
Gut Microbiome in Chronic Coronary Syndrome Patients

 , , , , , , , , ,
, , , , , , , , ,  , and
, and  add
Show full author list
add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Quantitation of Metabolites Using an AbsoluteIDQ p180 Kit
2.3. Statistical Analysis
3. Results
3.1. Description of the Study Population
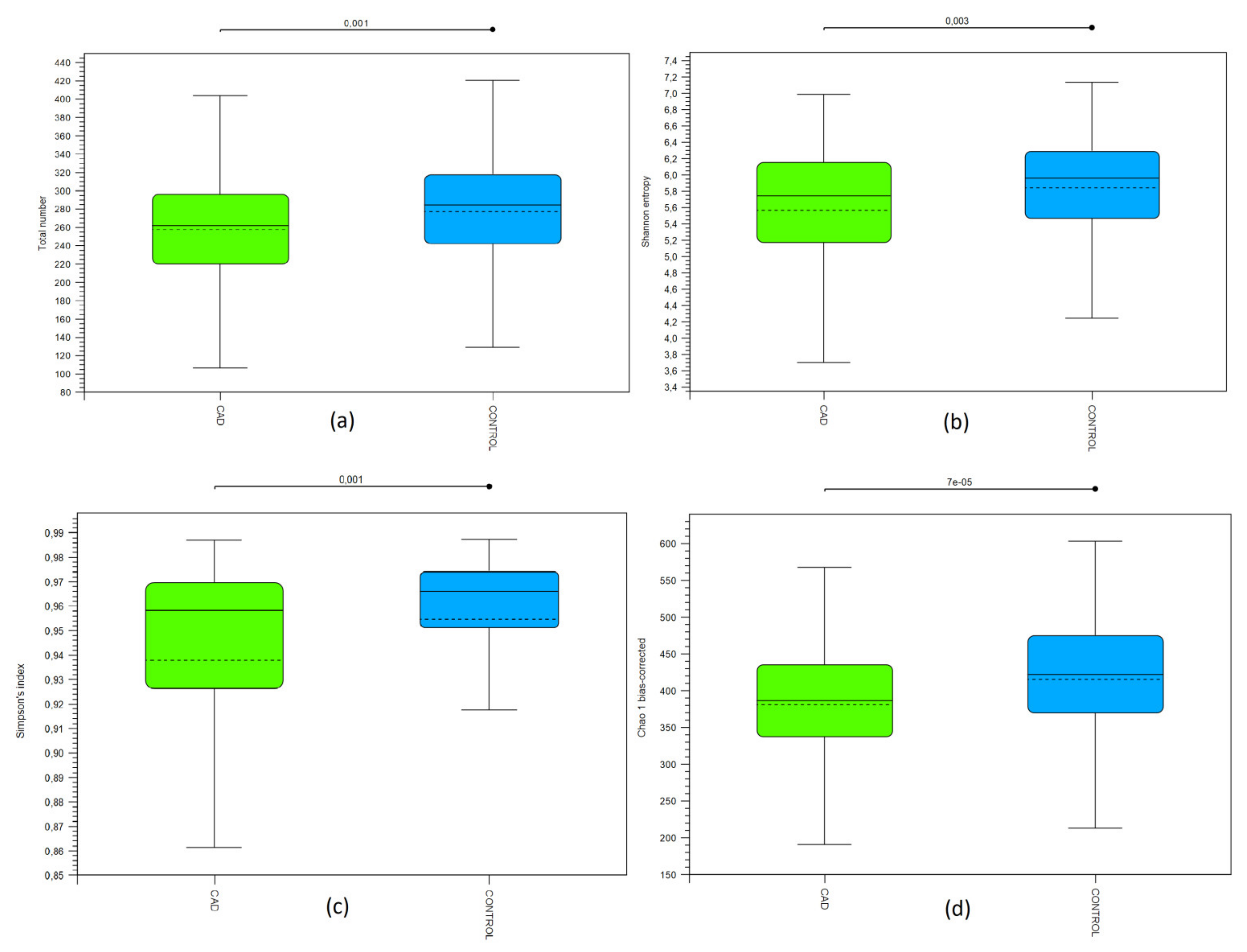
3.2. Bioinformatic Biodiversity Analysis
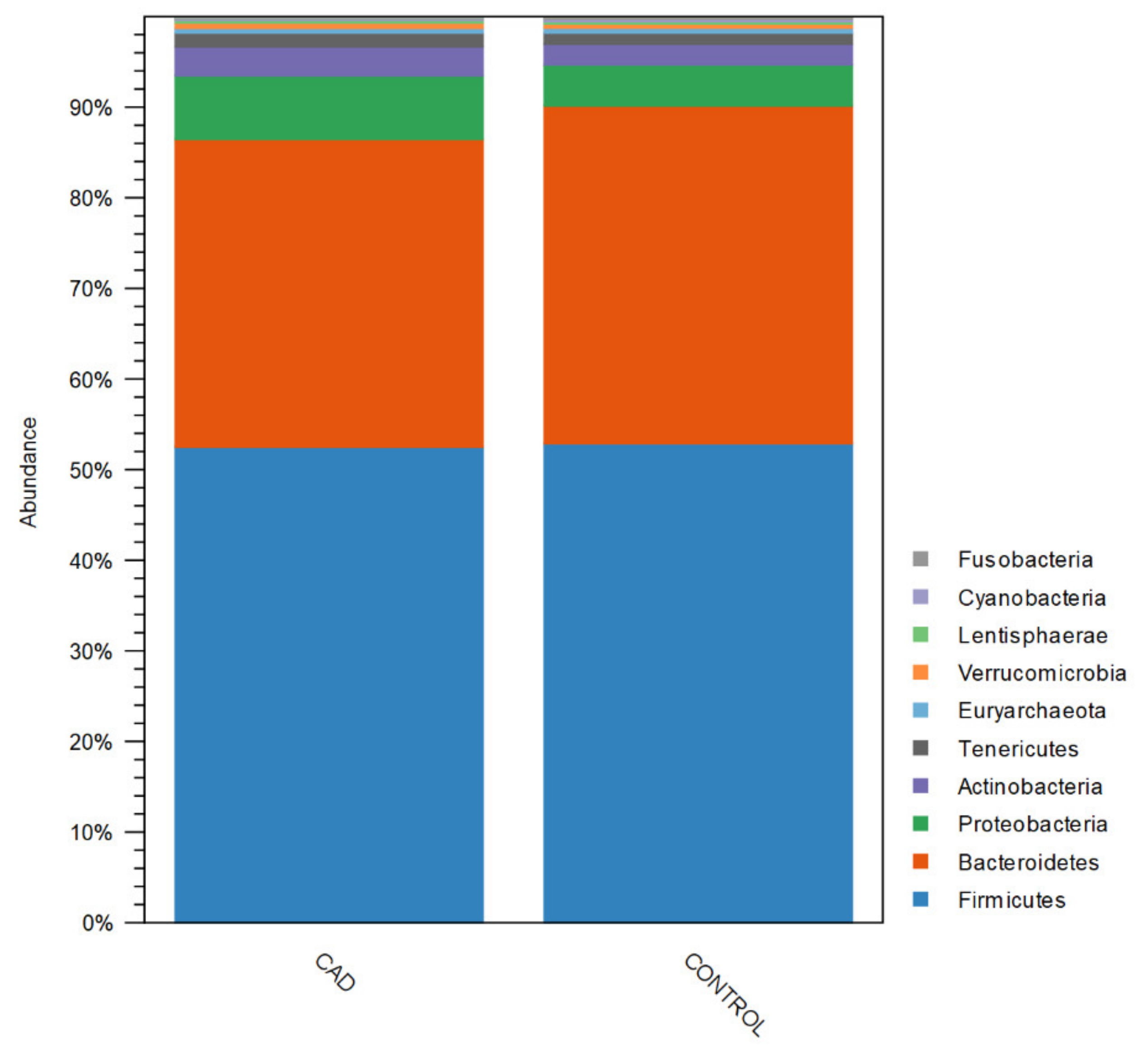
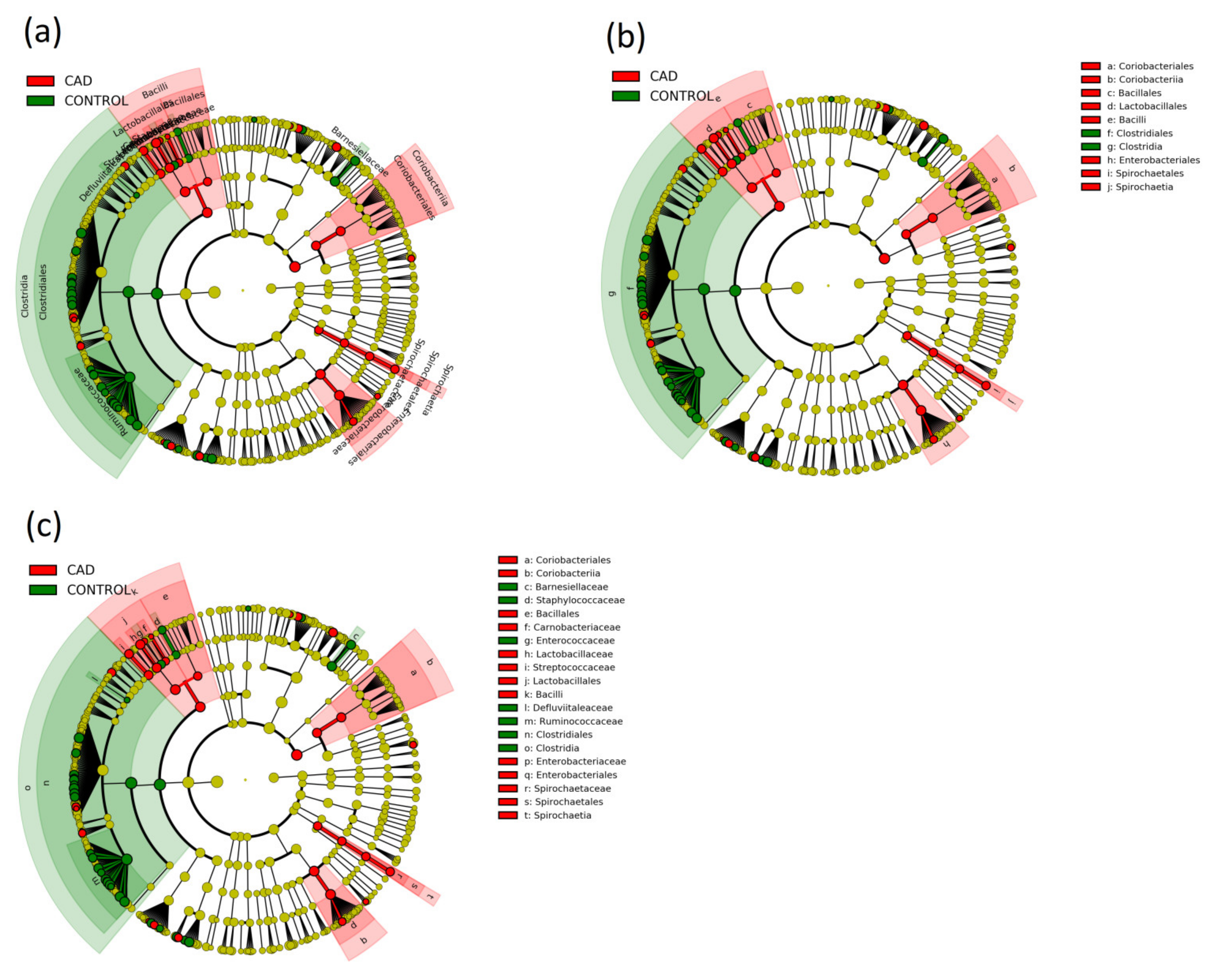
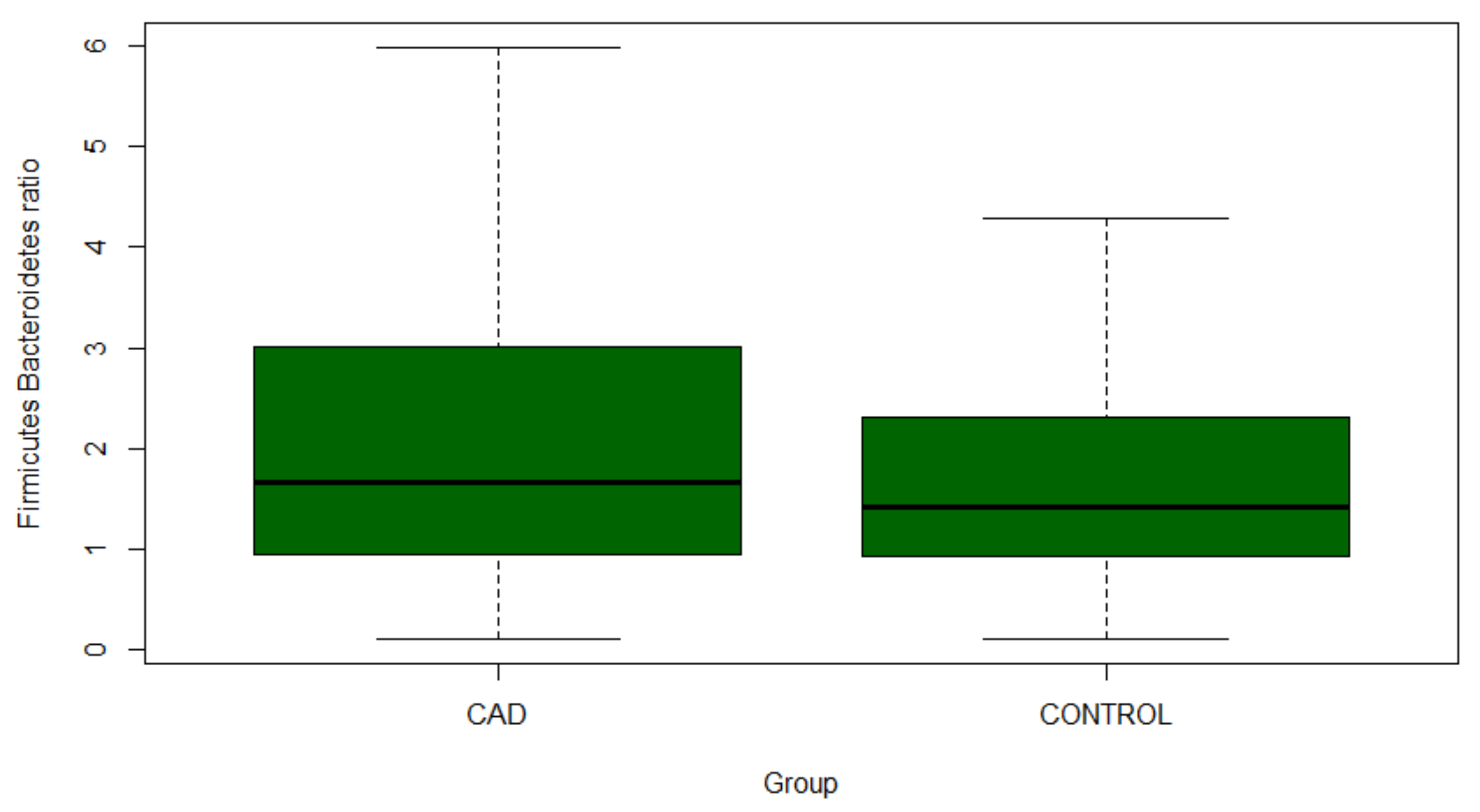
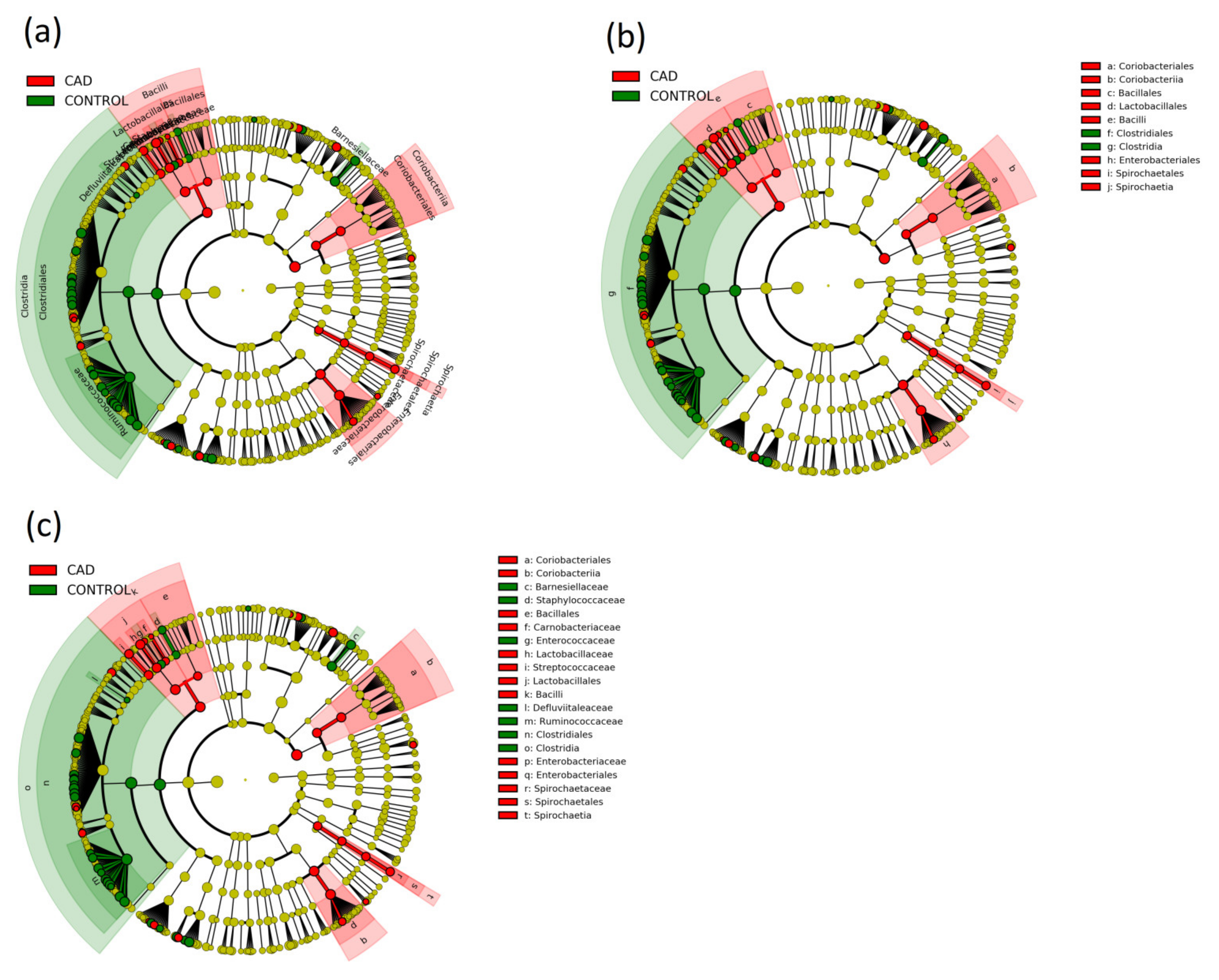
3.3. Gut Microbiome Composition Analysis
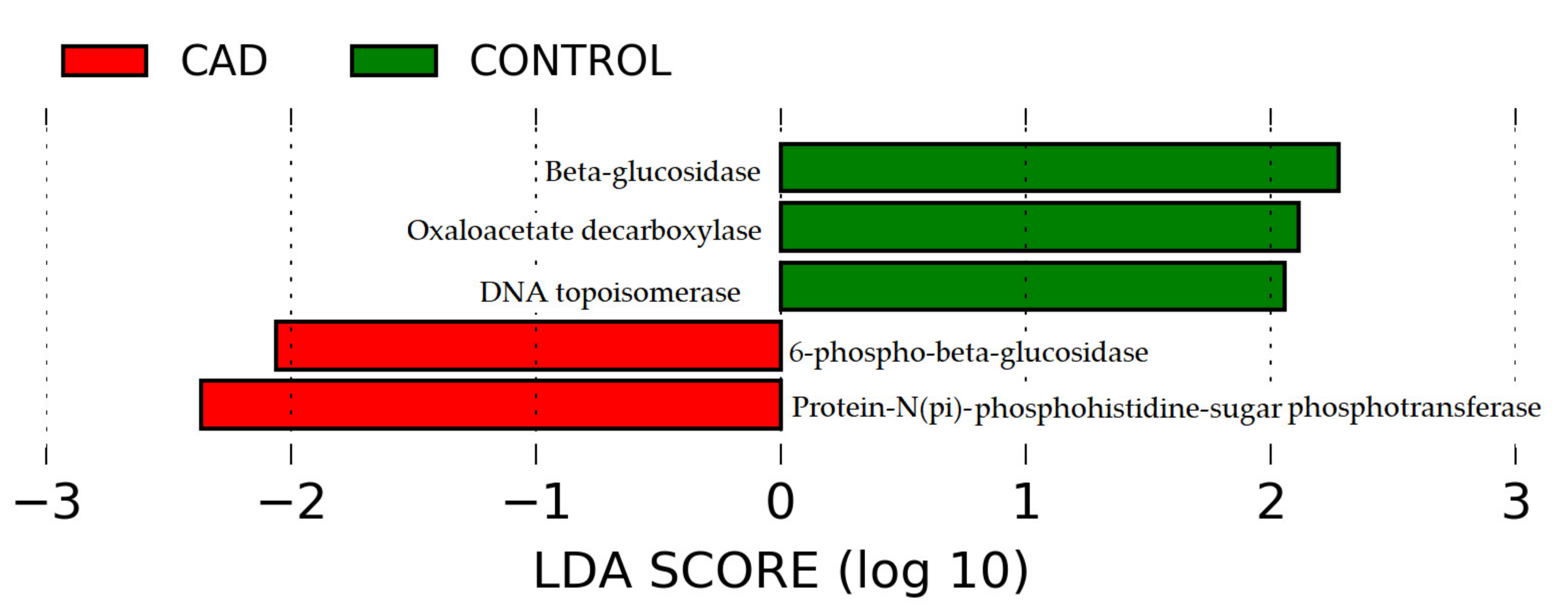
3.4. Prediction of the Functional Potential of the Intestinal Microbiome
3.5. Targeted Metabolomics and Biochemical Parameters
4. Discussion
5. Limitation of the Study
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wojtyniak, B.; Zdrojewski, T. Mortality due to cardiovascular diseases in Poland-analysis of changes in 1999–2013. In Epidemiology and Prevention of Cardiovascular Diseases, 1st ed.; Kopec, G., Jankowski, P., Pajak, A., Drygas, W., Eds.; Medycyna Praktyczna: Krakow, Poland, 2015; pp. 29–39. [Google Scholar]
- European Health Examination Survey. Available online: http://www.ehes.info/manuals/national_manuals/national_manual_Poland_EN.pdf (accessed on 25 August 2021).
- Jozwiak, J.J.; Studzinski, K.; Tomasik, T.; Windak, A.; Banach, M. LIPIDOGRAM 2015 Investigators. The prevalence of cardiovascular risk factors and cardiovascular disease among primary care patients in Poland: Results from the LIPIDOGRAM2015 study. Atheroscler. Suppl. 2020, 42, e15–e24. [Google Scholar] [CrossRef] [PubMed]
- Kannel, W.B.; Dawber, T.R.; Kagan, A.; Revotskie, N.; Stokes, J.O.S.E.P.H., III. Factors of risk in the development of coronary heart disease-six year follow-up experience: The Framingham Study. Ann. Intern. Med. 1961, 55, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Marcinkiewicz-Siemion, M.; Ciborowski, M.; Kretowski, A.; Musial, W.J.; Kaminski, K.A. Metabolomics-A wide-open door to personalized treatment in chronic heart failure? Int. J. Cardiol. 2016, 219, 156–163. [Google Scholar] [CrossRef]
- Emoto, T.; Sasaki, N.; Yamashita, T.; Kasahara, K.; Yodoi, K.; Sasaki, Y.; Matsumoto, T.; Mizoguchi, T.; Hirata, K. Regulatory/effector T-cell ratio is reduced in coronary artery disease. Circ. J. 2014, 78, 2935–2941. [Google Scholar] [CrossRef] [Green Version]
- Atarashi, K.; Tanoue, T.; Oshima, K.; Suda, W.; Nagano, Y.; Nishikawa, H.; Fukuda, S.; Saito, T.; Narushima, S.; Hase, K.; et al. Treg induction by a rationally selected mixture of Clostridia strains from the human micro biota. Nature 2013, 500, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Emoto, T.; Sasaki, N.; Hirata, K.I. Gut microbiota and coronary artery disease. Int. Heart J. 2016, 57, 663–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salamon, D.; Gosiewski, T.; Krawczyk, A.; Sroka-Oleksiak, A.; Duplaga, M.; Fyderek, K.; Kowalska-Duplaga, K. Quantitative changes in selected bacteria in the stool during the treatment of Crohn’s disease. Adv. Med. Sci. 2020, 65, 348–353. [Google Scholar] [CrossRef]
- Fukudo, S. Irritable bowel syndrome, emotion regulation, and gut microbiota. Brain Nerve 2016, 68, 607–615. [Google Scholar]
- Wozniak, D.; Cichy, W.; Przyslawski, J.; Drzymała-Czyz, S. The role of microbiota and enteroendocrine cells in maintaining homeostasis in the human digestive tract. Adv. Med. Sci. 2021, 66, 284–292. [Google Scholar] [CrossRef]
- Lynch, S.V. Gut microbiota and allergic disease. New Insights. Ann. Am. Thorac. Soc. 2016, 13, 51–54. [Google Scholar]
- Gülden, E.; Wong, F.S.; Wen, L. The gut microbiota and type 1 diabetes. Clin. Immunol. 2015, 159, 143–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ussar, S.; Griffin, N.W.; Bezy, O.; Fujisaka, S.; Vienberg, S.; Softic, S.; Deng, L.; Bry, L.; Gordon, J.I.; Kahn, C.R. Interactions between gut microbiota, host genetics and diet modulate the predisposition to obesity and metabolic syndrome. Cell Metab. 2015, 22, 516–530. [Google Scholar] [CrossRef] [Green Version]
- Kootte, R.S.; Vrieze, A.; Holleman, F.; Dallinga-Thie, G.M.; Zoetendal, E.G.; de Vos, W.M.; Groen, A.K.; Hoekstra, J.B.; Stroes, E.S.; Nieuwdorp, M. The therapeutic potential of manipulating gut microbiota in obesity and type 2 diabetes mellitus. Diabetes Obes. Metab. 2012, 14, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Bonder, M.J.; Cenit, M.C.; Tigchelaar, E.F.; Maatman, A.; Dekens, J.A.; Brandsma, E.; Marczynska, J.; Imhann, F.; Weersma, R.K.; et al. The gut microbiome contributes to a substantial proportion of the variation in blood lipids. Circ. Res. 2015, 117, 817–824. [Google Scholar] [CrossRef]
- Yang, T.; Santisteban, M.M.; Rodriguez, V.; Li, E.; Ahmari, N.; Carvajal, J.M.; Zadeh, M.; Gong, M.; Qi, Y.; Zubcevic, J.; et al. Gut dysbiosis is linked to hypertension. Hypertension 2015, 65, 1331–1340. [Google Scholar] [CrossRef] [Green Version]
- Emoto, T.; Yamashita, T.; Sasaki, N.; Hirota, Y.; Hayashi, T.; So, A.; Kasahara, K.; Yodoi, K.; Matsumoto, T.; Mizoguchi, T.; et al. Analysis of gut microbiota in coronary artery disease patients: A possible link between gut microbiota and coronary artery disease. J. Atheroscler. Thromb. 2016, 23, 908–921. [Google Scholar] [CrossRef] [Green Version]
- Emoto, T.; Yamashita, T.; Kobayashi, T.; Sasaki, N.; Hirota, Y.; Hayashi, T.; So, A.; Kasahara, K.; Yodoi, K.; Matsumoto, T.; et al. Characterization of gut microbiota profiles in coronary artery disease patients using data mining analysis of terminal restriction fragment length polymorphism: Gut microbiota could be a diagnostic marker of coronary artery disease. Heart Vessel. 2017, 32, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Gao, R.; Zhang, Y.; Pan, D.; Zhu, Y.; Zhang, X.; Yang, R.; Jiang, R.; Xu, Y.; Qin, H. Dysbiosis signatures of gut microbiota in coronary artery disease. Physiol. Genom. 2018, 50, 893–903. [Google Scholar] [CrossRef]
- Liu, H.; Chen, X.; Hu, X.; Niu, H.; Tian, R.; Wang, H.; Pang, H.; Jiang, L.; Qiu, B.; Chen, X.; et al. Alterations in the gut microbiome and metabolism with coronary artery disease severity. Microbiome 2019, 7, 68. [Google Scholar] [CrossRef] [PubMed]
- Tian, R.; Liu, H.; Feng, S.; Wang, H.; Wang, Y.; Wang, Y.; Liang, L.; Xu, H.; Xing, H.; Zhang, S. Gut microbiota dysbiosis in stable coronary artery disease combined with type 2 diabetes mellitus influences cardiovascular prognosis. Nutr. Metab. Cardiovasc. Dis. 2021, 31, 1454–1466. [Google Scholar] [CrossRef]
- Cui, L.; Zhao, T.; Hu, H.; Zhang, W.; Hua, X. Association Study of Gut Flora in Coronary Heart Disease through High-Throughput Sequencing. Biomed Res. Int. 2017, 2017, 3796359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paniczko, M.; Chlabicz, M.; Jamiolkowski, J.; Sowa, P.; Szpakowicz, M.; Lapinska, M.; Kondraciuk, M.; Ptaszynska-Kopczynska, K.; Raczkowski, A.; Szpakowicz, A.; et al. Impact of Pulse Wave Velocity and Parameters Reflecting Android Type Fat Distribution on Left Ventricular Diastolic Dysfunction in Patients with Chronic Coronary Syndromes. J. Clin. Med. 2020, 9, 3924. [Google Scholar] [CrossRef] [PubMed]
- Chlabicz, M.; Jamiolkowski, J.; Paniczko, M.; Sowa, P.; Lapinska, M.; Szpakowicz, M.; Jurczuk, N.; Kondraciuk, M.; Raczkowski, A.; Sawicka, E.; et al. Independent Impact of Gynoid Fat Distribution and Free Testosterone on Circulating Levels of N-Terminal Pro-Brain Natriuretic Peptide (NT-proBNP) in Humans. J. Clin. Med. 2019, 9, 74. [Google Scholar] [CrossRef] [Green Version]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.Y.; Wu, T.T.; Liu, Z.Q.; Li, A.; Guo, Q.Q.; Ma, Y.Y.; Zhang, Z.L.; Xun, Y.L.; Zhang, J.C.; Wang, W.R.; et al. Gut Microbiome-Based Diagnostic Model to Predict Coronary Artery Disease. J. Agric. Food Chem. 2020, 68, 3548–3557. [Google Scholar] [CrossRef]
- Wexler, H.M. Bacteroides: The good, the bad, and the nitty-gritty. Clin. Microbiol. Rev. 2007, 20, 593–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisanz, J.E.; Upadhyay, V.; Turnbaugh, J.A.; Ly, K.; Turnbaugh, P.J. Meta-Analysis Reveals Reproducible Gut Microbiome Alterations in Response to a High-Fat Diet. Cell Host Microbe 2019, 26, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef] [PubMed]
- Schwiertz, A.; Taras, D.; Schafer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.D. Microbiota and SCFA in lean and overweight healthy subjects. Obesity 2010, 18, 190–195. [Google Scholar] [CrossRef]
- Gomes, A.C.; Hoffmann, C.; Mota, J.F. The human gut microbiota: Metabolism and perspective in obesity. Gut Microbes 2018, 9, 308–325. [Google Scholar] [CrossRef] [Green Version]
- Chambers, E.S.; Preston, T.; Frost, G.; Morrison, D.J. Role of Gut Microbiota-Generated Short-Chain Fatty Acids in Metabolic and Cardiovascular Health. Curr. Nutr. Rep. 2018, 7, 198–206. [Google Scholar] [CrossRef] [Green Version]
- Miquel, S.; Martin, R.; Rossi, O.; Bermudez-Humaran, L.G.; Chatel, J.M.; Sokol, H.; Thomas, M.; Wells, J.M.; Langella, P. Faecalibacterium prausnitzii and human intestinal health. Curr. Opin. Microbiol. 2013, 16, 255–261. [Google Scholar] [CrossRef]
- Harrison, C.A.; Laubitz, D.; Ohland, C.L.; Midura-Kiela, M.T.; Patil, K.; Besselsen, D.G.; Jamwal, D.R.; Jobin, C.; Ghishan, F.K.; Kiela, P.R. Microbial Dysbiosis Associated with Impaired Intestinal Na+/H+ Exchange Accelerates and Exacerbates Colitis in Ex-germ Free Mice. Mucosal Immunol. 2018, 11, 1329–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, W.; Zhu, H.; Chen, J.; Kwek, E.; He, Z.; Liu, J.; Ma, N.; Ma, K.Y.; Chen, Z.Y. Wild Melon Seed Oil Reduces Plasma Cholesterol and Modulates Gut Microbiota in Hypercholesterolemic Hamsters. J. Agric. Food Chem. 2020, 68, 2071–2081. [Google Scholar] [CrossRef] [PubMed]
- Shang, Q.; Song, G.; Zhang, M.; Shi, J.; Xu, C.; Hao, J.; Li, G.; Yu, G. Dietary Fucoidan Improves Metabolic Syndrome in Association with Increased Akkermansia Population in the Gut Microbiota of High-fat Diet-fed Mice. J. Funct. Foods 2017, 28, 138–146. [Google Scholar] [CrossRef]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef] [Green Version]
- Gao, B.; Zhong, M.; Shen, Q.; Wu, Y.; Cao, M.; Ju, S.; Chen, L. Gut microbiota in early pregnancy among women with Hyperglycaemia vs. Normal blood glucose. BMC Pregnancy Childbirth 2020, 20, 284. [Google Scholar]
- Tang, W.H.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.B.; Gu, X.; Buffa, J.A.; Hurd, A.G.; Wang, Z.; Zhu, W.; Gupta, N.; Skye, S.M.; Cody, D.B.; Levison, B.S.; et al. Development of a gut microbe-targeted nonlethal therapeutic to inhibit thrombosis potential. Nat. Med. 2018, 24, 1407–1417. [Google Scholar] [CrossRef]
- Alakomi, H.L.; Skytta, E.; Saarela, M.; Mattila-Sandholm, T.; Latva-Kala, K.; Helander, I.M. Lactic acid permeabilizes Gram-negative bacteria by disrupting the outer membrane. Appl. Environ. Microbiol. 2000, 66, 2001–2005. [Google Scholar] [CrossRef] [Green Version]
- Faith, J.J.; McNulty, N.P.; Rey, F.E.; Gordon, J.I. Predicting a human gut microbiota’s response to diet in gnotobiotic mice. Science 2011, 333, 101–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Jie, Z.; Xia, H.; Zhong, S.L.; Feng, Q.; Li, S.; Liang, S.; Zhong, H.; Liu, Z.; Gao, Y.; Zhao, H.; et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat. Commun. 2017, 8, 845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, N.; Emoto, T.; Yamashita, T.; Watanabe, H.; Hayashi, T.; Tabata, T.; Hoshi, N.; Hatano, N.; Ozawa, G.; Sasaki, N.; et al. Bacteroides vulgatus and Bacteroides dorei Reduce Gut Microbial Lipopolysaccharide Production and Inhibit Atherosclerosis. Circulation 2018, 138, 2486–2498. [Google Scholar] [CrossRef]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhao, F.; Wang, Y.; Chen, J.; Tao, J.; Tian, G.; Wu, S.; Liu, W.; Cui, Q.; Geng, B.; et al. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome 2017, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Kelly, T.N.; Bazzano, L.A.; Ajami, N.J.; He, H.; Zhao, J.; Petrosino, J.F.; Correa, A.; He, J. Gut Microbiome Associates With Lifetime Cardiovascular Disease Risk Profile Among Bogalusa Heart Study Participants. Circ. Res. 2016, 119, 956–964. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Li, J.; Liu, H.; Tang, Y.; Zhan, Q.; Lai, W.; Ao, L.; Meng, X.; Ren, H.; Xu, D.; et al. The intestinal microbiota associated with cardiac valve calcification differs from that of coronary artery disease. Atherosclerosis 2019, 284, 121–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.F.; Zhang, W.Y.; Wen, Q.; Chen, W.J.; Wang, Z.M.; Chen, J.; Zhu, F.; Liu, K.; Cheng, L.X.; Yang, J.; et al. Fecal microbiota transplantation alleviates myocardial damage in myocarditis by restoring the microbiota composition. Pharmacol. Res. 2019, 139, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Toya, T.; Corban, M.T.; Marrietta, E.; Horwath, I.E.; Lerman, L.O.; Murray, J.A.; Lerman, A. Coronary artery disease is associated with an altered gut microbiome composition. PLoS ONE 2020, 15, e0227147. [Google Scholar]
- Holscher, H.D.; Caporaso, J.G.; Hooda, S.; Brulc, J.M.; Fahey, G.C.; Swanson, K.S. Fiber supplementation influences phylogenetic structure and functional capacity of the human intestinal microbiome: Follow-up of a randomized controlled trial. Am. J. Clin. Nutr. 2015, 101, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Dominianni, C.; Sinha, R.; Goedert, J.J.; Pei, Z.; Yang, L.; Hayes, R.B.; Ahn, J. Sex, body mass index, and dietary fiber intake influence the human gut microbiome. PLoS ONE 2015, 10, e0124599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Wang, B.; Zhang, M.; Rantalainen, M.; Wang, S.; Zhou, H.; Zhang, Y.; Shen, J.; Pang, X.; Zhang, M.; et al. Symbiotic gut microbes modulate human metabolic phenotypes. Proc. Natl. Acad. Sci. USA 2008, 105, 2117–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, S.; Saunier, K.; Hanisch, C.; Norin, E.; Alm, L.; Midtvedt, T.; Cresci, A.; Silvi, S.; Orpianesi, C.; Verdenelli, M.C.; et al. Differences in fecal microbiota in different European study populations in relation to age, gender, and country: A cross-sectional study. Appl. Environ. Microbiol. 2006, 72, 1027–1033. [Google Scholar] [CrossRef] [Green Version]
- Mariat, D.; Firmesse, O.; Levenez, F.; Guimarăes, V.; Sokol, H.; Dore, J.; Corthier, G.; Furet, J.P. The Firmicutes/Bacteroidetes ratio of the human microbota changes with age. BMC Microbiol. 2009, 9, 123. [Google Scholar] [CrossRef]
- Canfora, E.E.; van der Beek, C.M.; Hermes, G.D.A.; Goossens, G.H.; Jocken, J.W.E.; Holst, J.J.; van Eijk, H.M.; Venema, K.; Smidt, H.; Zoetendal, E.G.; et al. Supplementation of Diet With Galacto-oligosaccharides Increases Bifidobacteria, but Not Insulin Sensitivity, in Obese Prediabetic Individuals. Gastroenterology 2017, 153, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Stratmann, B.; Richter, K.; Wang, R.; Yu, Z.; Xu, T.; Prehn, C.; Adamski, J.; Illig, T.; Tschoepe, D.; Wang-Sattler, R. Metabolomic Signature of Coronary Artery Disease in Type 2 Diabetes Mellitus. Int. J. Endocrinol. 2017, 2017, 7938216. [Google Scholar] [CrossRef]
- Lin, H.; Zhang, J.; Gao, P. Silent myocardial ischemia is associated with altered plasma phospholipids. J. Clin. Lab. Anal. 2009, 23, 45–50. [Google Scholar] [CrossRef]
- Poszytek, K. Mikrobiologiczna utylizacja celulozy. Post. Mikrobiol. 2016, 55, 132. [Google Scholar]
- Sauer, U.; Eikmanns, B.J. The PEP-pyruvate-oxaloacetate node as the switch point for carbon flux distribution in bacteria. FEMS Microbiol. Rev. 2005, 29, 765–794. [Google Scholar] [CrossRef] [Green Version]
- Metacyc. Available online: https://biocyc.org/META/NEW-IMAGE?type=PATHWAY&object=GLYCOLYSIS-TCA-GLYOX-BYPASS (accessed on 28 August 2021).
- Szafran, M.; Zakrzewska-Czerwinska, J.; Jakimowicz, D. Bacterial type I topoisomerases-biological function and potential use as targets for antibiotic treatments. Postepy Hig. Med. Dosw. 2013, 67, 130–142. [Google Scholar] [CrossRef]
- Stepper, J.; Dabin, J.; Eklof, J.M.; Thongpoo, P.; Kongsaeree, P.; Taylor, E.J.; Turkenburg, J.P.; Brumer, H.; Davies, G.J. Structure and activity of the Streptococcus pyogenes family GH1 6-phospho-beta-glucosidase SPy1599. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 16–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.L.; Jiang, Y.L.; Pikis, A.; Cheng, W.; Bai, X.H.; Ren, Y.M.; Thompson, J.; Zhou, C.Z.; Chen, Y. Structural insights into the substrate specificity of a 6-phospho-β-glucosidase BglA-2 from Streptococcus pneumoniae TIGR4. J. Biol. Chem. 2013, 288, 14949–14958. [Google Scholar] [CrossRef] [Green Version]
- Metacyc. Available online: https://www.genome.jp/dbget-bin/www_bget?ec:3.2.1.86 (accessed on 25 August 2021).
- Saier, M.H., Jr. The Bacterial Phosphotransferase System: New Frontiers 50 Years after Its Discovery. J. Mol. Microbiol. Biotechnol. 2015, 25, 73–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metacyc. Available online: https://www.genome.jp/dbget-bin/www_bget?enzyme+2.7.1.69 (accessed on 25 August 2021).
- Gast, G.C.; de Roos, N.M.; Sluijs, I.; Bots, M.L.; Beulens, J.W.; Geleijnse, J.M.; Witteman, J.C.; Grobbee, D.E.; Peeters, P.H.; van der Schouw, Y.T. A high menaquinone intake reduces the incidence of coronary heart disease. Nutr. Metab. Cardiovasc. Dis. 2009, 19, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Geleijnse, J.M.; Vermeer, C.; Grobbee, D.E.; Schurgers, L.J.; Knapen, M.H.; van der Meer, I.M.; Hofman, A.; Witteman, J.C. Dietary intake of menaquinone is associated with a reduced risk of coronary heart disease: The Rotterdam study. J. Nutr. 2004, 134, 3100–3105. [Google Scholar] [CrossRef]
- Chen, C.L.; Mermoud, J.C.; Paul, L.N.; Steussy, C.N.; Stauffacher, C.V. Mevalonate 5-diphosphate mediates ATP binding to the mevalonate diphosphate decarboxylase from the bacterial pathogen Enterococcus faecalis. J. Biol. Chem. 2017, 29, 21340–21351. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CAD Group | Control Group | |
|---|---|---|
| Inclusion criteria |
| |
|
| |
| Exclusion criteria |
| |
| ||
| CAD Group (n = 169) | Control Group (n = 166) | p Value | |
|---|---|---|---|
| Age, years | 64.1 ± 7.7 | 62.4 ± 10.5 | 0.08 |
| Sex, male, n (%) | 124 (73%) | 108 (65%) | 0.1 |
| BMI, kg/m2 | 30.52 ± 5.3 | 28.46 ± 4.73 | 0.001 |
| WHR | 0.96 ± 0.08 | 0.92 ± 0.09 | 0.001 |
| Arterial hypertension, n (%) | 138 (83%) | 82 (56%) | 0.001 |
| Diabetes, n (%) | 50 (30%) | 16 (15%) | 0.003 |
| Current tobacco smoker, n (%) | 28 (22%) | 26 (27%) | 0.435 |
| History of tobacco smoking, n (%) | 125 (74%) | 98 (60%) | 0.005 |
| Lower limb atherosclerosis or intermittent claudication, n (%) | 12 (80%) | 3 (3%) | 0.001 |
| Hypercholesterolemia, n (%) | 137 (82%) | 69 (50%) | 0.001 |
| History of kidney disease, n (%) | 14 (9%) | 14 (11%) | 0.435 |
| Family history of the acute coronary syndrome, n (%) | 44 (30%) | 10 (7%) | 0.001 |
| Family history of stroke, n (%) | 33 (22%) | 17 (13%) | 0.037 |
| Pharmacotherapy | |||
| Use of ACE-I/ARB, n (%) | 147 (88%) | 56 (44%) | <0.001 |
| Use of beta-blockers, n (%) | 53 (31%) | 23 (18%) | 0.012 |
| Use of statins, n (%) | 150 (90%) | 38 (30%) | <0.001 |
| Use of diuretics, n (%) | 84 (50%) | 18 (14%) | <0.001 |
| Use of antiplatelet and anticoagulant treatment, n (%) | 164 (98%) | 30 (24%) | <0.001 |
| Assessments | |||
| Atherosclerotic plaques in the carotid arteries, n (%) | 163 (96%) | 123 (74%) | 0.001 |
| Heart rate in electrocardiogram, beats/minute | 62 IQR: 58–68.5 | 64.5 IQR: 58–72 | 0.044 |
| Bone Densitometry | |||
| Total fat mass, g | 29667.93 IQR: 23652.98–35995.84 | 28604.12 IQR: 20777.52–33493.96 | 0.047 |
| Android fat mass, g | 3220.41 IQR: 2510.92–4005.02 | 2849.09 IQR: 1991.26–3752.75 | 0.002 |
| Echocardiography | |||
| Left ventricle ejection fraction, % | 52.3 IQR: 47.6–57.9 | 57 IQR: 53.8–60.4 | <0.001 |
| Left atrium diameter, mm | 39.8 IQR: 36–43.9 | 37.3 IQR: 33.2–39.7 | <0.001 |
| Biochemical Test | |||
|---|---|---|---|
| NTproBNP, pg/mL | 154.5 IQR: 78.1–329 | 65.2 IQR: 32.6–110.1 | 0.001 |
| WBC, tys/μL | 6.4 IQR: 5.2–7.5 | 5.9 IQR: 5.1–6.9 | 0.119 |
| RBC, mln/mm3 | 4.7 IQR: 4.4–5 | 4.9 IQR: 4.5–5.08 | 0.012 |
| Hb, g/dL | 14.1 IQR: 13.1–14.6 | 14.7 IQR: 13.7–15.4 | <0.001 |
| PLT, tys/μL | 217 IQR: 185–256.5 | 214 IQR: 185–249 | 0.985 |
| Creatinine, μmol/L | 85.4 IQR: 74.4–97.2 | 75.8 IQR: 64.9–83.7 | <0.001 |
| Urea, mg/dL | 34.4 IQR: 30–41 | 32.6 IQR: 28.4–38.7 | 0.011 |
| Fasting glucose, mg/dL | 105 IQR: 97–119.5 | 102 IQR: 96–109 | 0.027 |
| 120 min glucose, mg/dL | 130.5 IQR: 102–161 | 134 IQR: 106–174 | 0.398 |
| HbA1c, % | 5.9 IQR: 5.6–6.2 | 5.7 IQR: 5.3–5.9 | <0.001 |
| Total cholesterol, mg/dL | 154 IQR: 129.5–179 | 195 IQR: 166–227 | <0.001 |
| Triglycerides, mg/dL | 103 IQR: 73–157 | 113 IQR: 82–149 | 0.394 |
| Low-density lipoprotein cholesterol, mg/dL | 85.8 IQR: 69.4–108.8 | 132.6 IQR: 99.9–160.8 | <0.001 |
| High-density lipoprotein cholesterol, mg/dL | 48 IQR: 41.5–61.5 | 58 IQR: 47.4–68.3 | <0.001 |
| hs-CRP, mg/L | 1.1 IQR: 0.5–2.1 | 0.9 IQR: 0.5–1.7 | 0.138 |
| ALT, IU/L | 23 IQR: 18.3–32.3 | 21.1 IQR: 16.9–29.6 | 0.05 |
| AST, IU/L | 23 IQR: 18.8–27.9 | 22.3 IQR: 19.4–26.9 | 0.167 |
| GGT, IU/L | 23.9 IQR: 16.3–39.1 | 20.2 IQR: 13.6–34.7 | 0.033 |
| Iron, μg/mL | 99.7 IQR: 82–119.7 | 113.8 IQR: 88.7–133.8 | 0.006 |
| Taxonomic Unit | Relative Abundance in CAD Group | Relative Abundance in Control Group | p-Value (Adjusted for Sex and Age) |
|---|---|---|---|
| Phyla | |||
| Proteobacteria | 7.4695 (5.1935–9.7455) | 4.6714 (3.4979–5.8449) | 0.032 |
| Bacteroidetes | 33.5334 (30.8517–36.2151) | 37.1957 (35.1636–39.2278) | 0.032 |
| Classes | |||
| Bacteroidetes, Bacteroidia | 33.5334 (30.8517–36.2151) | 37.1957 (35.1636–39.2278) | 0.032 |
| Firmicutes, Bacilli | 4.1117 (2.9382–5.2852) | 1.2206 (0.7898–1.6514) | <0.001 |
| Proteobacteria, Gammaproteobacteria | 6.0816 (3.8101–8.3531) | 3.4073 (2.2397–4.5749) | 0.04 |
| Order | |||
| Actinobacteria, Actinobacteria, Actinomycetales | 0.0281 (0.0164–0.0398) | 0.0135 (0.0095–0.0175) | 0.021 |
| Actinobacteria, Actinobacteria, Micrococcales | 0.0152 (0.0108–0.0196) | 0.0083 (0.0043–0.0123) | 0.021 |
| Bacteroidetes, Bacteroidia, Bacteroidales | 33.4714 (30.7887–36.1541) | 37.1229 (35.0883–39.1575) | 0.033 |
| Firmicutes, Bacilli, Lactobacillales | 3.891 (2.7367–5.0453) | 1.1638 (0.745–1.5826) | <0.001 |
| Proteobacteria, Gammaproteobacteria, Enterobacteriales | 5.4589 (3.1754–7.7424) | 2.5555 (1.4063–3.7047) | 0.026 |
| Phylum | Order | Family | Genus | CAD Group (n = 169) | Control Group (n = 166) | Adjusted p-Value (FDR) CAD vs. Control |
|---|---|---|---|---|---|---|
| Firmicutes | Clostridiales | Clostridiales vadinBB60 group | Uncultured organism | 0.3797 (0.1217–0.6377) | 0.4296 (0.2595–0.5997) | <0.001 |
| Fusobacteria | Fusobacteriales | Fusobacteriaceae | Fusobacterium | 0.159 (0.0099–0.3081) | 0.2248 (−0.0595–0.5091) | <0.001 |
| Firmicutes | Lactobacillales | Lactobacillaceae | Lactobacillus sp., Gut metagenome | 0.1493 (−0.1451–0.4437) | 0.0002 (0–0.0004) | <0.001 |
| Firmicutes | Clostridiales | Christensenellaceae | Christensenellaceae R-7 group | 1.3802 (1.0337–1.7267) | 1.1589 (0.8781–1.4397) | <0.001 |
| Firmicutes | Lactobacillales | Lactobacillaceae | Lactobacillus sp, Ambiguous taxa | 0.7075 (0.2925–1.1225) | 0.0626 (0.0121–0.1131) | <0.001 |
| Spirochaetes | Spirochaetales | Spirochaetaceae | Treponema 2 | 0.0699 (−0.0293–0.1691) | 0 (0–0) | <0.001 |
| Bacteroidetes | Bacteroidales | Prevotellaceae | Prevotellaceae UCG-003 | 0.0434 (−0.0297–0.1165) | 0.0002 (−0.0002–0.0006) | <0.001 |
| Firmicutes | Clostridiales | Ruminococcaceae | Ruminococcaceae UCG-014 | 4.6675 (3.6518–5.6832) | 3.5864 (2.8496–4.3232) | <0.001 |
| Bacteroidetes | Bacteroidales | Muribaculaceae | Uncultured bacterium | 0.108 (0.027–0.189) | 0.1365 (0.0385–0.2345) | <0.001 |
| Proteobacteria | Enterobacteriales | Enterobacteriaceae | Escherichia-Shigella | 4.5528 (2.398–6.7076) | 1.9492 (0.9105–2.9879) | <0.001 |
| Metabolite or Biochemical Test | Group with F/B Ratio ≤ 1.54 (n = 167) | Group with F/B Ratio > 1.54 (n = 167) | p Value |
|---|---|---|---|
| Phosphatidylcholine with diacyl residue sum C36:4, μmol/L | 145,308.5375 (95% CI 136,165.244–154,451.831) | 131,525.3521 (95% CI 122,485.2512–140,565.4529) | 0.037 * |
| Phosphatidylcholine with diacyl residue sum C38:5, μmol/L | 46,853.2875 (95% CI 43,791.1361–49,915.4389) | 41,838.3521 (95% CI 39,090.319–44,586.3852) | 0.019 * |
| Phosphatidylcholine with diacyl residue sum C38:6, μmol/L | 68,122.05 (95% CI 62,784.975–73,459.125) | 58,899.7887 (95% CI 54,099.1744–63,700.403) | 0.013 * |
| Phosphatidylcholine with diacyl residue sum C40:6, μmol/L | 26,837.9747 (95% CI 24,778.9683–28,896.9811) | 23,893.6056 (95% CI 21,873.7272–25,913.484) | 0.047 * |
| Phosphatidylcholine with acyl-alkyl residue sum C30:0, μmol/L | 165.6974 (95% CI 153.7441–177.6507) | 209.942 (95% CI 178.6529–241.2311) | 0.008 * |
| Phosphatidylcholine with acyl-alkyl residue sum C34:0, μmol/L | 839.25 (95% CI 782.9649–895.5351) | 958.2254 (95% CI 852.6987–1063.7521) | 0.045 * |
| Phosphatidylcholine with acyl-alkyl residue sum C38:0, μmol/L | 1663.75 (95% CI 1520.5806–1806.9194) | 1465.4225 (95% CI 1353.087–1577.758) | 0.034 * |
| Sphingomyelin with acyl residue sum C24:1, μmol/L | 69,876.9375 (95% CI 65,633.3358–74,120.5392) | 63,321.1268 (95% CI 59,226.511–67,415.7426) | 0.028 * |
| Hematocrit, % | 41.3928 (95% CI 40.8434–41.9422) | 41.8814 (95% CI 41.3236–42.4392) | 0.047 * |
| Hemoglobin, g/dL | 14.1371 (95% CI 13.9429–14.3313) | 14.3413 (95% CI 14.1502–14.5324) | 0.023 * |
| Total cholesterol, mg/dL | 188.9341 (95% CI 181.2163–196.6519) | 171.2455 (95% CI 164.3927–178.0983) | 0.006 ** |
| LDL cholesterol, mg/dL | 118.8796 (95% CI 112.3616–125.3976) | 104.9257 (95% CI 98.4497–111.4017) | 0.026 ** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sawicka-Smiarowska, E.; Bondarczuk, K.; Bauer, W.; Niemira, M.; Szalkowska, A.; Raczkowska, J.; Kwasniewski, M.; Tarasiuk, E.; Dubatowka, M.; Lapinska, M.; et al. Gut Microbiome in Chronic Coronary Syndrome Patients. J. Clin. Med. 2021, 10, 5074. https://doi.org/10.3390/jcm10215074
Sawicka-Smiarowska E, Bondarczuk K, Bauer W, Niemira M, Szalkowska A, Raczkowska J, Kwasniewski M, Tarasiuk E, Dubatowka M, Lapinska M, et al. Gut Microbiome in Chronic Coronary Syndrome Patients. Journal of Clinical Medicine. 2021; 10(21):5074. https://doi.org/10.3390/jcm10215074
Chicago/Turabian StyleSawicka-Smiarowska, Emilia, Kinga Bondarczuk, Witold Bauer, Magdalena Niemira, Anna Szalkowska, Justyna Raczkowska, Miroslaw Kwasniewski, Ewa Tarasiuk, Marlena Dubatowka, Magda Lapinska, and et al. 2021. "Gut Microbiome in Chronic Coronary Syndrome Patients" Journal of Clinical Medicine 10, no. 21: 5074. https://doi.org/10.3390/jcm10215074
APA StyleSawicka-Smiarowska, E., Bondarczuk, K., Bauer, W., Niemira, M., Szalkowska, A., Raczkowska, J., Kwasniewski, M., Tarasiuk, E., Dubatowka, M., Lapinska, M., Szpakowicz, M., Stachurska, Z., Szpakowicz, A., Sowa, P., Raczkowski, A., Kondraciuk, M., Gierej, M., Motyka, J., Jamiolkowski, J., ... Kaminski, K. A. (2021). Gut Microbiome in Chronic Coronary Syndrome Patients. Journal of Clinical Medicine, 10(21), 5074. https://doi.org/10.3390/jcm10215074