
Viscoelastic Testing and Coagulopathy of Traumatic Brain Injury


 , ,
, ,  ,
,  ,
,  ,
,
Abstract
1. Introduction
1.1. Incidence of Coagulopathy of Traumatic Brain Injury
1.2. Implications of CTBI and Relation to VET-Based Definition
1.3. Inadequacy of Conventional Coagulation Assays in the Diagnosis of CTBI
2. Pathophysiology in CTBI and Its Relation to VETs
3. Basics of TEG®/ROTEM®
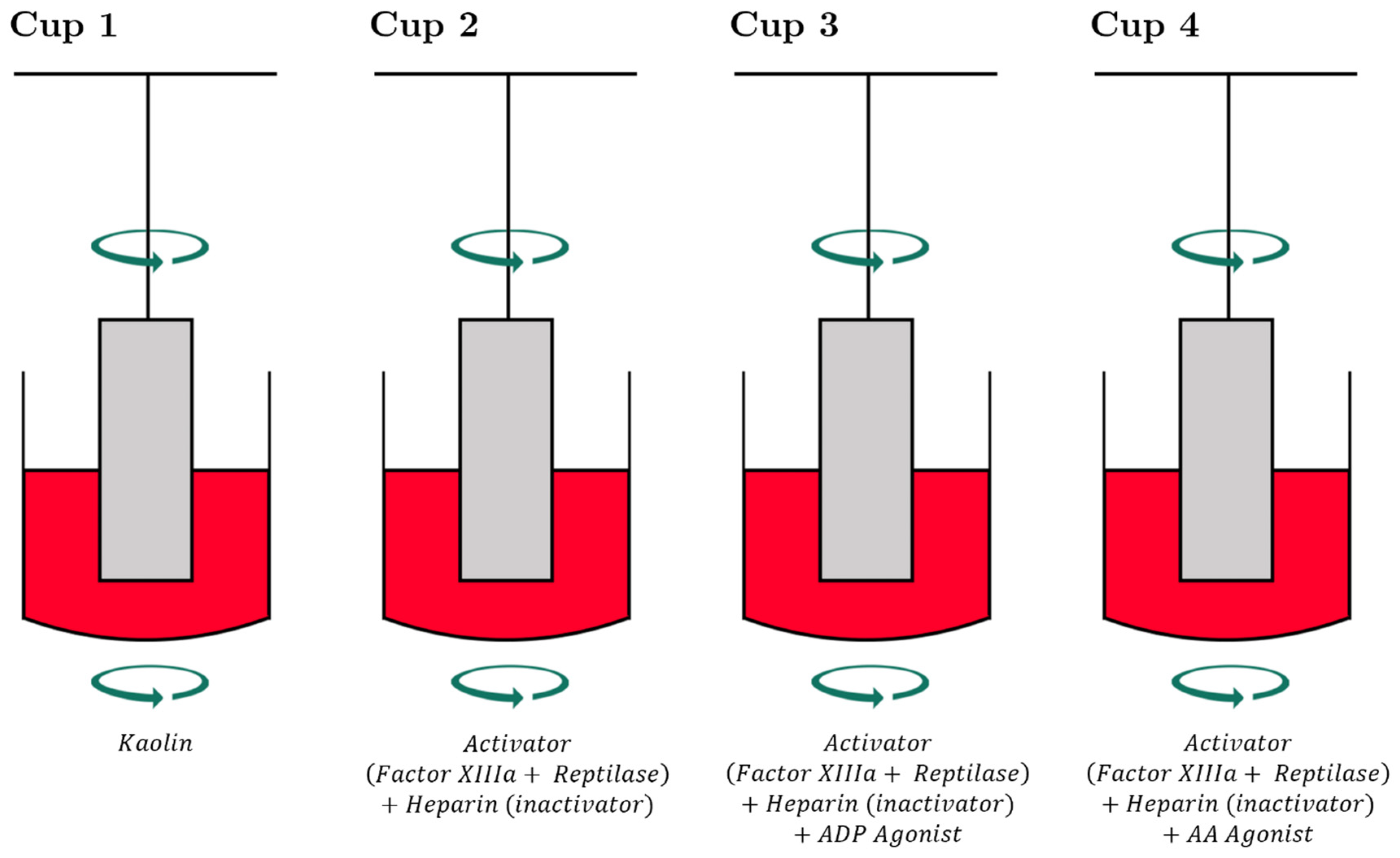
3.1. Description of the Cup and Pin
3.2. TEG-PM® and ROTEM® with Specialized Platelet Function Testing to Diagnose and Guide Platelet Transfusion in Patients with CTBI
4. Utilizing VETs for the Diagnosis and Treatment of TBI
4.1. VETs to Diagnose, Treat, and Prognosticate CTBI
4.1.1. Diagnosis of CTBI
4.1.2. Treatment of CTBI

4.1.3. Prediction of Morbidity and Mortality in CTBI
4.2. Basic TEG®/ROTEM® Parameters Triggering BCT and HAT
4.3. Guiding Blood Products with VETs in TBI
4.3.1. VET-Guided FFP, Fibrinogen Concentrate, and Cryoprecipitate in TBI
4.3.2. VET-Guided Diagnosis and Treatment of Platelet Dysfunction in TBI
4.4. Preinjury Antithrombotic Use
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| A10EX | EXTEM A10 |
| A10FIB | FIBTEM A10 |
| AA | Arachidonic acid |
| ACT | Activated coagulation time |
| ADP | Adenosine diphosphate |
| AIS | Abbreviated injury severity score |
| AU | Aggregation units |
| BBB | Blood-brain barrier |
| CCA | Common coagulation assay |
| CFT | Clot formation time (ROTEM parameter) |
| CT | Clotting time (ROTEM parameter) |
| CTBI | Coagulopathy of traumatic brain injury |
| DDAVP | Desmopressin |
| DOAC | Direct oral anticoagulant |
| FFP | Fresh frozen plasma |
| GCS | Glasgow Coma Scale |
| ICH | Intracerebral hemorrhage |
| INR | International normalized ratio |
| ISS | Injury severity score |
| K | Clot formation time (TEG parameter) |
| LI30 | Lysis index at 30 min (ROTEM parameter) |
| LY30 | Lysis at 30 min (TEG parameter) |
| MA | Maximum amplitude (TEG parameter) |
| MCF | Maximum clot firmness (ROTEM parameter) |
| MEA | Multiple electrode aggregometry |
| ML | Maximum lysis |
| PAMPer | Prehospital Air Medical Plasma trial |
| PAR-1 | Protease Activated Receptor-1 |
| PFA | Platelet Function Analyzer |
| POC | Point of care |
| pRBCs | Packed red blood cells |
| PT | Prothrombin time |
| PTT | Partial thromboplastin time |
| R | Reaction time (TEG parameter) |
| ROTEM | Rotational thromboelastometry |
| TBI | Traumatic brain injury |
| TEG | Thromboelastography |
| TEG-PM | Thrombelastography PlateletMapping |
| TF | Tissue factor |
| TRAP | Thrombin receptor activating peptides |
| VET | Viscoelastic test |
| vWF | von Willebrand factor |
References
- Davis, P.K.; Musunuru, H.; Walsh, M.; Cassady, R.; Yount, R.; Losiniecki, A.; Moore, E.E.; Wohlauer, M.V.; Howard, J.; Ploplis, V.A.; et al. Platelet dysfunction is an early marker for traumatic brain injury-induced coagulopathy. Neurocrit. Care 2013, 18, 201–208. [Google Scholar] [CrossRef]
- Gozal, Y.M.; Carroll, C.P.; Krueger, B.M.; Khoury, J.; Andaluz, N.O. Point-of-care testing in the acute management of traumatic brain injury: Identifying the coagulopathic patient. Surg. Neurol. Int. 2017, 8, 48. [Google Scholar] [PubMed]
- Kunio, N.R.; Differding, J.A.; Watson, K.M.; Stucke, R.S.; Schreiber, M.A. Thrombelastography-identified coagulopathy is associated with increased morbidity and mortality after traumatic brain injury. Am. J. Surg. 2012, 203, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Kvint, S.; Schuster, J.; Kumar, M.A. Neurosurgical applications of viscoelastic hemostatic assays. Neurosurg. Focus 2017, 43, E9. [Google Scholar] [CrossRef] [PubMed]
- Massaro, A.M.; Doerfler, S.; Nawalinski, K.; Michel, B.; Driscoll, N.; Ju, C.; Patel, H.; Quattrone, F.; Frangos, S.; Maloney-Wilensky, E. Thromboelastography defines late hypercoagulability after TBI: A pilot study. Neurocrit. Care 2015, 22, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Schöchl, H.; Solomon, C.; Traintinger, S.; Nienaber, U.; Tacacs-Tolnai, A.; Windhofer, C.; Bahrami, S.; Voelckel, W. Thromboelastometric (ROTEM) findings in patients suffering from isolated severe traumatic brain injury. J. Neurotrauma 2011, 28, 2033–2041. [Google Scholar] [CrossRef]
- Harhangi, B.S.; Kompanje, E.J.O.; Leebeek, F.; Maas, A.I. Coagulation disorders after traumatic brain injury. Acta Neurochir. 2008, 150, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Auer, L. Disturbances of the coagulatory system in patients with severe cerebral trauma I. Acta Neurochir. 1978, 43, 51–59. [Google Scholar] [CrossRef]
- Auer, L.; Ott, E. Disturbances of the coagulatory system in patients with severe cerebral trauma II. Acta Neurochir. 1979, 49, 219–226. [Google Scholar] [CrossRef]
- Cortiana, M.; Zagara, G.; Fava, S.; Seveso, M. Coagulation abnormalities in patients with head injury. J. Neurosurg. Sci. 1986, 30, 133–138. [Google Scholar]
- Hoyt, D.B. A clinical review of bleeding dilemmas in trauma. Semin. Hematol. 2004, 41, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Brohi, K.; Singh, J.; Heron, M.; Coats, T. Acute traumatic coagulopathy. J. Trauma Acute Care Surg. 2003, 54, 1127–1130. [Google Scholar] [CrossRef] [PubMed]
- Carrick, M.M.; Tyroch, A.H.; Youens, C.A.; Handley, T. Subsequent development of thrombocytopenia and coagulopathy in moderate and severe head injury: Support for serial laboratory examination. J. Trauma Acute Care Surg. 2005, 58, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Hulka, F.; Mullins, R.J.; Frank, E.H. Blunt brain injury activates the coagulation process. Arch. Surg. 1996, 131, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Hymel, K.P.; Abshire, T.C.; Luckey, D.W.; Jenny, C. Coagulopathy in pediatric abusive head trauma. Pediatrics 1997, 99, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.S.; Fendya, D.G.; Weber, T.R. Glasgow Coma Scale predicts coagulopathy in pediatric trauma patients. Semin. Pediatr. Surg. 2001, 10, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Kumura, E.; Sato, M.; Fukuda, A.; Takemoto, Y.; Tanaka, S.; Kohama, A. Coagulation disorders following acute head injury. Acta Neurochir. 1987, 85, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Kuo, J.-R.; Chou, T.-J.; Chio, C.-C. Coagulopathy as a parameter to predict the outcome in head injury patients–analysis of 61 cases. J. Clin. Neurosci. 2004, 11, 710–714. [Google Scholar] [CrossRef] [PubMed]
- Miner, M.E.; Kaufman, H.H.; Graham, S.H.; Haar, F.H.; Gildenberg, P.L. Disseminated intravascular coagulation fibrinolytic syndrome following head injury in children: Frequency and prognostic implications. J. Pediatr. 1982, 100, 687–691. [Google Scholar] [CrossRef]
- Olson, J.D.; Kaufman, H.H.; Moake, J.; O’Gorman, T.W.; Hoots, K.; Wagner, K.; Brown, K.C.; Gildenberg, P.L. The incidence and significance of hemostatic abnormalities in patients with head injuries. Neurosurgery 1989, 24, 825–832. [Google Scholar] [CrossRef] [PubMed]
- Pondaag, W. Disseminated intravascular coagulation related to outcome in head injury. Acta Neurochir. Suppl. 1979, 28, 98–102. [Google Scholar] [PubMed]
- Selladurai, B.M.; Vickneswaran, M.; Duraisamy, S.; Atan, M. Coagulopathy in acute head injury-a study of its role as a prognostic indicator. Br. J. Neurosurg. 1997, 11, 398–404. [Google Scholar] [CrossRef]
- Maegele, M.; Schöchl, H.; Menovsky, T.; Maréchal, H.; Marklund, N.; Buki, A.; Stanworth, S. Coagulopathy and haemorrhagic progression in traumatic brain injury: Advances in mechanisms, diagnosis, and management. Lancet Neurol. 2017, 16, 630–647. [Google Scholar] [CrossRef]
- Shehata, M.; Afify, M.I.; El-Shafie, M.; Khaled, M. Prevalence and Clinical Implications of Coagulopathy in Patients with Isolated Head Traum. Med. J. Cairo Univ. 2011, 79, 131–137. [Google Scholar]
- Chhabra, G.; Rangarajan, K.; Subramanian, A.; Agrawal, D.; Sharma, S.; Mukhopadhayay, A. Hypofibrinogenemia in isolated traumatic brain injury in Indian patients. Neurol. India 2010, 58, 756–757. [Google Scholar]
- Epstein, D.S.; Mitra, B.; O’Reilly, G.; Rosenfeld, J.V.; Cameron, P.A. Acute traumatic coagulopathy in the setting of isolated traumatic brain injury: A systematic review and meta-analysis. Injury 2014, 45, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Zehtabchi, S.; Baki, S.G.A.; Falzon, L.; Nishijima, D.K. Tranexamic acid for traumatic brain injury: A systematic review and meta-analysis. Am. J. Emerg. Med. 2014, 32, 1503–1509. [Google Scholar] [CrossRef] [PubMed]
- Talving, P.; Benfield, R.; Hadjizacharia, P.; Inaba, K.; Chan, L.S.; Demetriades, D. Coagulopathy in severe traumatic brain injury: A prospective study. J. Trauma Acute Care Surg. 2009, 66, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Lustenberger, T.; Talving, P.; Kobayashi, L.; Barmparas, G.; Inaba, K.; Lam, L.; Branco, B.C.; Demetriades, D. Early coagulopathy after isolated severe traumatic brain injury: Relationship with hypoperfusion challenged. J. Trauma 2010, 69, 1410–1414. [Google Scholar] [CrossRef]
- Lustenberger, T.; Talving, P.; Kobayashi, L.; Inaba, K.; Lam, L.; Plurad, D.; Demetriades, D. Time course of coagulopathy in isolated severe traumatic brain injury. Injury 2010, 41, 924–928. [Google Scholar] [CrossRef] [PubMed]
- Wafaisade, A.; Lefering, R.; Tjardes, T.; Wutzler, S.; Simanski, C.; Paffrath, T.; Fischer, P.; Bouillon, B.; Maegele, M.; Trauma Registry of DGU. Acute coagulopathy in isolated blunt traumatic brain injury. Neurocrit. Care 2010, 12, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Greuters, S.; van den Berg, A.; Franschman, G.; Viersen, V.A.; Beishuizen, A.; Peerdeman, S.M.; Boer, C. Acute and delayed mild coagulopathy are related to outcome in patients with isolated traumatic brain injury. Crit. Care 2011, 15, R2. [Google Scholar] [CrossRef] [PubMed]
- Franschman, G.; Greuters, S.; Jansen, W.H.; Posthuma, L.M.; Peerdeman, S.M.; Wattjes, M.P.; Loer, S.A.; Boer, C. Haemostatic and cranial computed tomography characteristics in patients with acute and delayed coagulopathy after isolated traumatic brain injury. Brain Inj. 2012, 26, 1464–1471. [Google Scholar] [CrossRef] [PubMed]
- Genét, G.F.; Johansson, P.I.; Meyer, M.A.S.; Sølbeck, S.; Sørensen, A.M.; Larsen, C.F.; Welling, K.L.; Windeløv, N.A.; Rasmussen, L.S.; Ostrowski, S.R. Trauma-induced coagulopathy: Standard coagulation tests, biomarkers of coagulopathy, and endothelial damage in patients with traumatic brain injury. J. Neurotrauma 2013, 30, 301–306. [Google Scholar] [CrossRef]
- Alexiou, G.A.; Lianos, G.; Fotakopoulos, G.; Michos, E.; Pachatouridis, D.; Voulgaris, S. Admission glucose and coagulopathy occurrence in patients with traumatic brain injury. Brain Inj. 2014, 28, 438–441. [Google Scholar] [CrossRef] [PubMed]
- Joseph, B.; Aziz, H.; Zangbar, B.; Kulvatunyou, N.; Pandit, V.; O’Keeffe, T.; Tang, A.; Wynne, J.; Friese, R.S.; Rhee, P. Acquired coagulopathy of traumatic brain injury defined by routine laboratory tests: Which laboratory values matter? J. Trauma Acute Care Surg. 2014, 76, 121–125. [Google Scholar] [CrossRef]
- de Oliveira Manoel, A.L.; Neto, A.C.; Veigas, P.V.; Rizoli, S. Traumatic brain injury associated coagulopathy. Neurocrit. Care 2015, 22, 34–44. [Google Scholar] [CrossRef]
- Dekker, S.E.; Duvekot, A.; de Vries, H.-M.; Geeraedts, L.M., Jr.; Peerdeman, S.M.; de Waard, M.C.; Boer, C.; Schober, P. Relationship between tissue perfusion and coagulopathy in traumatic brain injury. J. Surg. Res. 2016, 205, 147–154. [Google Scholar] [CrossRef]
- Dragan, B.; Adamik, B.; Burzynska, M.; Dragan, S.L.; Gozdzik, W. Platelet Receptor Activity for Predicting Survival in Patients with Intracranial Bleeding. J. Clin. Med. 2021, 10, 2205. [Google Scholar] [CrossRef]
- Fletcher-Sandersjöö, A.; Thelin, E.P.; Maegele, M.; Svensson, M.; Bellander, B.-M. Time course of hemostatic disruptions after traumatic brain injury: A systematic review of the literature. Neurocrit. Care 2021, 34, 635–656. [Google Scholar] [CrossRef]
- Yuan, Q.; Yu, J.; Wu, X.; Sun, Y.-r.; Li, Z.-q.; Du, Z.-y.; Wu, X.-h.; Hu, J. Prognostic value of coagulation tests for in-hospital mortality in patients with traumatic brain injury. Scand. J. Trauma Resusc. Emerg. Med. 2018, 26, 3. [Google Scholar] [CrossRef] [PubMed]
- Gratz, J.; Güting, H.; Thorn, S.; Brazinova, A.; Görlinger, K.; Schäfer, N.; Schöchl, H.; Stanworth, S.; Maegele, M. Protocolised thromboelastometric-guided haemostatic management in patients with traumatic brain injury: A pilot study. Anaesthesia 2019, 74, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Schochl, H.; Voelckel, W.; Grassetto, A.; Schlimp, C.J. Practical application of point-of-care coagulation testing to guide treatment decisions in trauma. J. Trauma Acute Care Surg. 2013, 74, 1587–1598. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, J.; Walsh, M.; Grisoli, A.; Thomas, A.V.; Shariff, F.; McCauley, R.; Lune, S.V.; Zackariya, N.; Patel, S.; Farrell, M.S.; et al. Diagnosis and Treatment of Trauma-Induced Coagulopathy by Viscoelastography. Semin. Thromb. Hemost. 2020, 46, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Rimaitis, M.; Bilskienė, D.; Tamošuitis, T.; Vilcinis, R.; Rimaitis, K.; Macas, A. Implementation of Thromboelastometry for Coagulation Management in Isolated Traumatic Brain Injury Patients Undergoing Craniotomy. Med. Sci. Monit. 2020, 26, e922879. [Google Scholar] [CrossRef] [PubMed]
- Rossaint, R.; Bouillon, B.; Cerny, V.; Coats, T.J.; Duranteau, J.; Fernandez-Mondejar, E.; Filipescu, D.; Hunt, B.J.; Komadina, R.; Nardi, G.; et al. The European guideline on management of major bleeding and coagulopathy following trauma: Fourth edition. Crit. Care 2016, 20, 100. [Google Scholar] [CrossRef] [PubMed]
- Spahn, D.R.; Bouillon, B.; Cerny, V.; Duranteau, J.; Filipescu, D.; Hunt, B.J.; Komadina, R.; Maegele, M.; Nardi, G.; Riddez, L. The European guideline on management of major bleeding and coagulopathy following trauma: Fifth edition. Crit. Care 2019, 23, 98. [Google Scholar] [CrossRef]
- Maegele, M.; Aversa, J.; Marsee, M.K.; McCauley, R.; Chitta, S.H.; Vyakaranam, S.; Walsh, M. Changes in coagulation following brain injury. Semin. Thromb. Hemost. 2020, 46, 155–166. [Google Scholar] [CrossRef]
- Thorn, S.; Güting, H.; Mathes, T.; Schäfer, N.; Maegele, M. The effect of platelet transfusion in patients with traumatic brain injury and concomitant antiplatelet use: A systematic review and meta-analysis. Transfusion 2019, 59, 3536–3544. [Google Scholar] [CrossRef]
- Böhm, J.K.; Güting, H.; Thorn, S.; Schäfer, N.; Rambach, V.; Schöchl, H.; Grottke, O.; Rossaint, R.; Stanworth, S.; Curry, N. Global characterisation of coagulopathy in isolated traumatic brain injury (iTBI): A CENTER-TBI analysis. Neurocrit. Care 2021, 35, 184–196. [Google Scholar] [CrossRef]
- Cap, A.P.; Spinella, P.C. Severity of head injury is associated with increased risk of coagulopathy in combat casualties. J. Trauma Acute Care Surg. 2011, 71, S78–S81. [Google Scholar] [CrossRef] [PubMed]
- Herbert, J.P.; Guillotte, A.R.; Hammer, R.D.; Litofsky, N.S. Coagulopathy in the setting of mild traumatic brain injury: Truths and consequences. Brain Sci. 2017, 7, 92. [Google Scholar] [CrossRef]
- Castellino, F.J.; Chapman, M.P.; Donahue, D.L.; Thomas, S.; Moore, E.E.; Wohlauer, M.V.; Fritz, B.; Yount, R.; Ploplis, V.; Davis, P.; et al. Traumatic brain injury causes platelet adenosine diphosphate and arachidonic acid receptor inhibition independent of hemorrhagic shock in humans and rats. J. Trauma Acute Care Surg. 2014, 76, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Salehpour, F.; Bazzazi, A.M.; Porhomayon, J.; Nader, N.D. Correlation between coagulopathy and outcome in severe head trauma in neurointensive care and trauma units. J. Crit. Care 2011, 26, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Maegele, M.; Lefering, R.; Yucel, N.; Tjardes, T.; Rixen, D.; Paffrath, T. Early Coagulopathy in mulitple injury: An analysis from the German Trauma Registry on 8724 patients. Injury 2007, 38, 298–304. [Google Scholar] [CrossRef]
- Sixta, S.L.; Cardenas, J.C.; Kitagawa, R.; Wade, C.E.; Holcomb, J.B.; Cotton, B.A. Hypocoagulability in traumatic brain injury as measured by traditional means and thrombelastography. J. Neurol. Neurophysiol. 2015, 6, 1–5. [Google Scholar]
- Thorn, S.; Lefering, R.; Maegele, M.; Gruen, R.L.; Mitra, B. Early prediction of acute traumatic coagulopathy: A validation of the COAST score using the German Trauma Registry. Eur. J. Trauma Emerg. Surg. 2019, 47, 333–341. [Google Scholar] [CrossRef]
- Cannon, J.W.D.; Dias, J.D.; Kumar, M.A.; Walsh, M.; Thomas, S.; Cotton, B.A.; Schuster, J.; Evans, S.; Schreiber, M.A.; Adam, E.; et al. Use of Thromboelastography in the Evaluation and Management of Patients With Traumatic Brain Injury: A Systematic Review and Meta-Analysis. Crit. Care Explor. 2021, 3, e0526. [Google Scholar] [CrossRef]
- Maegele, M. Coagulopathy after traumatic brain injury: Incidence, pathogenesis, and treatment options. Transfusion 2013, 53, 28S–37S. [Google Scholar] [CrossRef]
- Samuels, J.M.; Moore, E.E.; Silliman, C.C.; Banerjee, A.; Cohen, M.J.; Ghasabyan, A.; Chandler, J.; Coleman, J.R.; Sauaia, A. Severe traumatic brain injury is associated with a unique coagulopathy phenotype. J. Trauma Acute Care Surg. 2019, 86, 686–693. [Google Scholar] [CrossRef]
- Furay, E.; Daley, M.; Teixeira, P.G.; Coopwood, T.B.; Aydelotte, J.D.; Malesa, N.; Tellinghuisen, C.; Ali, S.; Brown, L.H.; Brown, C.V. Goal-directed platelet transfusions correct platelet dysfunction and may improve survival in patients with severe traumatic brain injury. J. Trauma Acute Care Surg. 2018, 85, 881–887. [Google Scholar] [CrossRef]
- Furay, E.J.; Daley, M.J.; Satarasinghe, P.; Lara, S.; Aydelotte, J.D.; Teixeira, P.G.; Coopwood, T.B.; Ali, S.; Brown, C.V. Desmopressin is a transfusion sparing option to reverse platelet dysfunction in patients with severe traumatic brain injury. J. Trauma Acute Care Surg. 2020, 88, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Riojas, C.M.; Ekaney, M.L.; Ross, S.W.; Cunningham, K.W.; Furay, E.J.; Brown, C.V.; Evans, S.L. Platelet Dysfunction after Traumatic Brain Injury: A Review. J. Neurotrauma 2021, 38, 819–829. [Google Scholar] [CrossRef]
- Guillotte, A.R.; Herbert, J.P.; Madsen, R.; Hammer, R.D.; Litofsky, N.S. Effects of platelet dysfunction and platelet transfusion on outcomes in traumatic brain injury patients. Brain Inj. 2018, 32, 1849–1857. [Google Scholar] [CrossRef]
- Solomon, C.; Traintinger, S.; Ziegler, B.; Hanke, A.; Rahe-Meyer, N.; Voelckel, W.; Schochl, H. Platelet function following trauma. A multiple electrode aggregometry study. Thromb. Haemost. 2011, 106, 322–330. [Google Scholar]
- Briggs, A.; Gates, J.D.; Kaufman, R.M.; Calahan, C.; Gormley, W.B.; Havens, J.M. Platelet dysfunction and platelet transfusion in traumatic brain injury. J. Surg. Res. 2015, 193, 802–806. [Google Scholar] [CrossRef] [PubMed]
- Parry, P.V.; Choi, P.A.; Bauer, J.S.; Panczykowski, D.M.; Puccio, A.M.; Okonkwo, D.O. Utility of the aspirin and P2Y12 response assays to determine the effect of antiplatelet agents on platelet reactivity in traumatic brain injury. Neurosurgery 2017, 80, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Sun, Y.-R.; Wu, X.; Yu, J.; Li, Z.-Q.; Du, Z.-Y.; Wu, X.-H.; Zhou, L.-F.; Hu, J. Coagulopathy in traumatic brain injury and its correlation with progressive hemorrhagic injury: A systematic review and meta-analysis. J. Neurotrauma 2016, 33, 1279–1291. [Google Scholar] [CrossRef] [PubMed]
- Neerman-Arbez, M.; de Moerloose, P.; Casini, A. Laboratory and Genetic Investigation of Mutations Accounting for Congenital Fibrinogen Disorders. Semin. Thromb. Hemost. 2016, 42, 356–365. [Google Scholar] [PubMed]
- Can, M.M.; Tanboğa, I.H.; Türkyilmaz, E.; Karabay, C.Y.; Akgun, T.; Koca, F.; Tokgoz, H.C.; Keles, N.; Ozkan, A.; Bezgin, T.; et al. The risk of false results in the assessment of platelet function in the absence of antiplatelet medication: Comparison of the PFA-100, multiplate electrical impedance aggregometry and verify now assays. Thromb. Res. 2010, 125, e132–e137. [Google Scholar] [CrossRef]
- Ma, L.; Chen, W.; Pan, Y.; Yan, H.; Li, H.; Meng, X.; Wang, Y.; Wang, Y. Comparison of VerifyNow, thromboelastography, and PL-12 in patients with minor ischemic stroke or transient ischemic attack. Aging 2021, 13, 8396–8407. [Google Scholar] [CrossRef]
- Connelly, C.R.; Yonge, J.D.; McCully, S.P.; Hart, K.D.; Hilliard, T.C.; Lape, D.E.; Watson, J.J.; Rick, B.; Houser, B.; Deloughery, T.G. Assessment of three point-of-care platelet function assays in adult trauma patients. J. Surg. Res. 2017, 212, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Dias, J.D.; Pottgiesser, T.; Hartmann, J.; Duerschmied, D.; Bode, C.; Achneck, H.E. Comparison of three common whole blood platelet function tests for in vitro P2Y12 induced platelet inhibition. J. Thromb. Thrombolysis 2020, 50, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Bachelani, A.M.; Bautz, J.T.; Sperry, J.L.; Corcos, A.; Zenati, M.; Billiar, T.R.; Peitzman, A.B.; Marshall, G.T. Assessment of platelet transfusion for reversal of aspirin after traumatic brain injury. Surgery 2011, 150, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Curry, N.S.; Davenport, R.; Pavord, S.; Mallett, S.V.; Kitchen, D.; Klein, A.A.; Maybury, H.; Collins, P.W.; Laffan, M. The use of viscoelastic haemostatic assays in the management of major bleeding: A British Society for Haematology Guideline. Br. J. Haematol. 2018, 182, 789–806. [Google Scholar] [CrossRef] [PubMed]
- Wikkelsø, A.; Wetterslev, J.; Møller, A.M.; Afshari, A. Thromboelastography (TEG) or thromboelastometry (ROTEM) to monitor haemostatic treatment versus usual care in adults or children with bleeding. Cochrane Database Syst. Rev. 2016, 2016, CD007871. [Google Scholar] [CrossRef] [PubMed]
- Davenport, R.; Manson, J.; De’Ath, H.; Platton, S.; Coates, A.; Allard, S.; Hart, D.; Pearse, R.; Pasi, K.J.; MacCallum, P.; et al. Functional definition and characterization of acute traumatic coagulopathy. Crit. Care Med. 2011, 39, 2652–2658. [Google Scholar] [CrossRef] [PubMed]
- Inaba, K.; Rizoli, S.; Veigas, P.V.; Callum, J.; Davenport, R.; Hess, J.; Maegele, M. 2014 Consensus conference on viscoelastic test–based transfusion guidelines for early trauma resuscitation: Report of the panel. J. Trauma Acute Care Surg. 2015, 78, 1220–1229. [Google Scholar] [CrossRef] [PubMed]
- Moore, H.B.; Moore, E.E.; Neal, M.D. Trauma Induced Coagulopathy, 2nd ed.; Spring Nature Switzerland AG: Cham, Switzerland, 2021. [Google Scholar]
- Johansson, P.I.; Stensballe, J.; Oliveri, R.; Wade, C.E.; Ostrowski, S.R.; Holcomb, J.B. How I treat patients with massive hemorrhage. Blood 2014, 124, 3052–3058. [Google Scholar] [CrossRef] [PubMed]
- Maegele, M. The diagnosis and treatment of acute traumatic bleeding and coagulopathy. Dtsch. Aerzteblatt Int. 2019, 116, 799. [Google Scholar] [CrossRef] [PubMed]
- Webb, A.J.; Brown, C.S.; Naylor, R.M.; Rabinstein, A.A.; Mara, K.C.; Nei, A.M. Thromboelastography is a Marker for Clinically Significant Progressive Hemorrhagic Injury in Severe Traumatic Brain Injury. Neurocrit. Care 2021. [Google Scholar] [CrossRef]
- Wohlauer, M.V.; Moore, E.E.; Thomas, S.; Sauaia, A.; Evans, E.; Harr, J.; Silliman, C.C.; Ploplis, V.; Castellino, F.J.; Walsh, M. Early platelet dysfunction: An unrecognized role in the acute coagulopathy of trauma. J. Am. Coll. Surg. 2012, 214, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Nekludov, M.; Bellander, B.-M.; Blombäck, M.; Wallen, H.N. Platelet dysfunction in patients with severe traumatic brain injury. J. Neurotrauma 2007, 24, 1699–1706. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, R.C.; Owings, J.T.; Holmes, J.; Battistella, F.D.; Gosselin, R.C.; Paglieroni, T.G. Platelet activation and function after trauma. J. Trauma 2001, 51, 639–647. [Google Scholar] [CrossRef]
- Windelv, N.A.; Welling, K.-L.; Ostrowski, S.R.; Johansson, P.I. The prognostic value of thrombelastography in identifying neurosurgical patients with worse prognosis. Blood Coagul. Fibrinolysis 2011, 22, 416–419. [Google Scholar] [CrossRef] [PubMed]
- Kutcher, M.E.; Redick, B.J.; McCreery, R.C.; Crane, I.M.; Greenberg, M.D.; Cachola, L.M.; Nelson, M.F.; Cohen, M.J. Characterization of platelet dysfunction after trauma. J. Trauma Acute Care Surg. 2012, 73, 13–19. [Google Scholar] [CrossRef]
- Xu, X.; Kozar, R.; Zhang, J.; Dong, J.F. Diverse activities of von Willebrand factor in traumatic brain injury and associated coagulopathy. J. Thromb. Haemost. 2020, 18, 3154–3162. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Liu, W.; Zhou, Y.; Hilton, T.; Zhao, Z.; Liu, W.; Wang, M.; Yeon, J.; Houck, K.; Thiagarajan, P. von Willebrand factor enhances microvesicle-induced vascular leakage and coagulopathy in mice with traumatic brain injury. Blood J. Am. Soc. Hematol. 2018, 132, 1075–1084. [Google Scholar] [CrossRef]
- Kurland, D.; Hong, C.; Aarabi, B.; Gerzanich, V.; Simard, J.M. Hemorrhagic progression of a contusion after traumatic brain injury: A review. J. Neurotrauma 2012, 29, 19–31. [Google Scholar] [CrossRef]
- Simard, J.M.; Kilbourne, M.; Tsymbalyuk, O.; Tosun, C.; Caridi, J.; Ivanova, S.; Keledjian, K.; Bochicchio, G.; Gerzanich, V. Key role of sulfonylurea receptor 1 in progressive secondary hemorrhage after brain contusion. J. Neurotrauma 2009, 26, 2257–2267. [Google Scholar] [CrossRef]
- Di Battista, A.P.; Rizoli, S.B.; Lejnieks, B.; Min, A.; Shiu, M.Y.; Peng, H.T.; Baker, A.J.; Hutchison, M.G.; Churchill, N.; Inaba, K.; et al. Sympathoadrenal activation is associated with acute traumatic coagulopathy and endotheliopathy in isolated brain injury. Shock 2016, 46, 96. [Google Scholar] [CrossRef] [PubMed]
- Foley, J.H.; Conway, E.M. Cross talk pathways between coagulation and inflammation. Circ. Res. 2016, 118, 1392–1408. [Google Scholar] [CrossRef] [PubMed]
- Nekludov, M.; Mobarrez, F.; Gryth, D.; Bellander, B.-M.; Wallen, H. Formation of microparticles in the injured brain of patients with severe isolated traumatic brain injury. J. Neurotrauma 2014, 31, 1927–1933. [Google Scholar] [CrossRef]
- Medcalf, R.L. The traumatic side of fibrinolysis. Blood J. Am. Soc. Hematol. 2015, 125, 2457–2458. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fletcher-Sandersjöö, A.; Maegele, M.; Bellander, B.-M. Does complement-mediated hemostatic disturbance occur in traumatic brain injury? A literature review and observational study protocol. Int. J. Mol. Sci. 2020, 21, 1596. [Google Scholar] [CrossRef]
- Laroche, M.; Kutcher, M.E.; Huang, M.C.; Cohen, M.J.; Manley, G.T. Coagulopathy after traumatic brain injury. Neurosurgery 2012, 70, 1334–1345. [Google Scholar] [CrossRef] [PubMed]
- Hijazi, N.; Abu Fanne, R.; Abramovitch, R.; Yarovoi, S.; Higazi, M.; Abdeen, S.; Basheer, M.; Maraga, E.; Cines, D.B.; Al-Roof Higazi, A. Endogenous plasminogen activators mediate progressive intracerebral hemorrhage after traumatic brain injury in mice. Blood 2015, 125, 2558–2567. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.-L.; Chen, H.; Wu, B.-S.; Cao, H.-L.; Xu, T.; Hu, J.; Wang, G.; Gao, W.-W.; Lin, Z.-K.; Chen, S.-W. D-dimer as a predictor of progressive hemorrhagic injury in patients with traumatic brain injury: Analysis of 194 cases. Neurosurg. Rev. 2010, 33, 359–366. [Google Scholar] [CrossRef]
- Hoffman, M.; Monroe, D.M. Tissue factor in brain is not saturated with factor VIIa: Implications for factor VIIa dosing in intracerebral hemorrhage. Stroke 2009, 40, 2882–2884. [Google Scholar] [CrossRef]
- CRASH-3 Trial Collaborators. Effects of tranexamic acid on death, disability, vascular occlusive events and other morbidities in patients with acute traumatic brain injury (CRASH-3): A randomised, placebo-controlled trial. Lancet 2019, 394, 1713–1723. [Google Scholar] [CrossRef]
- Rowell, S.E.; Meier, E.N.; McKnight, B.; Kannas, D.; May, S.; Sheehan, K.; Bulger, E.M.; Idris, A.H.; Christenson, J.; Morrison, L.J.; et al. Effect of Out-of-Hospital Tranexamic Acid vs Placebo on 6-Month Functional Neurologic Outcomes in Patients with Moderate or Severe Traumatic Brain Injury. JAMA 2020, 324, 961–974. [Google Scholar] [CrossRef]
- Cotton, B.A.; Harvin, J.A.; Kostousouv, V.; Minei, K.M.; Radwan, Z.A.; Schöchl, H.; Wade, C.E.; Holcomb, J.B.; Matijevic, N. Hyperfibrinolysis at admission is an uncommon but highly lethal event associated with shock and prehospital fluid administration. J. Trauma Acute Care Surg. 2012, 73, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Moore, H.B.; Moore, E.E.; Liras, I.N.; Gonzalez, E.; Harvin, J.A.; Holcomb, J.B.; Sauaia, A.; Cotton, B.A. Acute fibrinolysis shutdown after injury occurs frequently and increases mortality: A multicenter evaluation of 2540 severely injured patients. J. Am. Coll. Surg. 2016, 222, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Johansson, P.I.; Stissing, T.; Bochsen, L.; Ostrowski, S.R. Thrombelastography and tromboelastometry in assessing coagulopathy in trauma. Scand J. Trauma Resusc. Emerg. Med. 2009, 17, 45. [Google Scholar] [CrossRef] [PubMed]
- Kashuk, J.L.; Moore, E.E.; Sawyer, M.; Le, T.; Johnson, J.; Biffl, W.L.; Cothren, C.C.; Barnett, C.; Stahel, P.; Sillman, C.C.; et al. Postinjury coagulopathy management: Goal directed resuscitation via POC thrombelastography. Ann. Surg. 2010, 251, 604–614. [Google Scholar] [CrossRef]
- Gonzalez, E.; Pieracci, F.M.; Moore, E.E.; Kashuk, J.L. Coagulation abnormalities in the trauma patient: The role of point-of-care thromboelastography. Semin. Thromb. Hemost. 2010, 36, 723–737. [Google Scholar] [CrossRef]
- MacDonald, S.G.; Luddington, R.J. Critical factors contributing to the thromboelastography trace. Semin. Thromb. Hemost. 2010, 36, 712–722. [Google Scholar] [CrossRef]
- Bochsen, L.; Wiinberg, B.; Kjelgaard-Hansen, M.; Steinbruchel, D.A.; Johansson, P.I. Evaluation of the TEG platelet mapping assay in blood donors. Thromb. J. 2007, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Bartels, A.N.; Johnson, C.; Lewis, J.; Clevenger, J.W.; Barnes, S.L.; Hammer, R.D.; Ahmad, S. Platelet adenosine diphosphate inhibition in trauma patients by thromboelastography correlates with paradoxical increase in platelet dense granule content by flow cytometry. Surgery 2016, 160, 954–959. [Google Scholar] [CrossRef] [PubMed]
- Solomon, C.; Schöchl, H.; Ranucci, M.; Schlimp, C. Can the Viscoelastic Parameter α-Angle Distinguish Fibrinogen from Platelet Deficiency and Guide Fibrinogen Supplementation? Anesth. Analg. 2015, 121, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.; Jbara, M.; Miller, A.; Lawson, J. Thromboelastographic Guided Blood Component Therapy for Severe Hemorrhage. In Blood Bulletin; Gresens, C., Ed.; America’s Blood Centers: Washington, DC, USA, 2014. [Google Scholar]
- Ranucci, M.; Baryshnikova, E. Sensitivity of viscoelastic tests to platelet function. J. Clin. Med. 2020, 9, 189. [Google Scholar] [CrossRef]
- Sayce, A.C.; Neal, M.D.; Leeper, C.M. Viscoelastic monitoring in trauma resuscitation. Transfusion 2020, 60, S33–S51. [Google Scholar] [CrossRef]
- Toffaletti, J.G.; Buckner, K.A. Use of Earlier-Reported Rotational Thromboelastometry Parameters to Evaluate Clotting Status, Fibrinogen, and Platelet Activities in Postpartum Hemorrhage Compared to Surgery and Intensive Care Patients. Anesth. Analg. 2019, 128, 414–423. [Google Scholar] [CrossRef]
- Görlinger, K.; Pérez-Ferrer, A.; Dirkmann, D.; Saner, F.; Maegele, M.; Calatayud, Á.A.P.; Kim, T.-Y. The role of evidence-based algorithms for rotational thromboelastometry-guided bleeding management. Korean J. Anesthesiol. 2019, 72, 297. [Google Scholar] [CrossRef] [PubMed]
- Kornblith, L.Z.; Moore, H.B.; Cohen, M.J. Trauma-induced coagulopathy: The past, present, and future. J. Thromb. Haemost. 2019, 17, 852–862. [Google Scholar] [CrossRef]
- Darlington, D.N.; Wu, X.; Keesee, J.D.; Cap, A.P. Severe trauma and hemorrhage leads to platelet dysfunction and changes in cyclic nucleotides in the rat. Shock 2020, 53, 468–475. [Google Scholar] [CrossRef]
- Tóth, O.; Calatzis, A.; Penz, S.; Losonczy, H.; Siess, W. Multiple electrode aggregometry: A new device to measure platelet aggregation in whole blood. Thromb. Haemost. 2006, 96, 781–788. [Google Scholar] [PubMed]
- George, M.J.; Burchfield, J.; MacFarlane, B.; Wang, Y.W.; Cardenas, J.C.; White, N.J.; Gill, B.S.; Wade, C.E. Multiplate and TEG platelet mapping in a population of severely injured trauma patients. Transfus. Med. 2018, 28, 224–230. [Google Scholar] [CrossRef]
- Walsh, M.; Fritz, S.; Hake, D.; Son, M.; Greve, S.; Jbara, M.; Chitta, S.; Fritz, B.; Miller, A.; Bader, M.K.; et al. Targeted Thromboelastographic (TEG) Blood Component and Pharmacologic Hemostatic Therapy in Traumatic and Acquired Coagulopathy. Curr. Drug Targets 2016, 17, 954–970. [Google Scholar] [CrossRef] [PubMed]
- Roozenbeek, B.; Maas, A.I.; Menon, D.K. Changing patterns in the epidemiology of traumatic brain injury. Nat. Rev. Neurol. 2013, 9, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Lindblad, C.; Thelin, E.P.; Nekludov, M.; Frostell, A.; Nelson, D.W.; Svensson, M.; Bellander, B.M. Assessment of Platelet Function in Traumatic Brain Injury-A Retrospective Observational Study in the Neuro-Critical Care Setting. Front. Neurol. 2018, 9, 15. [Google Scholar] [CrossRef]
- Nishijima, D.K.; Zehtabchi, S.; Berrong, J.; Legome, E. Utility of platelet transfusion in adult patients with traumatic intracranial hemorrhage and pre-injury anti-platelet use: A systematic review. J. Trauma Acute Care Surg. 2012, 72, 1658. [Google Scholar] [CrossRef] [PubMed]
- Anglin, C.O.; Spence, J.S.; Warner, M.A.; Paliotta, C.; Harper, C.; Moore, C.; Sarode, R.; Madden, C.; Diaz-Arrastia, R. Effects of platelet and plasma transfusion on outcome in traumatic brain injury patients with moderate bleeding diatheses. J. Neurosurg. 2013, 118, 676–686. [Google Scholar] [CrossRef] [PubMed]
- Baharoglu, M.I.; Cordonnier, C.; Salman, R.A.-S.; De Gans, K.; Koopman, M.M.; Brand, A.; Majoie, C.B.; Beenen, L.F.; Marquering, H.A.; Vermeulen, M. Platelet transfusion versus standard care after acute stroke due to spontaneous cerebral haemorrhage associated with antiplatelet therapy (PATCH): A randomised, open-label, phase 3 trial. Lancet 2016, 387, 2605–2613. [Google Scholar] [CrossRef]
- Dias, J.D.; Lopez-Espina, C.G.; Ippolito, J.; Hsiao, L.H.; Zaman, F.; Muresan, A.A.; Thomas, S.G.; Walsh, M.; Jones, A.J.; Grisoli, A.; et al. Rapid point-of-care detection and classification of direct-acting oral anticoagulants with the TEG 6s: Implications for trauma and acute care surgery. J. Trauma Acute Care Surg. 2019, 87, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Scorer, T.G.; FitzGibbon, L.; Aungraheeta, R.; Sharma, U.; Peltier, G.C.; McIntosh, C.S.; Reddoch-Cardenas, K.M.; Meyer, A.; Cap, A.P.; Mumford, A.D. TEG PlateletMapping assay results may be misleading in the presence of cold stored platelets. Transfusion 2020, 60, S119–S123. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, E.; Moore, E.E.; Moore, H.B.; Chapman, M.P.; Chin, T.L.; Ghasabyan, A.; Wohlauer, M.V.; Barnett, C.C.; Bensard, D.D.; Biffl, W.L.; et al. Goal-directed Hemostatic Resuscitation of Trauma-induced Coagulopathy: A Pragmatic Randomized Clinical Trial Comparing a Viscoelastic Assay to Conventional Coagulation Assays. Ann. Surg. 2016, 263, 1051–1059. [Google Scholar] [CrossRef] [PubMed]
- Brazinova, A.; Majdan, M.; Leitgeb, J.; Trimmel, H.; Mauritz, W.; Austrian Working Group on Improvement of Early, T.B.I.C. Factors that may improve outcomes of early traumatic brain injury care: Prospective multicenter study in Austria. Scand. J. Trauma Resusc. Emerg. Med. 2015, 23, 53. [Google Scholar] [CrossRef]
- Martini, W.Z. Coagulation complications following trauma. Mil. Med. Res. 2016, 3, 35. [Google Scholar] [CrossRef]
- Beynon, C.; Wessels, L.; Unterberg, A.W. Point-of-care testing in neurosurgery. Semin. Thromb. Hemost. 2017, 43, 416–422. [Google Scholar]
- Chang, R.; Cardenas, J.C.; Wade, C.E.; Holcomb, J.B. Advances in the understanding of trauma-induced coagulopathy. Blood 2016, 128, 1043–1049. [Google Scholar] [CrossRef]
- Cap, A.; Hunt, B. The pathogenesis of traumatic coagulopathy. Anaesthesia 2015, 70, 96–101. [Google Scholar] [CrossRef]
- Valle, E.J.; Van Haren, R.M.; Allen, C.J.; Jouria, J.M.; Bullock, M.R.; Schulman, C.I.; Namias, N.; Livingstone, A.S.; Proctor, K.G. Does traumatic brain injury increase the risk for venous thromboembolism in polytrauma patients? J. Trauma Acute Care Surg. 2014, 77, 243–250. [Google Scholar] [CrossRef]
- Liu, H.; Li, J.; Yu, J.; Yuan, T. Research into the predictive effect of TEG in the changes of coagulation functions of the patients with traumatic brain hemorrhage. Open Med. 2015, 10, 399–404. [Google Scholar] [CrossRef]
- Martin, G.; Shah, D.; Elson, N.; Boudreau, R.; Hanseman, D.; Pritts, T.A.; Makley, A.T.; Foreman, B.; Goodman, M.D. Relationship of coagulopathy and platelet dysfunction to transfusion needs after traumatic brain injury. Neurocrit. Care 2018, 28, 330–337. [Google Scholar] [CrossRef]
- Dunham, C.M.; Hoffman, D.A.; Huang, G.S.; Omert, L.A.; Gemmel, D.J.; Merrell, R. Traumatic intracranial hemorrhage correlates with preinjury brain atrophy, but not with antithrombotic agent use: A retrospective study. PLoS ONE 2014, 9, e109473. [Google Scholar] [CrossRef]
- Folkerson, L.E.; Sloan, D.; Davis, E.; Kitagawa, R.S.; Cotton, B.A.; Holcomb, J.B.; Tomasek, J.S.; Wade, C.E. Coagulopathy as a predictor of mortality after penetrating traumatic brain injury. Am. J. Emerg. Med. 2018, 36, 38–42. [Google Scholar] [CrossRef]
- Baksaas-Aasen, K.; Gall, L.S.; Stensballe, J.; Juffermans, N.P.; Curry, N.; Maegele, M.; Brooks, A.; Rourke, C.; Gillespie, S.; Murphy, J. Viscoelastic haemostatic assay augmented protocols for major trauma haemorrhage (ITACTIC): A randomized, controlled trial. Intensive Care Med. 2021, 47, 49–59. [Google Scholar] [CrossRef]
- Hota, S.; Ng, M.; Hilliard, D.; Burgess, J. Thromboelastogram-Guided Resuscitation for Patients with Traumatic Brain Injury on Novel Anticoagulants. Am. Surg. 2019, 85, 861–864. [Google Scholar] [CrossRef]
- Holzmacher, J.L.; Reynolds, C.; Patel, M.; Maluso, P.; Holland, S.; Gamsky, N.; Moore, H.; Acquista, E.; Carrick, M.; Amdur, R. Platelet transfusion does not improve outcomes in patients with brain injury on antiplatelet therapy. Brain Inj. 2018, 32, 325–330. [Google Scholar] [CrossRef]
- Kumar, M.; Ahmad, J.; Maiwall, R.; Choudhury, A.; Bajpai, M.; Mitra, L.G.; Saluja, V.; Mohan Agarwal, P.; Bihari, C.; Shasthry, S.M. Thromboelastography-Guided Blood Component Use in Patients With Cirrhosis With Nonvariceal Bleeding: A Randomized Controlled Trial. Hepatology 2019, 71, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Kay, A.B.; Morris, D.S.; Collingridge, D.S.; Majercik, S. Platelet dysfunction on thromboelastogram is associated with severity of blunt traumatic brain injury. Am. J. Surg. 2019, 218, 1134–1137. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J.T.; Petrone, P.; Axelrad, A.; Marini, C.P. Comparison between thromboelastography and conventional coagulation test: Should we abandon conventional coagulation tests in polytrauma patients? Cirugía Española 2018, 96, 443–449. [Google Scholar] [CrossRef]
- Ologun, G.O.; Pamula, A.; Alegbejo-Olarinoye, M.; Granet, P.; Behm, R. Rethinking the Use of Routine Platelet Transfusions for Head Injured Patients on Antiplatelet Therapy. Cureus 2019, 11, e6136. [Google Scholar] [CrossRef]
- Ganter, M.T.; Hofer, C.K. Coagulation monitoring: Current techniques and clinical use of viscoelastic point-of-care coagulation devices. Anesth. Analg. 2008, 106, 1366–1375. [Google Scholar] [CrossRef]
- Orban, M.; Sibbing, D. Chapter 10: Multiplate Analyzer. In Antiplatelet Therapy in Cardiovascular Disease; Waksman, R., Gurbel, P.A., Gaglia, M.A., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; pp. 82–91. ISBN 978-1-118-275-757. [Google Scholar]
- Bora, S.K.; Sahu, R.N. Extending Horizons of Extracorporeal Membrane Oxygenation: Can the Tight Rope of Anticoagulation Help Further? J. Card. Crit. Care TSS 2018, 2, 049–050. [Google Scholar] [CrossRef]
- Stettler, G.R.; Moore, E.E.; Moore, H.B.; Nunns, G.R.; Huebner, B.R.; Einersen, P.; Ghasabyan, A.; Silliman, C.C.; Banerjee, A.; Sauaia, A. Platelet adenosine diphosphate receptor inhibition provides no advantage in predicting need for platelet transfusion or massive transfusion. Surgery 2017, 162, 1286–1294. [Google Scholar] [CrossRef]
- Daley, M.; Enright, Z.; Nguyen, J.; Ali, S.; Clark, A.; Aydelotte, J.; Teixeira, P.; Coopwood, T.; Brown, C. Adenosine diphosphate platelet dysfunction on thromboelastogram is independently associated with increased morality in traumatic brain injury. Eur. J. Trauma Emerg. Surg. 2017, 43, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; Lin, A.; Hilliard, C.; Fu, R.; Lennox, T.; Barbosa, R.; Schreiber, M.; Rowell, S. The utility of thromboelastography for predicting the risk of progression of intracranial hemorrhage in traumatic brain injury patients. Neurosurgery 2017, 64, 182–187. [Google Scholar] [CrossRef]
- Folkerson, L.E.; Sloan, D.; Cotton, B.A.; Holcomb, J.B.; Tomasek, J.S.; Wade, C.E. Predicting progressive hemorrhagic injury from isolated traumatic brain injury and coagulation. Surgery 2015, 158, 655–661. [Google Scholar] [CrossRef]
- Holcomb, J.B.; Minei, K.M.; Scerbo, M.L.; Radwan, Z.A.; Wade, C.E.; Kozar, R.A.; Gill, B.S.; Albarado, R.; McNutt, M.K.; Khan, S. Admission rapid thrombelastography can replace conventional coagulation tests in the emergency department: Experience with 1974 consecutive trauma patients. Ann. Surg. 2012, 256, 476–486. [Google Scholar] [CrossRef]
- Tapia, N.; Chang, A.; Norman, M.; Welsh, F.; Scott, B.; Wall, M.; Mattox, K.; Suliburk, J. TEG-guided resuscitation is superior to standardized MTP resuscitation in massively transfused penetrating trauma patients. J. Trauma Acute Care Surg. 2013, 74, 378–386. [Google Scholar] [CrossRef]
- Plotkin, A.J.; Wade, C.E.; Jenkins, D.H.; Smith, K.A.; Noe, J.C.; Park, M.S.; Perkins, J.G.; Holcomb, J.B. A reduction in clot formation rate and strength assessed by thrombelastography is indicative of transfusion requirements in patients with penetrating injuries. J. Trauma 2008, 64, S64–S68. [Google Scholar] [CrossRef]
- Kaufmann, C.R.; Dwyer, K.M.; Crews, J.D.; Dols, S.J.; Trask, A.L. Usefulness of thrombelastography in assessment of trauma patient coagulation. J. Trauma 1997, 42, 716–720, discussion 720–712. [Google Scholar] [CrossRef]
- Spahn, D.R.; Bouillon, B.; Cerny, V.; Coats, T.J.; Duranteau, J.; Fernández-Mondéjar, E.; Filipescu, D.; Hunt, B.J.; Komadina, R.; Nardi, G. Management of bleeding and coagulopathy following major trauma: An updated European guideline. Crit. Care 2013, 17, R76. [Google Scholar] [CrossRef] [PubMed]
- Curry, N.S.; Davenport, R. Transfusion strategies for major haemorrhage in trauma. Br. J. Haematol. 2019, 184, 508–523. [Google Scholar] [CrossRef] [PubMed]
- Cryer, H.G.; Nathens, A.B.; Bulger, E.M.; Calland, J.F.; Cohen, M.J.; Cotton, B.A.; Davis, M.L.; Hemmila, M.R.; Hess, J.R.; Jawa, R.; et al. Massive Transfusion in Trauma Guidelines; American College of Surgeons: Chicago, IL, USA, 2013. [Google Scholar]
- Gruen, D.S.; Guyette, F.X.; Brown, J.B.; Okonkwo, D.O.; Puccio, A.M.; Campwala, I.K.; Tessmer, M.T.; Daley, B.J.; Miller, R.S.; Harbrecht, B.G. Association of prehospital plasma with survival in patients with traumatic brain injury: A secondary analysis of the PAMPer cluster randomized clinical trial. JAMA Netw. Open 2020, 3, e2016869. [Google Scholar] [CrossRef]
- Sperry, J.L.; Guyette, F.X.; Brown, J.B.; Yazer, M.H.; Triulzi, D.J.; Early-Young, B.J.; Adams, P.W.; Daley, B.J.; Miller, R.S.; Harbrecht, B.G. Prehospital plasma during air medical transport in trauma patients at risk for hemorrhagic shock. N. Engl. J. Med. 2018, 379, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.; Holcomb, J.B. Optimal fluid therapy for traumatic hemorrhagic shock. Crit. Care Clin. 2017, 33, 15–36. [Google Scholar] [CrossRef]
- Etemadrezaie, H.; Baharvahdat, H.; Shariati, Z.; Lari, S.M.; Shakeri, M.T.; Ganjeifar, B. The effect of fresh frozen plasma in severe closed head injury. Clin. Neurol. Neurosurg. 2007, 109, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Jokar, T.O.; Khalil, M.; Rhee, P.; Kulvatunyou, N.; Pandit, V.; O’Keeffe, T.; Tang, A.; Joseph, B. Ratio-based resuscitation in trauma patients with traumatic brain injury: Is there a similar effect? Am. Surg. 2016, 82, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.; Moore, E.E.; Moore, H.B.; Thomas, S.; Kwaan, H.C.; Speybroeck, J.; Marsee, M.; Bunch, C.M.; Stillson, J.; Thomas, A.V.; et al. Whole Blood, Fixed Ratio, or Goal-Directed Blood Component Therapy for the Initial Resuscitation of Severely Hemorrhaging Trauma Patients: A Narrative Review. J. Clin. Med. 2021, 10, 320. [Google Scholar] [CrossRef] [PubMed]
- Simurda, T.; Casini, A.; Stasko, J.; Hudecek, J.; Skornova, I.; Vilar, R.; Neerman-Arbez, M.; Kubisz, P. Perioperative management of a severe congenital hypofibrinogenemia with thrombotic phenotype. Thromb. Res. 2020, 188, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Simurda, T.; Caccia, S.; Asselta, R.; Zolkova, J.; Stasko, J.; Skornova, I.; Snahnicanova, Z.; Loderer, D.; Lasabova, Z.; Kubisz, P. Congenital hypofibrinogenemia associated with a novel heterozygous nonsense mutation in the globular C-terminal domain of the γ-chain (p.Glu275Stop). J. Thromb. Thrombolysis 2020, 50, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Pahatouridis, D.; Alexiou, G.A.; Zigouris, A.; Mihos, E.; Drosos, D.; Voulgaris, S. Coagulopathy in moderate head injury. The role of early administration of low molecular weight heparin. Brain Inj. 2010, 24, 1189–1192. [Google Scholar] [CrossRef] [PubMed]
- Rourke, C.; Curry, N.; Khan, S.; Taylor, R.; Raza, I.; Davenport, R.; Stanworth, S.; Brohi, K. Fibrinogen levels during trauma hemorrhage, response to replacement therapy, and association with patient outcomes. J. Thromb. Haemost. 2012, 10, 1342–1351. [Google Scholar] [CrossRef] [PubMed]
- Muradashvili, N.; Lominadze, D. Role of fibrinogen in cerebrovascular dysfunction after traumatic brain injury. Brain Inj. 2013, 27, 1508–1515. [Google Scholar] [CrossRef] [PubMed]
- Schnüriger, B.; Inaba, K.; Abdelsayed, G.A.; Lustenberger, T.; Eberle, B.M.; Barmparas, G.; Talving, P.; Demetriades, D. The impact of platelets on the progression of traumatic intracranial hemorrhage. J. Trauma Acute Care Surg. 2010, 68, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Watson, V.L.; Louis, N.; Seminara, B.V.; Muizelaar, J.P.; Alberico, A. Proposal for the rapid reversal of coagulopathy in patients with nonoperative head injuries on anticoagulants and/or antiplatelet agents: A case study and literature review. Neurosurgery 2017, 81, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Fiaschi, P.; Iaccarino, C.; Stefini, R.; Prior, E.; Prior, A.; Zona, G. Clinical practice for antiplatelet and anticoagulant therapy in neurosurgery: Data from an Italian survey and summary of current recommendations–part I, antiplatelet therapy. Neurosurg. Rev. 2020, 44, 485–493. [Google Scholar] [CrossRef]
- Keyes, M.; Alley, A.; Muertos, K.; Anderson, B.; Howerton, S.; Burns, A.; Pepe, A. The “Headstrike” Protocol: A Retrospective Review of a Single Trauma Center’s Operational Change in the Management of Anticoagulated Ground-Level Falls. Am. Surg. 2019, 85, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Godier, A.; Albaladejo, P.; On Perioperative Haemostasis Gihp Group, T. Management of Bleeding Events Associated with Antiplatelet Therapy: Evidence, Uncertainties and Pitfalls. J. Clin. Med. 2020, 9, 2318. [Google Scholar] [CrossRef] [PubMed]
- Naidech, A.M.; Liebling, S.M.; Rosenberg, N.F.; Lindholm, P.F.; Bernstein, R.A.; Batjer, H.H.; Alberts, M.J.; Kwaan, H.C. Early platelet transfusion improves platelet activity and may improve outcomes after intracerebral hemorrhage. Neurocrit. Care 2012, 16, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Fortuna, G.R.; Mueller, E.W.; James, L.E.; Shutter, L.A.; Butler, K.L. The impact of preinjury antiplatelet and anticoagulant pharmacotherapy on outcomes in elderly patients with hemorrhagic brain injury. Surgery 2008, 144, 598–603, discussion 603–595. [Google Scholar] [CrossRef]
- Washington, C.W.; Schuerer, D.J.; Grubb Jr, R.L. Platelet transfusion: An unnecessary risk for mild traumatic brain injury patients on antiplatelet therapy. J. Trauma Acute Care Surg. 2011, 71, 358–363. [Google Scholar] [CrossRef]
- Vargas, A.; Bleck, T.P. Hematological Challenges in Intensive Care Unit Patients with Neurological Disease. In Hematologic Challenges in the Critically Ill; Shander, A., Corwin, H.L., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 185–197. [Google Scholar]
- Kapapa, T.; Röhrer, S.; Struve, S.; Petscher, M.; König, R.; Wirtz, C.R.; Woischneck, D. Desmopressin acetate in intracranial haemorrhage. Neurol. Res. Int. 2014, 2014, 298767. [Google Scholar] [CrossRef]
- Naidech, A.M.; Maas, M.B.; Levasseur-Franklin, K.E.; Liotta, E.M.; Guth, J.C.; Berman, M.; Rosenow, J.M.; Lindholm, P.F.; Bendok, B.R.; Prabhakaran, S. Desmopressin improves platelet activity in acute intracerebral hemorrhage. Stroke 2014, 45, 2451–2453. [Google Scholar] [CrossRef] [PubMed]
- Wutzler, S.; Maegele, M.; Marzi, I.; Spanholtz, T.; Wafaisade, A.; Lefering, R.; Trauma Registry of the German Society for Trauma Surgery. Association of preexisting medical conditions with in-hospital mortality in multiple-trauma patients. J. Am. Coll. Surg. 2009, 209, 75–81. [Google Scholar] [CrossRef]
- Batchelor, J.S.; Grayson, A. A meta-analysis to determine the effect of anticoagulation on mortality in patients with blunt head trauma. Br. J. Neurosurg. 2012, 26, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, A.; Servadei, F.; Marchesini, G.; Bronzoni, C.; Montesi, D.; Arietta, L. Antiplatelet therapy and the outcome of subjects with intracranial injury: The Italian SIMEU study. Crit. Care 2013, 17, R53. [Google Scholar] [CrossRef] [PubMed]
- Grandhi, R.; Harrison, G.; Voronovich, Z.; Bauer, J.; Chen, S.H.; Nicholas, D.; Alarcon, L.H.; Okonkwo, D.O. Preinjury warfarin, but not antiplatelet medications, increases mortality in elderly traumatic brain injury patients. J. Trauma Acute Care Surg. 2015, 78, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Prinz, V.; Finger, T.; Bayerl, S.; Rosenthal, C.; Wolf, S.; Liman, T.; Vajkoczy, P. High prevalence of pharmacologically induced platelet dysfunction in the acute setting of brain injury. Acta Neurochir. 2016, 158, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, J.S.; Grayson, A. A meta-analysis to determine the effect on survival of platelet transfusions in patients with either spontaneous or traumatic antiplatelet medication-associated intracranial haemorrhage. BMJ Open 2012, 2, e000588. [Google Scholar] [CrossRef] [PubMed]
- Batchelor, J.S.; Grayson, A. A meta-analysis to determine the effect of preinjury antiplatelet agents on mortality in patients with blunt head trauma. Br. J. Neurosurg. 2013, 27, 12–18. [Google Scholar] [CrossRef]
- Tauber, M.; Koller, H.; Moroder, P.; Hitzl, W.; Resch, H. Secondary intracranial hemorrhage after mild head injury in patients with low-dose acetylsalicylate acid prophylaxis. J. Trauma Acute Care Surg. 2009, 67, 521–525. [Google Scholar] [CrossRef] [PubMed]
- Nishijima, D.K.; Shahlaie, K.; Sarkar, K.; Rudisill, N.; Holmes, J.F. Risk of unfavorable long-term outcome in older adults with traumatic intracranial hemorrhage and anticoagulant or antiplatelet use. Am. J. Emerg. Med. 2013, 31, 1244–1247. [Google Scholar] [CrossRef] [PubMed]
- Nishijima, D.K.; Offerman, S.R.; Ballard, D.W.; Vinson, D.R.; Chettipally, U.K.; Rauchwerger, A.S.; Reed, M.E.; Holmes, J.F.; Services, C.R.i.E.; Network, T. Risk of traumatic intracranial hemorrhage in patients with head injury and preinjury warfarin or clopidogrel use. Acad. Emerg. Med. 2013, 20, 140–145. [Google Scholar] [CrossRef]
- Joseph, B.; Pandit, V.; Aziz, H.; Kulvatunyou, N.; Hashmi, A.; Tang, A.; O’Keeffe, T.; Wynne, J.; Vercruysse, G.; Friese, R.S. Clinical outcomes in traumatic brain injury patients on preinjury clopidogrel: A prospective analysis. J. Trauma Acute Care Surg. 2014, 76, 817–820. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.P.; Trujillo, T.C.; Nordenholz, K.E. Practical considerations in emergency management of bleeding in the setting of target-specific oral anticoagulants. Am. J. Emerg. Med. 2014, 32, 375–382. [Google Scholar] [CrossRef]
- Feeney, J.M.; Santone, E.; DiFiori, M.; Kis, L.; Jayaraman, V.; Montgomery, S.C. Compared to warfarin, direct oral anticoagulants are associated with lower mortality in patients with blunt traumatic intracranial hemorrhage: A TQIP study. J. Trauma Acute Care Surg. 2016, 81, 843–848. [Google Scholar] [CrossRef]
- Maegele, M.; Grottke, O.; Schöchl, H.; Sakowitz, O.; Spannagl, M.; Koscielny, J. Direct Oral Anticoagulants in Emergency Trauma Admissions: Perioperative Management, and Handling Hemorrhage. Dtsch. Ärzteblatt Int. 2016, 113, 575. [Google Scholar]
- Hackam, D.G.; Mrkobrada, M. Selective serotonin reuptake inhibitors and brain hemorrhage: A meta-analysis. Neurology 2012, 79, 1862–1865. [Google Scholar] [CrossRef]
- Oberladstätter, D.; Schlimp, C.J.; Zipperle, J.; Osuchowski, M.F.; Voelckel, W.; Grottke, O.; Schöchl, H. Impact of Idarucizumab and Andexanet Alfa on DOAC Plasma Concentration and ClotPro(®) Clotting Time: An Ex Vivo Spiking Study in A Cohort of Trauma Patients. J. Clin. Med. 2021, 10, 3476. [Google Scholar] [CrossRef] [PubMed]
- McCully, S.P.; Schreiber, M.A. Traumatic brain injury and its effect on coagulopathy. Semin. Thromb. Hemost. 2013, 39, 896–901. [Google Scholar]
- Lawrie, R. Coagulopathy in severe, isolated traumatic brain injury: A prevalence study. Master’s Thesis, University of Cape Town, Cape Town, South Africa, 2018. [Google Scholar]
- Bugaev, N.; Como, J.J.; Golani, G.; Freeman, J.J.; Sawhney, J.S.; Vatsaas, C.J.; Yorkgitis, B.K.; Kreiner, L.A.; Garcia, N.M.; Aziz, H.A. Thromboelastography and Rotational Thromboelastometry in Bleeding Patients with Coagulopathy: Practice Management Guideline from the Eastern Association for the Surgery of Trauma. J. Trauma Acute Care Surg. 2020, 89, 999–1017. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.; Thomas, S.; Kwaan, H.; Aversa, J.; Anderson, S.; Sundararajan, R.; Zimmer, D.; Bunch, C.; Stillson, J.; Draxler, D. Modern methods for monitoring hemorrhagic resuscitation in the United States: Why the delay? J. Trauma Acute Care Surg. 2020, 89, 1018–1022. [Google Scholar] [CrossRef]
- Walsh, M.M.; Khan, R.; Kwaan, H.C.; Neal, M.D. Fibrinolysis Shutdown in COVID-19-Associated Coagulopathy: A Crosstalk among Immunity, Coagulation, and Specialists in Medicine and Surgery. J. Am. Coll. Surg. 2021, 232, 1003–1006. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, M.; Kaplan, L.J.; Cannon, J.W. Thromboelastography-Guided Resuscitation of the Trauma Patient. JAMA Surg. 2019, 154, 1152–1153. [Google Scholar] [CrossRef] [PubMed]
- Moore, E.E.; Thomas, S.G.; Mjaess, N.; Bunch, C.M.; Walsh, M.M. Guiding Hemorrhagic Resuscitation With Viscoelastic Tests in the Emergency Department: A Call to Action in Emergency Medicine Education. Ann. Emerg. Med. 2021, 78, 462–463. [Google Scholar] [CrossRef] [PubMed]
- Maegele, M. Coagulopathy and Progression of Intracranial Hemorrhage in Traumatic Brain Injury: Mechanisms, Impact, and Therapeutic Considerations. Neurosurgery 2021, 162, 329–336. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Study Design (VET Used) | No. of Patients | Conclusions |
|---|---|---|---|
| Nekdulov et al., 2007 [84] | Prospective Observational (TEG-PM) | 20 isolated TBI (GCS < 8, AIS-non-head ≤ 3) | TBI patients had 78% AA inhibition compared to 27% AA inhibition for healthy controls. The 8/20 TBI patients that bled had a significantly greater AA inhibition than nonbleeders. |
| Solomon et al., 2011 [65] | Retrospective Observational (ROTEM, MEA) | 163 polytrauma | Mortality was correlated with low platelet aggregation by ADPtest, TRAPtest, and ROTEM platelet component contribution. |
| Wohlauer et al., 2012 [83] | Retrospective Observational (TEG-PM) | 10 polytrauma TBI | Patients with TBI had a median ADP inhibition of 89.4% and median AA inhibition of 40.1% despite normal platelet counts and INR. |
| Davis et al., 2013 [1] | Retrospective Observational (TEG-PM) | 50 isolated TBI (AIShead ≥ 3, AIS-non-head < 2) | The median ADP inhibition was >91.7% for nonsurvivors vs. 48.2% for survivors; however, this difference was not statistically significant. |
| Castellino et al., 2014 [53] | Retrospective Observational (TEG-PM) | 70 isolated TBI (AIShead ≥ 3, AIS-non-head < 2) | The median ADP receptor inhibition of all TBI patients was 64.5% vs. 15.5% in controls. For GCS ≤ 8, the median ADP inhibition was 93.1% vs. 56.5% for those with GCS > 8. The median AA inhibition of all TBI patients was 25.6% vs. 2.2% in healthy controls. |
| Daley et al., 2017 [151] | Retrospective Observational (TEG-PM) | 90 isolated and polytrauma TBI (AIShead ≥ 3) | Patients with ADP inhibition on TEG-PM had a higher in-hospital mortality rate (8% vs. 32%). After controlling for age, gender, hypotension, GCS, ISS, and preinjury antiplatelet use, ADP inhibition > 60% demonstrated a significant odds ratio for mortality. AA inhibition > 60% was not found to be significant. |
| Furay et al., 2018 [61] | Retrospective Case-Control (TEG-PM) | 35 isolated and polytrauma blunt TBI (AIShead ≥ 3) | Patients who received TEG-PM guided goal-directed platelet transfusion for ADP inhibition > 60% had a significantly lower mortality compared to those who received no platelet transfusions (9% vs. 35%). |
| Guillotte et al., 2018 [64] | Retrospective Observational (TEG-PM) | 153 TBI | ADP inhibition was greater in moderate/severe TBI compared to mild TBI. ADP inhibition was not found to be associated with mortality or intracerebral lesion expansion. There was no significant difference in the reduction of ADP inhibition with platelet transfusion compared to patients who did not receive platelet transfusion. |
| Kay et al., 2019 [144] | Retrospective Observational (TEG-PM) | 119 isolated TBI (AIShead ≥ 3, AIS-non-head < 2) | The median ADP inhibition was 18.4 points higher in severe TBI (AIShead = 4 or 5) compared to moderate TBI (AIShead = 3). Increased degree of ADP inhibition was also associated with increased odds of in-hospital mortality. |
| Furay et al., 2020 [62] | Retrospective Observational (TEG-PM) | 57 isolated and polytrauma blunt TBI with ICH (AIShead ≥ 3) | There was no difference in post-treatment ADP inhibition levels whether DDAVP alone or platelets alone were administered, guided by TEG-PM ADP inhibition > 60% as threshold for therapy. There was no significant difference in all-cause mortality between the two treatment groups. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bradbury, J.L.; Thomas, S.G.; Sorg, N.R.; Mjaess, N.; Berquist, M.R.; Brenner, T.J.; Langford, J.H.; Marsee, M.K.; Moody, A.N.; Bunch, C.M.; et al. Viscoelastic Testing and Coagulopathy of Traumatic Brain Injury. J. Clin. Med. 2021, 10, 5039. https://doi.org/10.3390/jcm10215039
Bradbury JL, Thomas SG, Sorg NR, Mjaess N, Berquist MR, Brenner TJ, Langford JH, Marsee MK, Moody AN, Bunch CM, et al. Viscoelastic Testing and Coagulopathy of Traumatic Brain Injury. Journal of Clinical Medicine. 2021; 10(21):5039. https://doi.org/10.3390/jcm10215039
Chicago/Turabian StyleBradbury, Jamie L., Scott G. Thomas, Nikki R. Sorg, Nicolas Mjaess, Margaret R. Berquist, Toby J. Brenner, Jack H. Langford, Mathew K. Marsee, Ashton N. Moody, Connor M. Bunch, and et al. 2021. "Viscoelastic Testing and Coagulopathy of Traumatic Brain Injury" Journal of Clinical Medicine 10, no. 21: 5039. https://doi.org/10.3390/jcm10215039
APA StyleBradbury, J. L., Thomas, S. G., Sorg, N. R., Mjaess, N., Berquist, M. R., Brenner, T. J., Langford, J. H., Marsee, M. K., Moody, A. N., Bunch, C. M., Sing, S. R., Al-Fadhl, M. D., Salamah, Q., Saleh, T., Patel, N. B., Shaikh, K. A., Smith, S. M., Langheinrich, W. S., Fulkerson, D. H., & Sixta, S. (2021). Viscoelastic Testing and Coagulopathy of Traumatic Brain Injury. Journal of Clinical Medicine, 10(21), 5039. https://doi.org/10.3390/jcm10215039