.; Tanzi, R.E.; Rebeck, G.W.; Hyman, B.T.; Strickland, D.K. Modulation of β-Amyloid Precursor Protein Processing by the Low Density Lipoprotein Receptor-related Protein (LRP). Evidence that LRP contributes to the pathogenesis of Alzheimer’s disease. J. Biol. Chem. 2000, 275, 7410–7415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Einem, B.; Schwanzar, D.; Rehn, F.; Beyer, A.-S.; Weber, P.; Wagner, M.; Schneckenburger, H.; von Arnim, C.A. The role of low-density receptor-related protein 1 (LRP1) as a competitive substrate of the amyloid precursor protein (APP) for BACE1. Exp. Neurol. 2010, 225, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Hu, J.; Zhao, N.; Wang, J.; Wang, N.; Cirrito, J.R.; Kanekiyo, T.; Holtzman, D.M.; Bu, G. Astrocytic LRP1 Mediates Brain Aβ Clearance and Impacts Amyloid Deposition. J. Neurosci. 2017, 37, 4023–4031. [Google Scholar] [CrossRef] [Green Version]
- Storck, S.; Meister, S.; Nahrath, J.; Meißner, J.N.; Schubert, N.; Di Spiezio, A.; Baches, S.; Vandenbroucke, R.; Bouter, Y.; Prikulis, I.; et al. Endothelial LRP1 transports amyloid-β1–42 across the blood-brain barrier. J. Clin. Investig. 2015, 126, 123–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, X.; Xia, L.; Liu, L.; Jiang, H.; Shannahan, J.; Du, Y.; Zheng, W. Altered clearance of beta-amyloid from the cerebrospinal fluid following subchronic lead exposure in rats: Roles of RAGE and LRP1 in the choroid plexus. J. Trace Elem. Med. Biol. 2020, 61, 126520. [Google Scholar] [CrossRef]
- Kanekiyo, T.; Liu, C.-C.; Shinohara, M.; Li, J.; Bu, G. LRP1 in Brain Vascular Smooth Muscle Cells Mediates Local Clearance of Alzheimer’s Amyloid-β. J. Neurosci. 2012, 32, 16458–16465. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.; Zhao, Z.; Sagare, A.P.; Wu, Y.; Wang, M.; Owens, N.C.; Verghese, P.B.; Herz, J.; Holtzman, D.M.; Zlokovic, B.V. Blood-brain barrier-associated pericytes internalize and clear aggregated amyloid-β42 by LRP1-dependent apolipoprotein E isoform-specific mechanism. Mol. Neurodegener. 2018, 13, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Georgievska, B.; Gustavsson, S.; Lundkvist, J.; Neelissen, J.; Eketjäll, S.; Ramberg, V.; Bueters, T.; Agerman, K.; Jureus, A.; Svensson, S.; et al. Revisiting the peripheral sink hypothesis: Inhibiting BACE1 activity in the periphery does not alter β-amyloid levels in the CNS. J. Neurochem. 2014, 132, 477–486. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.E.; Priefer, R. Therapeutic Potential of Direct Clearance of the Amyloid-β in Alzheimer’s Disease. Brain Sci. 2020, 10, 93. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, C.; Kulik, A.; Frotscher, M.; Herz, J.; Schäfer, M.K.; Bock, H.H.; May, P. Low Density Lipoprotein Receptor-related Protein 1 (LRP1) Modulates N-Methyl-d-aspartate (NMDA) Receptor-dependent Intracellular Signaling and NMDA-induced Regulation of Postsynaptic Protein Complexes. J. Biol. Chem. 2013, 288, 21909–21923. [Google Scholar] [CrossRef] [Green Version]
- Dato, V.A.; Sánchez, M.C.; Chiabrando, G.A. LRP1 mediates the IGF-1-induced GLUT1 expression on the cell surface and glucose uptake in Müller glial cells. Sci. Rep. 2021, 11, 4742. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, J.; Zerbinatti, C.; Zhan, Y.; Kolber, B.J.; Herz, J.; Muglia, L.J.; Bu, G. Lipoprotein Receptor LRP1 Regulates Leptin Signaling and Energy Homeostasis in the Adult Central Nervous System. PLoS Biol. 2011, 11, e1000575. [Google Scholar] [CrossRef] [Green Version]
- Marzolo, M.-P.; Farfán, P. New Insights into the Roles of Megalin/LRP2 and the Regulation of its Functional Expression. Biol. Res. 2011, 44, 89–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, A.J.; Bertram, L.; Mullin, K.; Sampson, A.J.; Latifzai, K.; Basu, S.; Jones, J.; Kinney, D.; MacKenzie-Ingano, L.; Yu, S.; et al. Genetic association of Alzheimer’s disease with multiple polymorphisms in alpha-2-macroglobulin. Hum. Mol. Genet. 2003, 12, 2765–2776. [Google Scholar] [CrossRef]
- Xu, X.; Wang, Y.; Wang, L.; Liao, Q.; Chang, L.; Xu, L.; Huang, Y.; Ye, H.; Xu, L.; Chen, C.; et al. Meta-Analyses of 8 Polymorphisms Associated with the Risk of the Alzheimer’s Disease. PLoS ONE 2013, 8, e73129. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Jia, J. An Overview of Genome-Wide Association Studies in Alzheimer’s Disease. Neurosci. Bull. 2016, 32, 183–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krimbou, L.; Tremblay, M.; Davignon, J.; Cohn, J.S. Association of apolipoprotein E with alpha2-macroglobulin in human plasma. J. Lipid Res. 1998, 39, 2373–2386. [Google Scholar] [CrossRef]
- Whiten, D.R.; Cox, D.; Horrocks, M.H.; Taylor, C.G.; De, S.; Flagmeier, P.; Tosatto, L.; Kumita, J.R.; Ecroyd, H.; Dobson, C.M.; et al. Single-Molecule Characterization of the Interactions between Extracellular Chaperones and Toxic α-Synuclein Oligomers. Cell Rep. 2018, 23, 3492–3500. [Google Scholar] [CrossRef] [PubMed]
- Malito, E.; Hulse, R.E.; Tang, W.-J. Amyloid β-degrading cryptidases: Insulin degrading enzyme, presequence peptidase, and neprilysin. Cell. Mol. Life Sci. 2008, 65, 2574–2585. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Joachimiak, A.; Rosner, M.R.; Tang, W.-J. Structures of human insulin-degrading enzyme reveal a new substrate recognition mechanism. Nature 2006, 443, 870–874. [Google Scholar] [CrossRef] [Green Version]
- De Tullio, M.B.; Castelletto, V.; Hamley, I.W.; Adami, P.V.M.; Morelli, L.; Castaño, E.M. Proteolytically Inactive Insulin-Degrading Enzyme Inhibits Amyloid Formation Yielding Non-Neurotoxic Aβ Peptide Aggregates. PLoS ONE 2013, 8, e59113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Z.; Liu, N.; Wang, C.; Qin, B.; Zhou, Y.; Xiao, M.; Chang, L.; Yan, L.-J.; Zhao, B. Role of RAGE in Alzheimer’s Disease. Cell. Mol. Neurobiol. 2015, 36, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liu, G.; Wang, Y.; Yao, Y.; Wang, G.; Lei, X.; Zhang, N.; Dong, X. Protective Effect of Buyang Huanwu Decoction on Neurovascular Unit in Alzheimer’s Disease Cell Model via Inflammation and RAGE/LRP1 Pathway. Med. Sci. Monit. 2019, 25, 7813–7825. [Google Scholar] [CrossRef]
- Kook, S.-Y.; Hong, H.S.; Moon, M.; Ha, C.M.; Chang, S.; Mook-Jung, I. A 1-42-RAGE Interaction Disrupts Tight Junctions of the Blood-Brain Barrier Via Ca2+-Calcineurin Signaling. J. Neurosci. 2012, 32, 8845–8854. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Son, S.M.; Jin, S.M.; Hong, H.S.; Shin, D.H.; Kim, S.J.; Huh, K.; Mook-Jung, I. RAGE regulates BACE1 and Aβ generationviaNFAT1 activation in Alzheimer’s disease animal model. FASEB J. 2009, 23, 2639–2649. [Google Scholar] [CrossRef]
- Tobon-Velasco, J.C.; Cuevas, E.; Torres-Ramos, M.A. Receptor for AGEs (RAGE) as Mediator of NF-kB Pathway Activation in Neuroinflammation and Oxidative Stress. CNS Neurol. Disord.-Drug Targets 2014, 13, 1615–1626. [Google Scholar] [CrossRef]
- Hladky, S.B.; Barrand, M.A. Mechanisms of fluid movement into, through and out of the brain: Evaluation of the evidence. Fluids Barriers CNS 2014, 11, 1–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szentistvanyi, I.; Patlak, C.S.; Ellis, R.A.; Cserr, H.F. Drainage of interstitial fluid from different regions of rat brain. Am. J. Physiol. Physiol. 1984, 246, F835–F844. [Google Scholar] [CrossRef]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A Paravascular Pathway Facilitates CSF Flow through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, J.R.; Iliff, J.J. Vasomotor influences on glymphatic-lymphatic coupling and solute trafficking in the central nervous system. J. Cereb. Blood Flow Metab. 2019, 40, 1724–1734. [Google Scholar] [CrossRef]
- Hablitz, L.M.; Plá, V.; Giannetto, M.; Vinitsky, H.S.; Stæger, F.F.; Metcalfe, T.; Nguyen, R.; Benrais, A.; Nedergaard, M. Circadian control of brain glymphatic and lymphatic fluid flow. Nat. Commun. 2020, 11, 4411. [Google Scholar] [CrossRef]
- Proulx, S.T. Cerebrospinal fluid outflow: A review of the historical and contemporary evidence for arachnoid villi, perineural routes, and dural lymphatics. Cell. Mol. Life Sci. 2021, 78, 2429–2457. [Google Scholar] [CrossRef]
- Reiber, H. Blood-cerebrospinal fluid (CSF) barrier dysfunction means reduced CSF flow not barrier leakage-conclusions from CSF protein data. Arq. Neuro-Psiquiatr. 2021, 79, 56–67. [Google Scholar] [CrossRef]
- Pollay, M. The function and structure of the cerebrospinal fluid outflow system. Cereb. Fluid Res. 2010, 7, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coimbra, J.; Marques, D.F.F.; Baptista, S.J.; Pereira, C.M.F.; Moreira, P.I.; Dinis, T.C.P.; Santos, A.E.; Salvador, J.A.R. Highlights in BACE1 Inhibitors for Alzheimer’s Disease Treatment. Front. Chem. 2018, 6, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imbimbo, B.P.; Watling, M. Investigational BACE inhibitors for the treatment of Alzheimer’s disease. Expert Opin. Investig. Drugs 2019, 28, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Keskin, A.D.; Kekuš, M.; Adelsberger, H.; Neumann, U.; Shimshek, D.R.; Song, B.; Zott, B.; Peng, T.; Förstl, H.; Staufenbiel, M.; et al. BACE inhibition-dependent repair of Alzheimer’s pathophysiology. Proc. Natl. Acad. Sci. USA 2017, 114, 8631–8636. [Google Scholar] [CrossRef] [Green Version]
- Fukumoto, H.; Takahashi, H.; Tarui, N.; Matsui, J.; Tomita, T.; Hirode, M.; Sagayama, M.; Maeda, R.; Kawamoto, M.; Hirai, K.; et al. A Noncompetitive BACE1 Inhibitor TAK-070 Ameliorates Aβ Pathology and Behavioral Deficits in a Mouse Model of Alzheimer’s Disease. J. Neurosci. 2010, 30, 11157–11166. [Google Scholar] [CrossRef]
- Sperling, R.; Henley, D.; Aisen, P.S.; Raman, R.; Donohue, M.C.; Ernstrom, K.; Rafii, M.S.; Streffer, J.; Shi, Y.; Karcher, K.; et al. Findings of Efficacy, Safety, and Biomarker Outcomes of Atabecestat in Preclinical Alzheimer Disease. JAMA Neurol. 2021, 78, 293–301. [Google Scholar] [CrossRef]
- Egan, M.F.; Kost, J.; Tariot, P.N.; Aisen, P.S.; Cummings, J.L.; Vellas, B.; Sur, C.; Mukai, Y.; Voss, T.; Furtek, C.; et al. Randomized Trial of Verubecestat for Mild-to-Moderate Alzheimer’s Disease. N. Engl. J. Med. 2018, 378, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Shugart, J.; Strobel, G. Cognitive decline trips up API trials of BACE inhibitor. Alzheimer Research Forum, 12 July 2019. [Google Scholar]
- Menendez-Gonzalez, M.; Perez-Pinera, P.; Martinez-Rivera, M.; Muniz, A.L.; Vega, J.A. Immunotherapy for Alzheimer’s Disease: Rational Basis in Ongoing Clinical Trials. Curr. Pharm. Des. 2011, 17, 508–520. [Google Scholar] [CrossRef]
- Morrone, C.D.; Liu, M.; Black, S.E.; McLaurin, J. Interaction between therapeutic interventions for Alzheimer’s disease and physiological Aβ clearance mechanisms. Front. Aging Neurosci. 2015, 7, 64. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Y.; Bu, X.-L.; Liu, Y.-H.; Zhu, C.; Shen, L.-L.; Jiao, S.-S.; Zhu, X.-Y.; Giunta, B.; Tan, J.; Song, W.; et al. Physiological amyloid-beta clearance in the periphery and its therapeutic potential for Alzheimer’s disease. Acta Neuropathol. 2015, 130, 487–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, S.J.; Andersson, C.; Narwal, R.; Janson, J.; Goldschmidt, T.J.; Appelkvist, P.; Bogstedt, A.; Steffen, A.-C.; Haupts, U.; Tebbe, J.; et al. Sustained peripheral depletion of amyloid-β with a novel form of neprilysin does not affect central levels of amyloid-β. Brain 2014, 137, 553–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, J.; Xu, F.; Deane, R.; Romanov, G.; Previti, M.L.; Zeigler, K.; Zlokovic, B.V.; Van Nostrand, W.E. Early-onset and Robust Cerebral Microvascular Accumulation of Amyloid β-Protein in Transgenic Mice Expressing Low Levels of a Vasculotropic Dutch/Iowa Mutant Form of Amyloid β-Protein Precursor. J. Biol. Chem. 2004, 279, 20296–20306. [Google Scholar] [CrossRef] [Green Version]
- Moreira, P.I.; Sayre, L.M.; Zhu, X.; Nunomura, A.; Smith, M.A.; Perry, G. Detection and Localization of Markers of Oxidative Stress by In Situ Methods: Application in the Study of Alzheimer Disease. Methods Mol. Biol. 2010, 610, 419–434. [Google Scholar] [CrossRef] [Green Version]
- Boada, M.; López, O.L.; Olazarán, J.; Núñez, L.; Pfeffer, M.; Paricio, M.; Lorites, J.; Piñol-Ripoll, G.; Gámez, J.E.; Anaya, F.; et al. A randomized, controlled clinical trial of plasma exchange with albumin replacement for Alzheimer’s disease: Primary results of the AMBAR Study. Alzheimer’s Dement. 2020, 16, 1412–1425. [Google Scholar] [CrossRef]
- Jin, W.-S.; Shen, L.-L.; Bu, X.-L.; Zhang, W.-W.; Chen, S.-H.; Huang, Z.-L.; Xiong, J.-X.; Gao, C.-Y.; Dong, Z.; He, Y.-N.; et al. Peritoneal dialysis reduces amyloid-beta plasma levels in humans and attenuates Alzheimer-associated phenotypes in an APP/PS1 mouse model. Acta Neuropathol. 2017, 134, 207–220. [Google Scholar] [CrossRef]
- Kitaguchi, N.; Tatebe, H.; Sakai, K.; Kawaguchi, K.; Matsunaga, S.; Kitajima, T.; Tomizawa, H.; Kato, M.; Sugiyama, S.; Suzuki, N.; et al. Influx of Tau and Amyloid-β Proteins into the Blood During Hemodialysis as a Therapeutic Extracorporeal Blood Amyloid-β Removal System for Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 69, 687–707. [Google Scholar] [CrossRef] [PubMed]
- González, M.M. Implantable Systems for Continuous Liquorpheresis and CSF Replacement. Cureus 2017, 9, e1022. [Google Scholar] [CrossRef] [Green Version]
- Bolash, R.; Mekhail, N. Intrathecal Pain Pumps: Indications, patient selection, techniques, and outcomes. Neurosurg. Clin. N. Am. 2014, 25, 735–742. [Google Scholar] [CrossRef]
- Shirotani, K.; Tsubuki, S.; Iwata, N.; Takaki, Y.; Harigaya, W.; Maruyama, K.; Kiryu-Seo, S.; Kiyama, H.; Iwata, H.; Tomita, T.; et al. Neprilysin Degrades Both Amyloid β Peptides 1–40 and 1–42 Most Rapidly and Efficiently among Thiorphan- and Phosphoramidon-sensitive Endopeptidases. J. Biol. Chem. 2001, 276, 21895–21901. [Google Scholar] [CrossRef] [Green Version]
- Nalivaeva, N.N.; Zhuravin, I.A.; Turner, A.J. Neprilysin expression and functions in development, ageing and disease. Mech. Ageing Dev. 2020, 192, 111363. [Google Scholar] [CrossRef]
- Salazar, J.; Rojas-Quintero, J.; Cano, C.; Pérez, J.L.; Ramirez, P.P.; Carrasquero, R.; Torres, W.; Espinoza, C.; Chacín-González, M.; Bermudez, V. Neprilysin: A Potential Therapeutic Target of Arterial Hypertension? Curr. Cardiol. Rev. 2020, 16, 25–35. [Google Scholar] [CrossRef]
- Marr, R.A.; Rockenstein, E.; Mukherjee, A.; Kindy, M.S.; Hersh, L.B.; Gage, F.H.; Verma, I.M.; Masliah, E. Neprilysin Gene Transfer Reduces Human Amyloid Pathology in Transgenic Mice. J. Neurosci. 2003, 23, 1992–1996. [Google Scholar] [CrossRef] [Green Version]
- Leissring, M.A.; Farris, W.; Chang, A.Y.; Walsh, D.M.; Wu, X.; Sun, X.; Frosch, M.P.; Selkoe, D.J. Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron 2003, 40, 1087–1093. [Google Scholar] [CrossRef] [Green Version]
- Iwata, N.; Tsubuki, S.; Takaki, Y.; Shirotani, K.; Lu, B.; Gerard, N.P.; Gerard, C.; Hama, E.; Lee, H.J.; Saido, T.C. Metabolic regulation of brain Abeta by neprilysin. Science 2001, 292, 1550–1552. [Google Scholar] [CrossRef]
- Hemming, M.L.; Patterson, M.; Reske-Nielsen, C.; Lin, L.; Isacson, O.; Selkoe, D.J. Reducing Amyloid Plaque Burden via Ex Vivo Gene Delivery of an Aβ-Degrading Protease: A Novel Therapeutic Approach to Alzheimer Disease. PLoS Med. 2007, 4, e262. [Google Scholar] [CrossRef] [Green Version]
- El-Amouri, S.S.; Zhu, H.; Yu, J.; Marr, R.; Verma, I.M.; Kindy, M.S. Neprilysin: An Enzyme Candidate to Slow the Progression of Alzheimer’s Disease. Am. J. Pathol. 2008, 172, 1342–1354. [Google Scholar] [CrossRef] [Green Version]
- Webster, C.I.; Burrell, M.; Olsson, L.-L.; Fowler, S.B.; Digby, S.; Sandercock, A.; Snijder, A.; Tebbe, J.; Haupts, U.; Grudzinska, J.; et al. Engineering Neprilysin Activity and Specificity to Create a Novel Therapeutic for Alzheimer’s Disease. PLoS ONE 2014, 9, e104001. [Google Scholar] [CrossRef] [Green Version]
- Kamble, S.; Barale, S.; Dhanavade, M.; Sonawane, K. Structural significance of Neprylysin from Streptococcus suis GZ1 in the degradation of Aβ peptides, a causative agent in Alzheimer’s disease. Comput. Biol. Med. 2021, 136, 104691. [Google Scholar] [CrossRef] [PubMed]
- Barua, N.U.; Miners, J.S.; Bienemann, A.S.; Wyatt, M.J.; Welser, K.; Tabor, A.B.; Hailes, H.C.; Love, S.; Gill, S.S. Convection-Enhanced Delivery of Neprilysin: A Novel Amyloid-β-Degrading Therapeutic Strategy. J. Alzheimer’s Dis. 2012, 32, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Campos, C.R.; Kemble, A.M.; Niewoehner, J.; Freskgård, P.-O.; Urich, E. Brain Shuttle Neprilysin reduces central Amyloid-β levels. PLoS ONE 2020, 15, e0229850. [Google Scholar] [CrossRef] [PubMed]
- Rofo, F.; Yilmaz, C.U.; Metzendorf, N.; Gustavsson, T.; Beretta, C.; Erlandsson, A.; Sehlin, D.; Syvänen, S.; Nilsson, P.; Hultqvist, G. Enhanced neprilysin-mediated degradation of hippocampal Aβ42 with a somatostatin peptide that enters the brain. Theranostics 2021, 11, 789–804. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.A.; Hadziselimovic, A.; Sanders, C.R. The vexing complexity of the amyloidogenic pathway. Protein Sci. 2019, 28, 1177–1193. [Google Scholar] [CrossRef] [PubMed]
- Nedergaard, M.; Goldman, S.A. Glymphatic failure as a final common pathway to dementia. Science 2020, 370, 50–56. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Clearance Pathway | Direction/Biological Compartment | Key Players | Alterations in Pathological Conditions (AD) |
|---|---|---|---|
| Blood–brain barrier | ISF to peripheral circulation | LRP1, LRP2, ABCB1, ABCA1, α2-macroglobulin, IDE, ApoE, RAGE | Reduced efflux Increased RAGE-influx |
| Intracellular degradation | Microglia, Astrocyte | ubiquitin–proteasome pathway, autophagy–lysosome pathway, endosome–lysosome pathway | Reduced |
| Extracellular degradation | ISF | Proteases, phagocytosis (microglia/astrocyte uptake) | Reduced |
| Perivascular drainage | ISF to CSF | Diffusion Transporter-mediated active mechanism | Reduced diffusion |
| Glymphatic system | ISF to CSF | Bulk flow AQP4 Sleep | Unknown (probably reduced) |
| CSF absorption | CSF to peripheral circulation CSF to peripheral lymphatic system | Brain–CSF barrier Arachnoid villi Lymphatic absorption | Reduced flow via brain-CSF barrier and arachnoid villi |
| Type of Treatment | Pathophysiological Mechanism | Use/Efficiency |
|---|---|---|
| BACE inhibitors | Inhibit BACE1 and BACE2 in order to minimize Aβ production | Inefficient Several important adverse effects (brain atrophy, weight loss) Currently not recommended |
| Aβ monoclonal antibodies | Immunotherapy (Antigen-antibody complex)—favors Aβ elimination | Inconsistent results Recently approved Adacunumab for clinical use |
| Aβ vaccine | DNA vaccination for anti-Aβ immunotherapy | Phase III clinical trials ongoing |
| RAGE inhibitors | Inhibition of RAGE Inhibition of Aβ influx to CNS Reduction in oxidative stress and neuroinflammation | Azeliragon tested in phase 2/3 trials—missed endpoints Research in progress |
| Plasmapheresis | Reduction in Aβ peripheral level The “peripheral sink therapeutic strategy” | Positive preliminary results |
| Peritoneal dialysis | Reduction in Aβ peripheral level The “peripheral sink therapeutic strategy” | Positive preliminary results |
| Implantable intrathecal pumps | Reduction in Aβ CSF level The “CSF sink therapeutic strategy” | Near future approach |
| Aβ cleavage | Degradation of Aβ at both CNS and peripheral level | Intracerebral delivery of neprilysin—positive preliminary results Peripheral delivery of neprilysin—no impact on Aβ at brain level |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schreiner, T.G.; Popescu, B.O. Amyloid Beta Dynamics in Biological Fluids—Therapeutic Impact. J. Clin. Med. 2021, 10, 5986. https://doi.org/10.3390/jcm10245986
Schreiner TG, Popescu BO. Amyloid Beta Dynamics in Biological Fluids—Therapeutic Impact. Journal of Clinical Medicine. 2021; 10(24):5986. https://doi.org/10.3390/jcm10245986
Chicago/Turabian StyleSchreiner, Thomas Gabriel, and Bogdan Ovidiu Popescu. 2021. "Amyloid Beta Dynamics in Biological Fluids—Therapeutic Impact" Journal of Clinical Medicine 10, no. 24: 5986. https://doi.org/10.3390/jcm10245986
APA StyleSchreiner, T. G., & Popescu, B. O. (2021). Amyloid Beta Dynamics in Biological Fluids—Therapeutic Impact. Journal of Clinical Medicine, 10(24), 5986. https://doi.org/10.3390/jcm10245986