
Targeting Inflammatory Signaling in Prostate Cancer Castration Resistance


Abstract
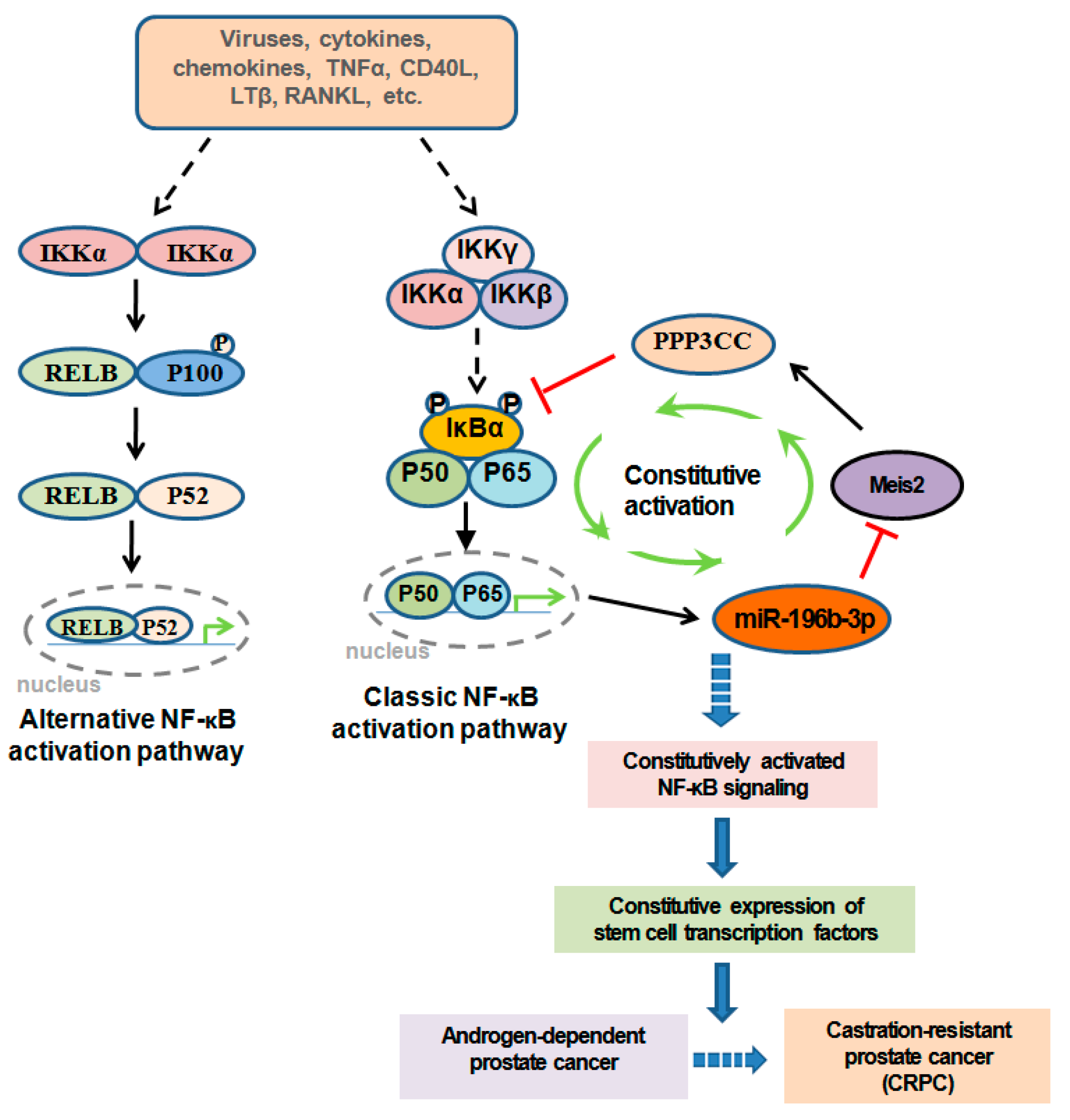
1. Introduction
2. Inflammatory Signaling in PCa Cells in PCa Castration Resistance
3. Inflammatory Signaling in the TME in PCa Castration Resistance
3.1. Inflammatory Signaling and Myeloid Cells
3.2. Inflammatory Signaling and Infiltrated Lymphocytes
3.3. Inflammatory Signaling and the ECM Network of the TME
4. Therapeutic Strategies for Targeting Inflammatory Signaling in CRPC
4.1. Imipramine
4.2. Artesunate
4.3. Pao Pereira Extract
4.4. Polyphyllin I (PPI)
4.5. CmpdA
4.6. EC-70124
4.7. Ursolic Acid
4.8. Apigenin
4.9. Retigeric Acid B
4.10. α-Tomatine
4.11. Simvastatin
4.12. Acacetin
4.13. Metformin
4.14. 3,3′-Diindolylmethane (DIM)
4.15. Betulinic Acid
4.16. Diosgenin
5. Conclusions and Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.J. Treatment effects in prostate cancer. Mod. Pathol. 2018, 31, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Jafari, S.; Molavi, O.; Kahroba, H.; Hejazi, M.S.; Maleki-Dizaji, N.; Barghi, S.; Kiaie, S.H.; Jadidi-Niaragh, F. Clinical application of immune checkpoints in targeted immunotherapy of prostate cancer. Cell. Mol. Life Sci. 2020, 77, 3693–3710. [Google Scholar] [CrossRef] [PubMed]
- Karantanos, T.; Corn, P.G.; Thompson, T.C. Prostate cancer progression after androgen deprivation therapy: Mechanisms of castrate resistance and novel therapeutic approaches. Oncogene 2013, 32, 5501–5511. [Google Scholar] [CrossRef]
- Amaral, T.M.S.; Macedo, D.; Fernandes, I.; Costa, L. Castration-Resistant Prostate Cancer: Mechanisms, Targets, and Treatment. Prostate Cancer 2012, 2012, 327253. [Google Scholar] [CrossRef] [PubMed]
- Thomas-Jardin, S.; Dahl, H.; Nawas, A.F.; Bautista, M.; Delk, N.A. NF-κB signaling promotes castration-resistant prostate cancer initiation and progression. Pharmacol. Ther. 2020, 211, 107538. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; He, B. Androgen Receptor Signaling in the Development of Castration-Resistant Prostate Cancer. Front. Oncol. 2019, 9, 858. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Jiang, X.; Liang, X.; Jiang, G. Molecular and cellular mechanisms of castration resistant prostate cancer (Review). Oncol. Lett. 2018, 15, 6063–6076. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Jeong, J.-H.; Huang, C.; Chen, X.; Dickinson, S.I.; Dhillon, J.; Yang, L.; Luo, J.-L. Targeting INMT and interrupting its methylation pathway for the treatment of castration resistant prostate cancer. J. Exp. Clin. Cancer Res. 2021, 40, 307. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Armstrong, C.M.; Ning, S.; Yang, J.C.; Lou, W.; Lombard, A.P.; Zhao, J.; Wu, C.-Y.; Yu, A.; Evans, C.P.; et al. ARVib suppresses growth of advanced prostate cancer via inhibition of androgen receptor signaling. Oncogene 2021, 40, 5379–5392. [Google Scholar] [CrossRef] [PubMed]
- Formaggio, N.; Rubin, M.A.; Theurillat, J.-P. Loss and revival of androgen receptor signaling in advanced prostate cancer. Oncogene 2021, 40, 1205–1216. [Google Scholar] [CrossRef]
- Ammirante, M.; Luo, J.-L.; Grivennikov, S.; Nedospasov, S.; Karin, M. B-cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nat. Cell Biol. 2010, 464, 302–305. [Google Scholar] [CrossRef]
- Rokavec, M.; Luo, J.-L. The transient and constitutive inflammatory signaling in tumorigenesis. Cell Cycle 2012, 11, 2587–2588. [Google Scholar] [CrossRef][Green Version]
- Rokavec, M.; Wu, W.; Luo, J.-L. IL6-Mediated Suppression of miR-200c Directs Constitutive Activation of Inflammatory Signaling Circuit Driving Transformation and Tumorigenesis. Mol. Cell 2012, 45, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Karin, M.; Sun, B. Targeting cancer-promoting inflammation–have anti-inflammatory therapies come of age? Nat. Rev. Clin. Oncol. 2021, 18, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.; Li, F.; Zhao, Z.; Zhang, Z.; Hu, J.; Zhang, Y. Tumor-Associated Macrophage Promotes the Survival of Cancer Cells upon Docetaxel Chemotherapy via the CSF1/CSF1R–CXCL12/CXCR4 Axis in Castration-Resistant Prostate Cancer. Genes 2021, 12, 773. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.-L.; Kamata, H.; Karin, M. The Anti-Death Machinery in IKK/NF-κB Signaling. J. Clin. Immunol. 2005, 25, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.-L.; Kamata, H.; Karin, M. IKK/NF- B signaling: Balancing life and death—A new approach to cancer therapy. J. Clin. Investig. 2005, 115, 2625–2632. [Google Scholar] [CrossRef] [PubMed]
- Archer, M.; Dogra, N.; Kyprianou, N. Inflammation as a Driver of Prostate Cancer Metastasis and Therapeutic Resistance. Cancers 2020, 12, 2984. [Google Scholar] [CrossRef] [PubMed]
- Kiely, M.; Ambs, S. Immune Inflammation Pathways as Therapeutic Targets to Reduce Lethal Prostate Cancer in African American Men. Cancers 2021, 13, 2874. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.-H.; Park, S.-J.; Dickinson, S.I.; Luo, J.-L. A Constitutive Intrinsic Inflammatory Signaling Circuit Composed of miR-196b, Meis2, PPP3CC, and p65 Drives Prostate Cancer Castration Resistance. Mol. Cell 2017, 65, 154–167. [Google Scholar] [CrossRef]
- Jeong, J.-H.; Dickinson, S.I.; Luo, J.-L. Targeting constitutive NF-κB specifically in tumor cells. Oncotarget 2017, 8, 93305–93306. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.-L.; Tan, W.; Ricono, J.M.; Korchynskyi, O.; Zhang, M.; Gonias, S.L.; Cheresh, D.A.; Karin, M. Nuclear cytokine-activated IKKα controls prostate cancer metastasis by repressing Maspin. Nat. Cell Biol. 2007, 446, 690–694. [Google Scholar] [CrossRef]
- Huang, H.; Du, T.; Xu, G.; Lai, Y.; Fan, X.; Chen, X.; Li, W.; Yue, F.; Li, Q.; Liu, L.; et al. Matrine suppresses invasion of castration-resistant prostate cancer cells by downregulating MMP-2/9 via NF-κB signaling pathway. Int. J. Oncol. 2017, 50, 640–648. [Google Scholar] [CrossRef]
- Gasparian, A.V.; Yao, Y.J.; Kowalczyk, D.; Lyakh, L.A.; Karseladze, A.; Slaga, T.J.; Budunova, I.V. The role of IKK in consti-tutive activation of NF-kappaB transcription factor in prostate carcinoma cells. J. Cell Sci. 2002, 115, 141–151. [Google Scholar] [CrossRef]
- Jin, R.J.; Lho, Y.; Connelly, L.; Wang, Y.; Yu, X.; Jean, L.S.; Case, T.C.; Ellwood-Yen, K.; Sawyers, C.L.; Bhowmick, N.A.; et al. The Nuclear Factor-κB Pathway Controls the Progression of Prostate Cancer to Androgen-Independent Growth. Cancer Res. 2008, 68, 6762–6769. [Google Scholar] [CrossRef]
- Meng, L.; Tian, Z.; Wang, J.; Liu, X.; Zhang, W.; Hu, M.; Wang, M.; Zhang, Y. Effect of myeloid ecotropic viral integration site (MEIS) family genes on tumor microenvironment remodeling and its potential therapeutic effect. Transl. Androl. Urol. 2021, 10, 594–608. [Google Scholar] [CrossRef] [PubMed]
- Jung, A.R.; Kim, G.E.; Kim, M.Y.; Ha, U.S.; Hong, S.H.; Lee, J.Y.; Kim, S.W.; Park, Y.H. HMGB1 promotes tumor progression and invasion through HMGB1/TNFR1/NF-kappaB axis in castration-resistant prostate cancer. Am. J. Cancer Res. 2021, 11, 2215–2227. [Google Scholar] [PubMed]
- Thapa, D.; Meng, P.; Bedolla, R.G.; Reddick, R.L.; Kumar, A.P.; Ghosh, R. NQO1 Suppresses NF-κB–p300 Interaction to Regulate Inflammatory Mediators Associated with Prostate Tumorigenesis. Cancer Res. 2014, 74, 5644–5655. [Google Scholar] [CrossRef]
- Kim, Y.-R.; Oh, K.-J.; Park, R.-Y.; Xuan, N.T.; Kang, T.-W.; Kwon, D.-D.; Choi, C.; Kim, M.S.; Nam, K.I.; Ahn, K.Y.; et al. HOXB13 promotes androgen independent growth of LNCaP prostate cancer cells by the activation of E2F signaling. Mol. Cancer 2010, 9, 124. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-R.; Kim, I.-J.; Kang, T.W.; Choi, C.; Kim, K.K.; Kim, M.S.; Nam, K.I.; Jung, C. HOXB13 downregulates intracellular zinc and increases NF-κB signaling to promote prostate cancer metastasis. Oncogene 2013, 33, 4558–4567. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Pitchiaya, S.; Cieślik, M.; Niknafs, Y.S.; Tien, J.C.-Y.; Hosono, Y.; Iyer, M.K.; Yazdani, S.; Subramaniam, S.; Shukla, S.; et al. Analysis of the androgen receptor–regulated lncRNA landscape identifies a role for ARLNC1 in prostate cancer progression. Nat. Genet. 2018, 50, 814–824. [Google Scholar] [CrossRef]
- Saha, S.; Kiran, M.; Kuscu, C.; Chatrath, A.; Wotton, D.; Mayo, M.W.; Dutta, A. Long Noncoding RNA DRAIC Inhibits Prostate Cancer Progression by Interacting with IKK to Inhibit NF-κB Activation. Cancer Res. 2020, 80, 950–963. [Google Scholar] [CrossRef] [PubMed]
- Shang, Z.; Yu, J.; Sun, L.; Tian, J.; Zhu, S.; Zhang, B.; Dong, Q.; Jiang, N.; Flores-Morales, A.; Chang, C.; et al. LncRNA PCAT1 activates AKT and NF-κB signaling in castration-resistant prostate cancer by regulating the PHLPP/FKBP51/IKKα complex. Nucleic Acids Res. 2019, 47, 4211–4225. [Google Scholar] [CrossRef]
- Thapa, D.; Ghosh, R. Chronic inflammatory mediators enhance prostate cancer development and progression. Biochem. Pharmacol. 2015, 94, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Araki, S.; Omori, Y.; Lyn, D.; Singh, R.K.; Meinbach, D.M.; Sandman, Y.; Lokeshwar, V.B.; Lokeshwar, B.L. Interleukin-8 Is a Molecular Determinant of Androgen Independence and Progression in Prostate Cancer. Cancer Res. 2007, 67, 6854–6862. [Google Scholar] [CrossRef]
- Wu, C.-T.; Hsieh, C.-C.; Lin, C.-C.; Chen, W.-C.; Hong, J.-H.; Chen, M.-F. Significance of IL-6 in the transition of hormone-resistant prostate cancer and the induction of myeloid-derived suppressor cells. J. Mol. Med. 2012, 90, 1343–1355. [Google Scholar] [CrossRef]
- Killian, P.H.; Kronski, E.; Michalik, K.M.; Barbieri, O.; Astigiano, S.; Sommerhoff, C.P.; Pfeffer, U.; Nerlich, A.G.; Bachmeier, B. Curcumin inhibits prostate cancer metastasis in vivo by targeting the inflammatory cytokines CXCL1 and -2. Carcinogenesis 2012, 33, 2507–2519. [Google Scholar] [CrossRef]
- Sharma, J.; Gray, K.P.; Harshman, L.C.; Evan, C.; Nakabayashi, M.; Fichorova, R.; Rider, J.; Mucci, L.; Kantoff, P.; Sweeney, C.J. Elevated IL-8, TNF-α, and MCP-1 in men with metastatic prostate cancer starting androgen-deprivation therapy (ADT) are associated with shorter time to castration-resistance and overall survival. Prostate 2014, 74, 820–828. [Google Scholar] [CrossRef]
- Nunes, J.J.; Pandey, S.K.; Yadav, A.; Goel, S.; Ateeq, B. Targeting NF-kappa B Signaling by Artesunate Restores Sensitivity of Castrate-Resistant Prostate Cancer Cells to Antiandrogens. Neoplasia 2017, 19, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Zhong, S.; Jeong, J.-H.; Chen, Z.; Chen, Z.; Luo, J.-L. Targeting Tumor Microenvironment by Small-Molecule Inhibitors. Transl. Oncol. 2020, 13, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Fujita, K.; Matsushita, M.; Nonomura, N. Main Inflammatory Cells and Potentials of Anti-Inflammatory Agents in Prostate Cancer. Cancers 2019, 11, 1153. [Google Scholar] [CrossRef] [PubMed]
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting Tumor-Associated Macrophages in Cancer. Trends Immunol. 2019, 40, 310–327. [Google Scholar] [CrossRef]
- Cheng, N.; Bai, X.; Shu, Y.; Ahmad, O.; Shen, P. Targeting tumor-associated macrophages as an antitumor strategy. Biochem. Pharmacol. 2021, 183, 114354. [Google Scholar] [CrossRef]
- Sica, A.; Allavena, P.; Mantovani, A. Cancer related inflammation: The macrophage connection. Cancer Lett. 2008, 267, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, D.; Cang, H.; Guo, B. Crosstalk between cancer and immune cells: Role of tumor-associated macrophages in the tumor microenvironment. Cancer Med. 2019, 8, 4709–4721. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Luo, Z.; Li, X.; Han, X.; Shi, S.; Zhang, T. Tumor-associated macrophages: Role in tumorigenesis and immunotherapy implications. J. Cancer 2021, 12, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Nonomura, N.; Satoh, E.; Harada, Y.; Nakayama, M.; Tokizane, T.; Fukui, T.; Ono, Y.; Inoue, H.; Shin, M.; et al. Potential mechanism for the effects of dexamethasone on growth of androgen-independent prostate cancer. J. Natl. Cancer Inst. 2001, 93, 1739–1746. [Google Scholar] [CrossRef]
- Nonomura, N.; Takayama, H.; Nakayama, M.; Nakai, Y.; Kawashima, A.; Mukai, M.; Nagahara, A.; Aozasa, K.; Tsujimura, A. Infiltration of tumour-associated macrophages in prostate biopsy specimens is predictive of disease progression after hormonal therapy for prostate cancer. BJU Int. 2010, 107, 1918–1922. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Qian, Y.; Yu, F.; Liu, W.; Wu, Y.; Fang, X.; Hao, W. Alternatively activated macrophages are associated with metastasis and poor prognosis in prostate adenocarcinoma. Oncol. Lett. 2015, 10, 1390–1396. [Google Scholar] [CrossRef] [PubMed]
- Di Mitri, D.; Mirenda, M.; Vasilevska, J.; Calcinotto, A.; Delaleu, N.; Revandkar, A.; Gil, V.; Boysen, G.; Losa, M.; Mosole, S.; et al. Re-education of Tumor-Associated Macrophages by CXCR2 Blockade Drives Senescence and Tumor Inhibition in Advanced Prostate Cancer. Cell Rep. 2019, 28, 2156–2168. [Google Scholar] [CrossRef]
- Hagemann, T.; Lawrence, T.; McNeish, I.; Charles, K.A.; Kulbe, H.; Thompson, R.G.; Robinson, S.C.; Balkwill, F. “Re-educating” tumor-associated macrophages by targeting NF-κB. J. Exp. Med. 2008, 205, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Xue, W.; Wang, Q.-M.; Lian, G.-Y.; Huang, X.-R.; Lee, T.-L.; To, K.-F.; Tang, P.M.-K.; Lan, H.-Y. The Mincle/Syk/NF-κB Signaling Circuit Is Essential for Maintaining the Protumoral Activities of Tumor-Associated Macrophages. Cancer Immunol. Res. 2020, 8, 1004–1017. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, B.; Li, X.; Zhao, X.; Wan, L.; Lin, G.; Yu, M.; Wang, J.; Jiang, X.; Feng, W.; et al. Transmembrane TNF-α Promotes Suppressive Activities of Myeloid-Derived Suppressor Cells via TNFR2. J. Immunol. 2014, 192, 1320–1331. [Google Scholar] [CrossRef]
- Craig, M.; Ying, C.; Loberg, R.D. Co-inoculation of prostate cancer cells with U937 enhances tumor growth and angiogenesis in vivo. J. Cell. Biochem. 2008, 103, 1–8. [Google Scholar] [CrossRef]
- Watanabe, M.; Kanao, K.; Suzuki, S.; Muramatsu, H.; Morinaga, S.; Kajikawa, K.; Kobayashi, I.; Nishikawa, G.; Kato, Y.; Zennami, K.; et al. Increased infiltration of CCR4-positive regulatory T cells in prostate cancer tissue is associated with a poor prognosis. Prostate 2019, 79, 1658–1665. [Google Scholar] [CrossRef] [PubMed]
- Ji, Z.; Zhao, W.; Lin, H.-K.; Zhou, X. Systematically understanding the immunity leading to CRPC progression. PLoS Comput. Biol. 2019, 15, e1007344. [Google Scholar] [CrossRef] [PubMed]
- Obradovic, A.; Dallos, M.C.; Zahurak, M.L.; Partin, A.W.; Schaeffer, E.M.; Ross, A.E.; Allaf, M.E.; Nirschl, T.R.; Liu, D.; Chapman, C.G.; et al. T-Cell Infiltration and Adaptive Treg Resistance in Response to Androgen Deprivation With or Without Vaccination in Localized Prostate Cancer. Clin. Cancer Res. 2020, 26, 3182–3192. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Chen, H.; Luo, W.; Zhang, H.; Li, G.; Zeng, F.; Deng, F. The Landscape of Immune Cells Infiltrating in Prostate Cancer. Front. Oncol. 2020, 10, 517637. [Google Scholar] [CrossRef]
- Ammirante, M.; Kuraishy, A.I.; Shalapour, S.; Strasner, A.; Ramirez-Sanchez, C.; Zhang, W.; Shabaik, A.; Karin, M. An IKK -E2F1-BMI1 cascade activated by infiltrating B cells controls prostate regeneration and tumor recurrence. Genes Dev. 2013, 27, 1435–1440. [Google Scholar] [CrossRef] [PubMed]
- Shalapour, S.; Font-Burgada, J.; Di Caro, G.; Zhong, Z.; Sanchez-Lopez, E.; Dhar, D.; Willimsky, G.; Ammirante, M.; Strasner, A.; Hansel, D.E.; et al. Immunosuppressive plasma cells impede T-cell-dependent immunogenic chemotherapy. Nat. Cell Biol. 2015, 521, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Viselli, S.M.; Reese, K.R.; Fan, J.; Kovacsac, W.J.; Olsen, N.J. Androgens Alter B Cell Development in Normal Male Mice. Cell. Immunol. 1997, 182, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, B.C. Androgen Signaling in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2021, 1270, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Mrose, S.; Thomas, D. Enhanced production of B lymphocytes after castration. Blood 1995, 85, 1535–1539. [Google Scholar] [CrossRef]
- Ellis, T.M.; Moser, M.T.; Le, P.T.; Flanigan, R.C.; Kwon, E.D. Alterations in peripheral B cells and B cell progenitors following androgen ablation in mice. Int. Immunol. 2001, 13, 553–558. [Google Scholar] [CrossRef]
- Altuwaijri, S.; Chuang, K.-H.; Lai, K.-P.; Lai, J.-J.; Lin, H.-Y.; Young, F.M.; Bottaro, A.; Tsai, M.-Y.; Zeng, W.-P.; Chang, H.-C.; et al. Susceptibility to Autoimmunity and B Cell Resistance to Apoptosis in Mice Lacking Androgen Receptor in B Cells. Mol. Endocrinol. 2009, 23, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Mora, L.B.; Buettner, R.; Seigne, J.; Diaz, J.; Ahmad, N.; Garcia, R.; Bowman, T.; Falcone, R.; Fairclough, R.; Cantor, A.; et al. Constitutive activation of Stat3 in human prostate tumors and cell lines: Direct inhibition of Stat3 signaling induces apoptosis of prostate cancer cells. Cancer Res. 2002, 62, 6659–6666. [Google Scholar] [PubMed]
- Weiner, A.B.; Vidotto, T.; Liu, Y.; Mendes, A.A.; Salles, D.C.; Faisal, F.A.; Murali, S.; McFarlane, M.; Imada, E.L.; Zhao, X.; et al. Plasma cells are enriched in localized prostate cancer in Black men and are associated with improved outcomes. Nat. Commun. 2021, 12, 935. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.F.; Cui, J.W. The Role of Tumor-Infiltrating B Cells in Tumor Immunity. J. Oncol. 2019, 2019, 2592419. [Google Scholar] [CrossRef]
- Wang, S.-S.; Liu, W.; Ly, D.; Xu, H.; Qu, L.; Zhang, L. Tumor-infiltrating B cells: Their role and application in anti-tumor immunity in lung cancer. Cell. Mol. Immunol. 2019, 16, 6–18. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Barreto, J.B.; Andreu, P.; Vasquez, L.; Tawfik, D.; Kolhatkar, N.; Coussens, L.M. CD4+ T Cells Regulate Pulmonary Metastasis of Mammary Carcinomas by Enhancing Protumor Properties of Macrophages. Cancer Cell 2009, 16, 91–102. [Google Scholar] [CrossRef]
- Mousset, C.M.; Hobo, W.; Woestenenk, R.; Preijers, F.; Dolstra, H.; Van Der Waart, A.B. Comprehensive Phenotyping of T Cells Using Flow Cytometry. Cytom. Part A 2019, 95, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Malek, T.R.; Castro, I. Interleukin-2 Receptor Signaling: At the Interface between Tolerance and Immunity. Immunity 2010, 33, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Lescarbeau, R.M.; Seib, F.P.; Prewitz, M.; Werner, C.; Kaplan, D.L. In Vitro Model of Metastasis to Bone Marrow Mediates Prostate Cancer Castration Resistant Growth through Paracrine and Extracellular Matrix Factors. PLoS ONE 2012, 7, e40372. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Xie, R.; Zhang, Z.; Liu, Q.; Wu, S.; Cui, Y. Identification of SPP1 as an Extracellular Matrix Signature for Metastatic Castration-Resistant Prostate Cancer. Front. Oncol. 2019, 9, 924. [Google Scholar] [CrossRef]
- Kim, S.; Kang, M.; Ko, J. Small leucine zipper protein promotes the metastasis of castration-resistant prostate cancer through transcriptional regulation of matrix metalloproteinase-13. Carcinogenesis 2021, 42, 1089–1099. [Google Scholar] [CrossRef] [PubMed]
- Bonfil, R.D.; Dong, Z.; Filho, J.C.T.; Sabbota, A.; Osenkowski, P.; Nabha, S.; Yamamoto, H.; Chinni, S.R.; Zhao, H.; Mobashery, S.; et al. Prostate Cancer-Associated Membrane Type 1-Matrix Metalloproteinase: A Pivotal Role in Bone Response and Intraosseous Tumor Growth. Am. J. Pathol. 2007, 170, 2100–2111. [Google Scholar] [CrossRef]
- Jabłońska-Trypuć, A.; Matejczyk, M.; Rosochacki, S. Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. J. Enzym. Inhib. Med. Chem. 2016, 31, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Zhang, N.; Feng, Y.; Cao, J.; Chen, X.; Liu, B. Aspirin Inhibits IKK-β-mediated Prostate Cancer Cell Invasion by Targeting Matrix Metalloproteinase-9 and Urokinase-Type Plasminogen Activator. Cell. Physiol. Biochem. 2017, 41, 1313–1324. [Google Scholar] [CrossRef] [PubMed]
- Alshyarba, M.; Otifi, H.; Al Fayi, M.; Dera, A.A.; Rajagopalan, P. Thymoquinone inhibits IL-7-induced tumor progression and metastatic invasion in prostate cancer cells by attenuating matrix metalloproteinase activity and Akt/NF-κB signaling. Biotechnol. Appl. Biochem. 2020. [Google Scholar] [CrossRef]
- Lim, E.Y.; Park, J.; Kim, Y.T.; Kim, M.J. Imipramine Inhibits Migration and Invasion in Metastatic Castration-Resistant Prostate Cancer PC-3 Cells via AKT-Mediated NF-κB Signaling Pathway. Molecules 2020, 25, 4619. [Google Scholar] [CrossRef]
- Chang, C.; Zhao, W.; Xie, B.; Deng, Y.; Han, T.; Cui, Y.; Dai, Y.; Zhang, Z.; Gao, J.; Guo, H.; et al. Pao Pereira Extract Suppresses Castration-Resistant Prostate Cancer Cell Growth, Survival, and Invasion Through Inhibition of NFκB Signaling. Integr. Cancer Ther. 2014, 13, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.; Zou, P.; Wu, J.; Zheng, F.; Tang, Q.; Zhou, J.; Hann, S.S. Crosstalk of NF-κB/P65 and LncRNA HOTAIR-Mediated Repression of MUC1 Expression Contribute to Synergistic Inhibition of Castration-Resistant Prostate Cancer by Polyphyllin 1–Enzalutamide Combination Treatment. Cell. Physiol. Biochem. 2018, 47, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lapidus, R.G.; Liu, P.; Choi, E.Y.; Adediran, S.; Hussain, A.; Wang, X.; Liu, X.; Dan, H.C. Targeting IκB Kinase β/NF-κB Signaling in Human Prostate Cancer by a Novel IκB Kinase β Inhibitor CmpdA. Mol. Cancer Ther. 2016, 15, 1504–1514. [Google Scholar] [CrossRef] [PubMed]
- Civenni, G.; Longoni, N.; Costales, P.; Dallavalle, C.; Inclán, C.G.; Albino, D.; Nuñez, L.E.; Morís, F.; Carbone, G.M.; Catapano, C.V. EC-70124, a Novel Glycosylated Indolocarbazole Multikinase Inhibitor, Reverts Tumorigenic and Stem Cell Properties in Prostate Cancer by Inhibiting STAT3 and NF-κB. Mol. Cancer Ther. 2016, 15, 806–818. [Google Scholar] [CrossRef]
- Shanmugam, M.K.; Rajendran, P.; Li, F.; Nema, T.; Vali, S.; Abbasi, T.; Kapoor, S.; Sharma, A.; Kumar, A.P.; Ho, P.C.; et al. Ursolic acid inhibits multiple cell survival pathways leading to suppression of growth of prostate cancer xenograft in nude mice. J. Mol. Med. 2011, 89, 713–727. [Google Scholar] [CrossRef]
- Shukla, S.; Gupta, S. Suppression of Constitutive and Tumor Necrosis Factor α-Induced Nuclear Factor (NF)-κB Activation and Induction of Apoptosis by Apigenin in Human Prostate Carcinoma PC-3 Cells: Correlation with Down-Regulation of NF-κB-Responsive Genes. Clin. Cancer Res. 2004, 10, 3169–3178. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-Q.; Hu, X.-Y.; Lu, T.; Cheng, Y.-N.; Young, C.Y.F.; Yuan, H.-Q.; Lou, H.-X. Retigeric Acid B Exhibits Antitumor Activity through Suppression of Nuclear Factor-κB Signaling in Prostate Cancer Cells in Vitro and in Vivo. PLoS ONE 2012, 7, e38000. [Google Scholar] [CrossRef]
- Lee, S.-T.; Wong, P.-F.; He, H.; Hooper, J.D.; Mustafa, M.R. Alpha-Tomatine Attenuation of In Vivo Growth of Subcutaneous and Orthotopic Xenograft Tumors of Human Prostate Carcinoma PC-3 Cells Is Accompanied by Inactivation of Nuclear Factor-Kappa B Signaling. PLoS ONE 2013, 8, e57708. [Google Scholar] [CrossRef]
- Lee, S.-T.; Wong, P.-F.; Cheah, S.-C.; Mustafa, M.R. Alpha-Tomatine Induces Apoptosis and Inhibits Nuclear Factor-Kappa B Activation on Human Prostatic Adenocarcinoma PC-3 Cells. PLoS ONE 2011, 6, e18915. [Google Scholar] [CrossRef]
- Park, Y.H.; Seo, S.Y.; Lee, E.; Ku, J.H.; Kim, H.H.; Kwak, C. Simvastatin Induces Apoptosis in Castrate Resistant Prostate Cancer Cells by Deregulating Nuclear Factor-κB Pathway. J. Urol. 2013, 189, 1547–1552. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Park, C.G.; Jung, J.Y. Acacetin (5,7-dihydroxy-4′-methoxyflavone) exhibits in vitro and in vivo anticancer activity through the suppression of NF-κB/Akt signaling in prostate cancer cells. Int. J. Mol. Med. 2014, 33, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Ge, R.; Wang, Z.; Wu, S.; Zhuo, Y.; Otsetov, A.G.; Cai, C.; Zhong, W.; Wu, C.-L.; Olumi, A.F. Metformin represses cancer cells via alternate pathways in N-cadherin expressing vs. N-cadherin deficient cells. Oncotarget 2015, 6, 28973–28987. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, Q.; Tong, D.; Liu, G.; Gao, J.; Wang, L.-A.; Xu, J.; Yang, X.; Xie, Q.; Huang, Y.; Pang, J.; et al. Metformin Inhibits Prostate Cancer Progression by Targeting Tumor-Associated Inflammatory Infiltration. Clin. Cancer Res. 2018, 24, 5622–5634. [Google Scholar] [CrossRef]
- Rahman, K.W.; Banerjee, S.; Ali, S.; Ahmad, A.; Wang, Z.; Kong, D.; Sakr, W.A. 3,3′-Diindolylmethane Enhances Taxotere-Induced Apoptosis in Hormone-Refractory Prostate Cancer Cells through Survivin Down-regulation. Cancer Res. 2009, 69, 4468–4475. [Google Scholar] [CrossRef] [PubMed]
- Rabi, T.; Shukla, S.; Gupta, S. Betulinic acid suppresses constitutive and TNFα-induced NF-κB activation and induces apoptosis in human prostate carcinoma PC-3 cells. Mol. Carcinog. 2008, 47, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Bilusic, M.; Heery, C.R.; Collins, J.M.; Donahue, R.N.; Palena, C.; Madan, R.A.; Karzai, F.; Marté, J.L.; Strauss, J.; Gatti-Mays, M.E.; et al. Phase I trial of HuMax-IL8 (BMS-986253), an anti-IL-8 monoclonal antibody, in patients with metastatic or unresectable solid tumors. J. Immunother. Cancer 2019, 7, 240. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-S.; Shih, Y.-W.; Huang, H.-C.; Cheng, H.-W. Diosgenin, a Steroidal Saponin, Inhibits Migration and Invasion of Human Prostate Cancer PC-3 Cells by Reducing Matrix Metalloproteinases Expression. PLoS ONE 2011, 6, e20164. [Google Scholar] [CrossRef]
- Crespo-Ortiz, M.P.; Wei, M.Q. Antitumor Activity of Artemisinin and Its Derivatives: From a Well-Known Antimalarial Agent to a Potential Anticancer Drug. J. Biomed. Biotechnol. 2012, 2012, 247597. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.; Ganapathi, S.; Ster, I.C.; Saeed, M.E.; Cowan, M.; Finlayson, C.; Kovacsevics, H.; Jansen, H.; Kremsner, P.G.; Efferth, T.; et al. A Randomised, Double Blind, Placebo-Controlled Pilot Study of Oral Artesunate Therapy for Colorectal Cancer. EBioMedicine 2015, 2, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Von Hagens, C.; Walter-Sack, I.; Goeckenjan, M.; Storch-Hagenlocher, B.; Sertel, S.; Elsässer, M.; Remppis, B.A.; Munzinger, J.; Edler, L.; Efferth, T.; et al. Long-term add-on therapy (compassionate use) with oral artesunate in patients with metastatic breast cancer after participating in a phase I study (ARTIC M33/2). Phytomedicine 2019, 54, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Deeken, J.F.; Wang, H.; Hartley, M.; Cheema, A.K.; Smaglo, B.; He, A.R.; Weiner, L.M.; Marshall, J.L.; Giaccone, G.; Liu, S.; et al. A phase I study of intravenous artesunate in patients with advanced solid tumor malignancies. Cancer Chemother. Pharmacol. 2018, 81, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Cuenca-López, M.D.; Serrano-Heras, G.; Montero, J.C.; Sánchez, V.C.; Gomez-Juarez, M.; Gascón-Escribano, M.J.; Morales, J.C.; Voisin, V.; Núñez, L.E.; Morís, F.; et al. Antitumor activity of the novel multi-kinase inhibitor EC-70124 in triple negative breast cancer. Oncotarget 2015, 6, 27923–27937. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sánchez, C.; Salas, A.P.; Braña, A.F.; Palomino, M.; Pineda-Lucena, A.; Carbajo, R.J.; Méndez, C.; Moris, F.; Salas, J.A. Generation of potent and selective kinase inhibitors by combinatorial biosynthesis of glycosylated indolocarbazoles. Chem. Commun. 2009, 27, 4118–4120. [Google Scholar] [CrossRef] [PubMed]
- Kassi, E.; Papoutsi, Z.; Pratsinis, H.; Aligiannis, N.; Manoussakis, M.; Moutsatsou, P. Ursolic acid, a naturally occurring triterpenoid, demonstrates anticancer activity on human prostate cancer cells. J. Cancer Res. Clin. Oncol. 2007, 133, 493–500. [Google Scholar] [CrossRef]
- Manu, K.; Kuttan, G. Ursolic acid induces apoptosis by activating p53 and caspase-3 gene expressions and suppressing NF-κB mediated activation of bcl-2 in B16F-10 melanoma cells. Int. Immunopharmacol. 2008, 8, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Santockyte, R.; Yu, S.; Shen, R.-F.; Tekle, E.; Lee, C.; Yang, D.C.; Chock, P.B. FAT10 modifies p53 and upregulates its transcriptional activity. Arch. Biochem. Biophys. 2011, 509, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Lee, K.-H.; Lee, H.S.; Jeong, C.W.; Ku, J.H.; Kim, H.H.; Kwak, C. Concurrent treatment with simvastatin and NF-κB inhibitor in human castration-resistant prostate cancer cells exerts synergistic anti-cancer effects via control of the NF-κB/LIN28/let-7 miRNA signaling pathway. PLoS ONE 2017, 12, e0184644. [Google Scholar] [CrossRef]
- Kim, Y.; Kim, T.W.; Han, S.W.; Ahn, J.B.; Kim, S.T.; Lee, J.; Park, J.O.; Park, Y.S.; Lim, H.Y.; Kang, W.K. A Single Arm, Phase II Study of Simvastatin Plus XELOX and Bevacizumab as First-Line Chemotherapy in Metastatic Colorectal Cancer Patients. Cancer Res. Treat. 2019, 51, 1128–1134. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.T.; Kang, J.H.; Lee, J.; Park, S.H.; Park, J.O.; Park, Y.S.; Lim, H.Y.; Hwang, I.G.; Lee, S.-C.; Park, K.-W.; et al. Simvastatin plus capecitabine–cisplatin versus placebo plus capecitabine–cisplatin in patients with previously untreated advanced gastric cancer: A double-blind randomised phase 3 study. Eur. J. Cancer 2014, 50, 2822–2830. [Google Scholar] [CrossRef] [PubMed]
- Rothermundt, C.; Hayoz, S.; Templeton, A.J.; Winterhalder, R.; Strebel, R.T.; Bärtschi, D.; Pollak, M.; Lui, L.; Endt, K.; Schiess, R.; et al. Metformin in Chemotherapy-naive Castration-resistant Prostate Cancer: A Multicenter Phase 2 Trial (SAKK 08/09). Eur. Urol. 2014, 66, 468–474. [Google Scholar] [CrossRef]
- Heath, E.I.; Heilbrun, L.K.; Li, J.; Vaishampayan, U.; Harper, F.; Pemberton, P.; Sarkar, F.H. A phase I dose-escalation study of oral BR-DIM (BioResponse 3,3′- Diindolylmethane) in castrate-resistant, non-metastatic prostate cancer. Am. J. Transl. Res. 2010, 2, 402–411. [Google Scholar] [PubMed]
- Kaur, H.B.; Guedes, L.B.; Lu, J.; Maldonado, L.; Reitz, L.; Barber, J.R.; De Marzo, A.M.; Tosoian, J.J.; Tomlins, S.A.; Schaeffer, E.M.; et al. Association of tumor-infiltrating T-cell density with molecular subtype, racial ancestry and clinical outcomes in prostate cancer. Mod. Pathol. 2018, 31, 1539–1552. [Google Scholar] [CrossRef] [PubMed]
- Petitprez, F.; Fossati, N.; Vano, Y.; Freschi, M.; Becht, E.; Lucianò, R.; Calderaro, J.; Guédet, T.; Lacroix, L.; Rancoita, P.; et al. PD-L1 Expression and CD8+ T-cell Infiltrate are Associated with Clinical Progression in Patients with Node-positive Prostate Cancer. Eur. Urol. Focus 2019, 5, 192–196. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Target | Method or Inhibitor | Target Strategy | Mechanism of Action | Reference |
|---|---|---|---|---|
| NF-κB signaling pathway | Imipramine | attenuates PC-3 cell proliferation and inhibits migration and invasion in PC-3 cells | decrease p-IKK, p-IκBα, and p-p65 | [83] |
| NF-κB signaling and AR and/or AR-variant 7 expression | Artesunate (combined with bicalutamide) | sensitize CRPC cells to antagonists, lead to tumor regression and reduce lungs and bone metastases | inhibition of NF-κB signaling and decrease AR and/or AR-variant 7 expression | [41] |
| NF-κB signaling pathway | Pao pereira extract | suppress CRPC PC3 cell growth, migration, and invasion | inhibit relocation of NF-κB/p65 in cells and NF-κB/p65 transcription activity | [84] |
| NF-κB/p65 | Polyphyllin I (PPI) (combined with enzalutamide) | suppress CRPC cell growth and tumor development | decrease p65 and MUC1 protein expression as well as lncRNA HOX transcript antisense RNA (HOTAIR) expression | [85] |
| IKKβ | CmpdA (synergize with docetaxel) | inhibit cell proliferation, migration, and stemness and induction of apoptosis; suppress tumor growth | inhibit constitutively activated IKKβ/NF-κB signaling and IKKβ/Nanog signaling | [86] |
| NF-κB and STAT3 | EC-70124 | decrease cell proliferation, migration, and colony formation of DUI45; suppress tumor growth | block concomitant activation of NF-κB and STAT3 | [87] |
| IκB kinase (IKK) | Ursolic acid | decrease DUI45 cell proliferation, suppress tumor growth | inhibit TNFα-induced and constitutive IKK activation as well as NF-κB-dependent reporter activity | [88] |
| IKKα kinase | Apigenin | sensitize PC3 cells to TNFα-induced apoptosis | decrease IKKα kinase activity and inhibit IκBα degradation and IκBα phosphorylation | [89] |
| IκBα and p65 | Retigeric acid B | inhibit cell proliferation and tumor growth in PC3 and DU145 cells models | inhibit phosphorylation levels of IκBα and p65, and block the translocation of p65 to the nucleus and its DNA binding activity | [90] |
| IκBα kinase | α-tomatine | inhibit cell proliferation and attenuate the growth of subcutaneous and orthotopic xenograft tumors of PC3 | inhibit IKK kinase activity, resulting in sequential suppression of IκBα phosphorylation, IκBα degradation, NF-κB/p65 phosphorylation, and NF-κB p50/p65 nuclear translocation; reduce TNFα-induced activation of the pro-survival mediator Akt | [91,92] |
| IκBα | Simvastatin | inhibit cell growth and induce apoptosis in PC3 and DU145 cells | inhibit IκBα phosphorylation and degradation and reduce phosphorylated p65 protein levels in nuclear fractions | [93] |
| IκBα | Acacetin | decrease cell viability of DUI45; suppress tumor growth | inhibit the phosphorylation of IκBα to suppress Akt and NF-κB signaling pathways | [94] |
| p65 as well as tumor-associated inflammatory infiltration | Metformin | decrease cell proliferation of PC3; suppress tumor growth; repress PCa progression | suppress N-cadherin and p65 accumulation; inhibit the COX2/PGE2 axis | [95,96] |
| NF-κB DNA-binding activity | 3,3′-diindolylmethane (DIM) (synergize Taxotere) | inhibit cell growth and induce apoptosis in C4-2B, and inhibit C4-2B bone tumor growth | decrease in survivin expression and NF-κB DNA-binding activity | [97] |
| IκBα and NF-κB DNA-binding activity | Betulinic acid | inhibit cell growth and induce apoptosis in PC3 cells | decrease IKK activity and phosphorylation of IκBα; inhibit DNA binding and nuclear levels of the NF-κB/p65 | [98] |
| feedback loop between pro-inflammatory cytokines CXCL1/2 and NF-κB | Curcumin | inhibit cell growth and metastasis in a PC3 cell model | abolish the feedback loop between pro-inflammatory cytokines CXCL1/2 and NF-κB through inhibiting IKKβ activation and NF-κB nuclear translocation | [39] |
| CXCL2-CXCR2 pathway in TAMs | AZD5069 | lead to PCa cell senescence and inhibition of tumor progression | disrupt the CXCL2-CXCR2 pathway and trigger re-education of TAMs toward a pro-inflammatory state | [53] |
| IL8-stimulated NF-κB signaling | HuMaxIL8 (Anti-IL-8 antibodies) | inhibit PCa cell growth | attenuate activation of NF-κB (p65) | [37,99] |
| lncRNA-PCAT1-PHLPP/FKBP51/IKKα complex | lncRNA-PCAT1 shRNA | inhibit androgen-independentcell growth and CRPC progression | Prevent activation of AKT as well as NF-κB signaling | [35] |
| subunits of the IKK complex | lncRNA-DRAIC | inhibit cell invasion, soft agar colony formation, and tumor growth | inhibit interaction with the IKK complex, the phosphorylation of IκBa, and the activation of NF-κB | [34] |
| IKKβ-mediated MMP-9 and urokinase-type plasminogen activator | Aspirin | suppress invasion and attachment of DU145 and PC3 cells | decrease in inhibitors of κB IκBα phosphorylation, NF-κB p65 to nuclear translocation and IKKβ activation, leading to reduced MMP-9 activity and uPA and PAI-1 expression | [81] |
| IL-7/Akt/NF-κB signaling | Thymoquinone | inhibit IL-7-induced tumor progression and metastatic invasion in DU145 cells | down-regulate p-Akt and NF-κB, reduce the levels of MMP-3 and MMP-7 | [82] |
| MMP-2, MMP-9, and NF-κB activity | Diosgenin | inhibit proliferation, cell migration, and invasion in PC3 cells | reduce the activities and expression of matrix metalloproteinase-2 (MMP-2) and MMP-9; suppress JNK, ERK, and PI3K/Akt signaling as well as NF-κB activity | [100] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhong, S.; Huang, C.; Chen, Z.; Chen, Z.; Luo, J.-L. Targeting Inflammatory Signaling in Prostate Cancer Castration Resistance. J. Clin. Med. 2021, 10, 5000. https://doi.org/10.3390/jcm10215000
Zhong S, Huang C, Chen Z, Chen Z, Luo J-L. Targeting Inflammatory Signaling in Prostate Cancer Castration Resistance. Journal of Clinical Medicine. 2021; 10(21):5000. https://doi.org/10.3390/jcm10215000
Chicago/Turabian StyleZhong, Shangwei, Changhao Huang, Zhikang Chen, Zihua Chen, and Jun-Li Luo. 2021. "Targeting Inflammatory Signaling in Prostate Cancer Castration Resistance" Journal of Clinical Medicine 10, no. 21: 5000. https://doi.org/10.3390/jcm10215000
APA StyleZhong, S., Huang, C., Chen, Z., Chen, Z., & Luo, J.-L. (2021). Targeting Inflammatory Signaling in Prostate Cancer Castration Resistance. Journal of Clinical Medicine, 10(21), 5000. https://doi.org/10.3390/jcm10215000