
Autoimmunity in Primary Immunodeficiency Disorders: An Updated Review on Pathogenic and Clinical Implications



Abstract
1. Introduction
2. Autoimmunity in Primary Antibody Deficiency Disorders
2.1. Common Variable Immunodeficiency
2.2. Selective IgA Deficiency
2.3. Hyper IgM Syndrome
2.4. X-Linked Agammaglobulinemia
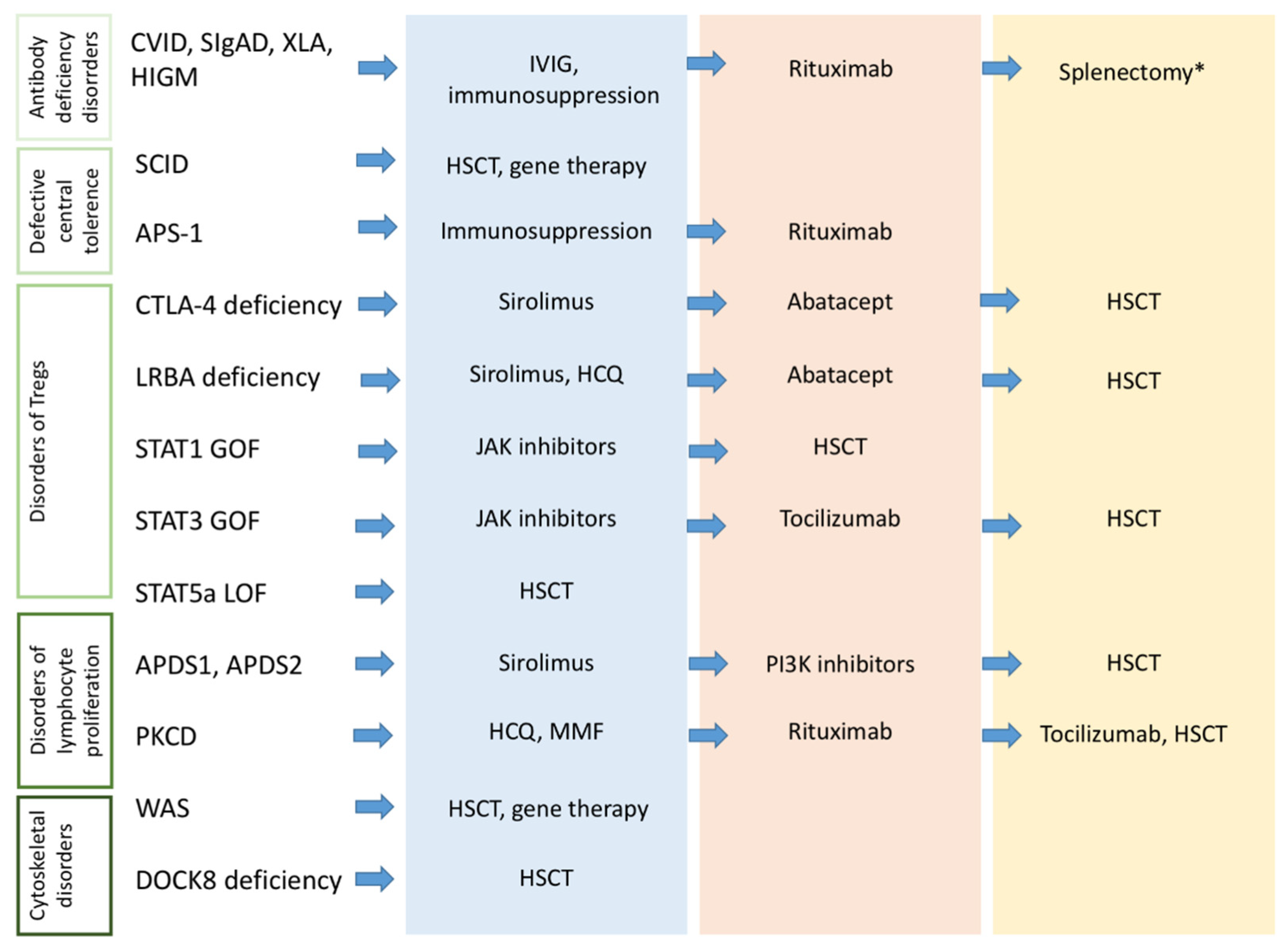
2.5. Therapeutic Approach to Autoimmunity in Primary Antibody Deficiencies
3. Autoimmunity in Severe Combined Immunodeficiency and Related Disorders
4. Autoimmunity in Disorders of T-Cell Central Tolerance
4.1. Autoimmune Polyendocrine Syndrome 1
4.2. 22q11.2 Deletion Syndrome
5. Autoimmunity in Disorders of T-Cell Peripheral Tolerance
5.1. CTLA-4 Deficiency
5.2. LRBA Deficiency
5.3. STAT-Related Disorders
5.4. Other Disorders of Regulatory T Cells
6. Autoimmunity in Disorders of Lymphocyte Differentiation and Proliferation
6.1. Activated Phosphoinositide 3-Kinase d Syndrome
6.2. Protein Kinase C δ Deficiency
7. Autoimmunity in Disorders of Cytoskeletal Function
8. Autoimmunity in Complement Deficiencies and Disorders of Innate Immunity
9. From Theory to Bedside
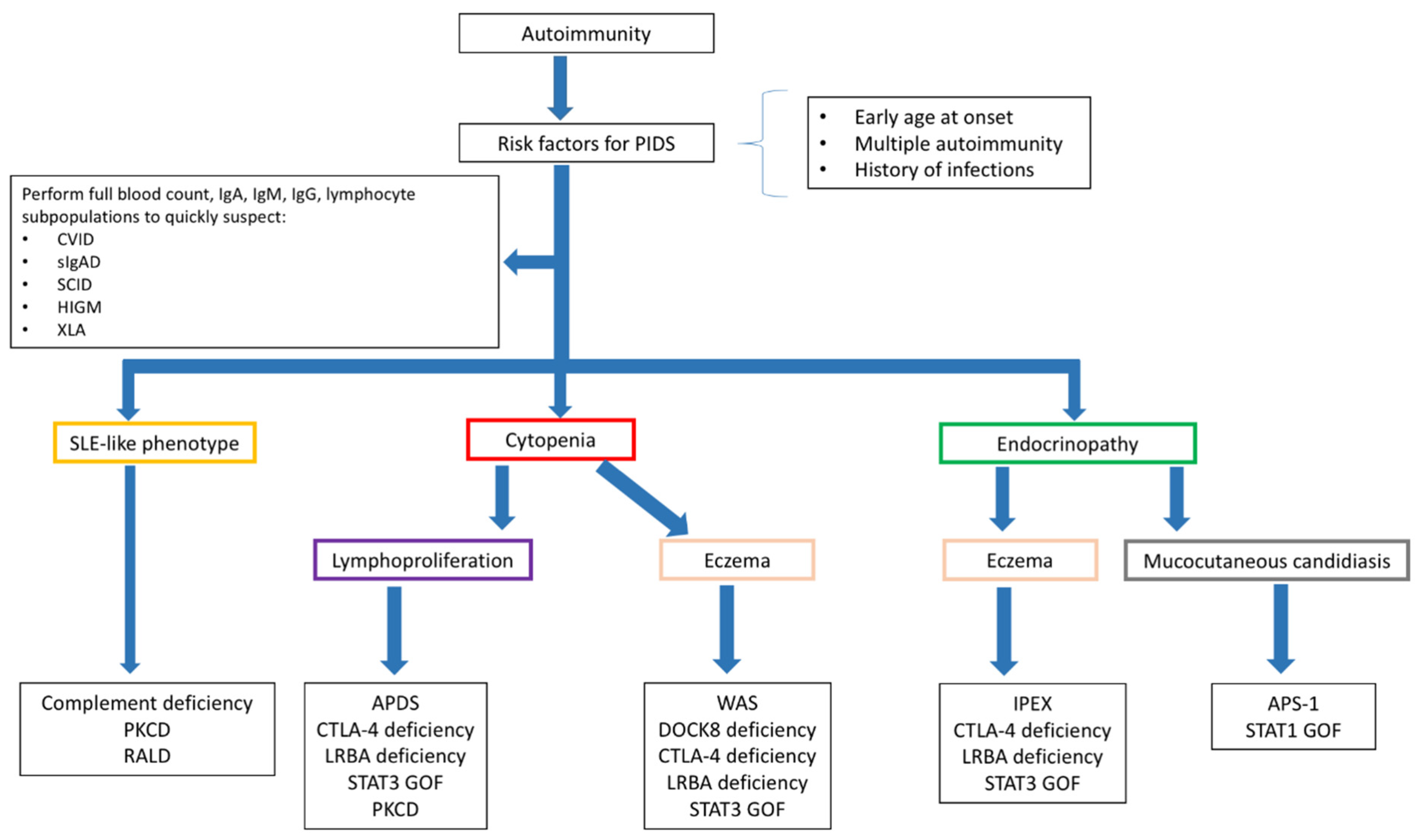
9.1. Diagnosing PIDs in Children Presenting with Autoimmunity
9.2. Diagnosing Autoimmunity in Children with PIDs
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Walter, J.E.; Ayala, I.A.; Milojevic, D. Autoimmunity as a continuum in primary immunodeficiency. Curr. Opin. Pediatr. 2019, 31, 851–862. [Google Scholar] [CrossRef]
- Amaya-Uribe, L.; Rojas, M.; Azizi, G.; Anaya, J.M.; Gershwin, M.E. Primary immunodeficiency and autoimmunity: A comprehensive review. J. Autoimmun. 2019, 99, 52–72. [Google Scholar] [CrossRef]
- Allenspach, E.; Torgerson, T.R. Autoimmunity and Primary Immunodeficiency Disorders. J. Clin. Immunol. 2016, 36 (Suppl 1), 57–67. [Google Scholar] [CrossRef]
- Notarangelo, L.D.; Uzel, G.; Rao, V.K. Primary immunodeficiencies: Novel genes and unusual presentations. Hematol. Am. Soc. Hematol. Educ. Program 2019, 2019, 443–448. [Google Scholar] [CrossRef]
- Bousfiha, A.; Jeddane, L.; Picard, C.; Ailal, F.; Bobby Gaspar, H.; Al-Herz, W. The 2017 IUIS Phenotypic Classification for Primary Immunodeficiencies. J. Clin. Immunol. 2018, 38, 129–143. [Google Scholar] [CrossRef]
- Seidel, M.G.; Kindle, G.; Gathmann, B.; Quinti, I.; Buckland, M.; van Montfrans, J. The European Society for Immunodeficiencies (ESID) Registry Working Definitions for the Clinical Diagnosis of Inborn Errors of Immunity. J. Allergy Clin. Immunol. Pract. 2019, 7, 1763–1770. [Google Scholar] [CrossRef]
- Chapel, H.; Lucas, M.; Lee, M.; Bjorkander, J.; Webster, D.; Grimbacher, B. Common variable immunodeficiency disorders: Division into distinct clinical phenotypes. Blood 2008, 112, 277–286. [Google Scholar] [CrossRef]
- Gathmann, B.; Mahlaoui, N.; Gérard, L.; Oksenhendler, E.; Warnatz, K.; Schulze, I. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J. Allergy Clin. Immunol. 2014, 134, 116–126. [Google Scholar] [CrossRef]
- Odnoletkova, I.; Kindle, G.; Quinti, I.; Grimbacher, B.; Knerr, V.; Gathmann, B. The burden of common variable immunodeficiency disorders: A retrospective analysis of the European Society for Immunodeficiency (ESID) registry data. Orphanet J. Rare Dis. 2018, 13, 201. [Google Scholar] [CrossRef]
- Resnick, E.S.; Moshier, E.L.; Godbold, J.H.; Cunningham-Rundles, C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood 2012, 119, 1650–1657. [Google Scholar] [CrossRef]
- Cunningham-Rundles, C. Autoimmunity in primary immune deficiency: Taking lessons from our patients. Clin. Exp. Immunol. 2011, 164 (Suppl. 2), 6–11. [Google Scholar] [CrossRef]
- Chapel, H.; Lucas, M.; Patel, S.; Lee, M.; Cunningham-Rundles, C.; Resnick, E. Confirmation and improvement of criteria for clinical phenotyping in common variable immunodeficiency disorders in replicate cohorts. J. Allergy Clin. Immunol. 2012, 130, 1197–1198.e9. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.J.; Sullivan, K.E.; Fuleihan, R.; Bingham, C.O. Phenotypic characterization of patients with rheumatologic manifestations of common variable immunodeficiency. Semin. Arthritis Rheum. 2018, 48, 318–326. [Google Scholar] [CrossRef]
- Maglione, P.J. Autoimmune and Lymphoproliferative Complications of Common Variable Immunodeficiency. Curr. Allergy Asthma. Rep. 2016, 16, 19. [Google Scholar] [CrossRef]
- Abolhassani, H.; Amirkashani, D.; Parvaneh, N.; Mohammadinejad, P.; Gharib, B.; Shahinpour, S. Autoimmune phenotype in patients with common variable immunodeficiency. J. Investig. Allergol. Clin. Immunol. 2013, 23, 323–329. [Google Scholar]
- Vlkova, M.; Ticha, O.; Nechvatalova, J.; Kalina, T.; Litzman, J.; Mauri, C. Regulatory B cells in CVID patients fail to suppress multifunctional IFN-γ+ TNF-α+ CD4+ T cells differentiation. Clin. Immunol. 2015, 160, 292–300. [Google Scholar] [CrossRef]
- Azizi, G.; Rezaei, N.; Kiaee, F.; Tavakolinia, N.; Yazdani, R.; Mirshafiey, A. T-Cell Abnormalities in Common Variable Immunodeficiency. J. Investig. Allergol. Clin. Immunol. 2016, 26, 233–243. [Google Scholar] [CrossRef]
- Warnatz, K.; Voll, R.E. Pathogenesis of autoimmunity in common variable immunodeficiency. Front Immunol. 2012, 3, 210. [Google Scholar] [CrossRef]
- Genre, J.; Errante, P.R.; Kokron, C.M.; Toledo-Barros, M.; Câmara, N.O.; Rizzo, L.V. Reduced frequency of CD4(+)CD25(HIGH)FOXP3(+) cells and diminished FOXP3 expression in patients with Common Variable Immunodeficiency: A link to autoimmunity? Clin. Immunol. 2009, 132, 215–221. [Google Scholar] [CrossRef]
- Kutukculer, N.; Azarsiz, E.; Aksu, G.; Karaca, N.E. CD4+CD25+Foxp3+ T regulatory cells, Th1 (CCR5, IL-2, IFN-γ) and Th2 (CCR4, IL-4, Il-13) type chemokine receptors and intracellular cytokines in children with common variable immunodeficiency. Int. J. Immunopathol. Pharmacol. 2016, 29, 241–251. [Google Scholar] [CrossRef]
- Crotty, S. T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity 2019, 50, 1132–1148. [Google Scholar] [CrossRef]
- De Valles-Ibáñez, G.; Esteve-Solé, A.; Piquer, M.; González-Navarro, E.A.; Hernandez-Rodriguez, J.; Laayouni, H. Evaluating the Genetics of Common Variable Immunodeficiency: Monogenetic Model and Beyond. Front Immunol. 2018, 9, 636. [Google Scholar] [CrossRef] [PubMed]
- Salzer, U.; Bacchelli, C.; Buckridge, S.; Pan-Hammarström, Q.; Jennings, S.; Lougaris, V. Relevance of biallelic versus monoallelic TNFRSF13B mutations in distinguishing disease-causing from risk-increasing TNFRSF13B variants in antibody deficiency syndromes. Blood 2009, 113, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Gereige, J.D.; Maglione, P.J. Current Understanding and Recent Developments in Common Variable Immunodeficiency Associated Autoimmunity. Front Immunol. 2019, 10, 2753. [Google Scholar] [CrossRef] [PubMed]
- Bogaert, D.J.; Dullaers, M.; Lambrecht, B.N.; Vermaelen, K.Y.; De Baere, E.; Haerynck, F. Genes associated with common variable immunodeficiency: One diagnosis to rule them all? J. Med. Genet. 2016, 53, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Milner, J.D. PLAID: A Syndrome of Complex Patterns of Disease and Unique Phenotypes. J. Clin. Immunol. 2015, 35, 527–530. [Google Scholar] [CrossRef]
- Yazdani, R.; Azizi, G.; Abolhassani, H.; Aghamohammadi, A. Selective IgA Deficiency: Epidemiology, Pathogenesis, Clinical Phenotype, Diagnosis, Prognosis and Management. Scand. J. Immunol. 2017, 85, 3–12. [Google Scholar] [CrossRef]
- Edwards, E.; Razvi, S.; Cunningham-Rundles, C. IgA deficiency: Clinical correlates and responses to pneumococcal vaccine. Clin. Immunol. 2004, 111, 93–97. [Google Scholar] [CrossRef]
- Jacob, C.M.; Pastorino, A.C.; Fahl, K.; Carneiro-Sampaio, M.; Monteiro, R.C. Autoimmunity in IgA deficiency: Revisiting the role of IgA as a silent housekeeper. J. Clin. Immunol. 2008, 28 (Suppl. 1), S56–S61. [Google Scholar] [CrossRef]
- Aghamohammadi, A.; Abolhassani, H.; Biglari, M.; Abolmaali, S.; Moazzami, K.; Tabatabaeiyan, M.; Asgarian-Omran, H.; Parvaneh, N.; Mirahmadian, M.; Rezaei, N. Analysis of switched memory B cells in patients with IgA deficiency. Int. Arch. Allergy Immunol. 2011, 156, 462–468. [Google Scholar] [CrossRef]
- Aytekin, C.; Tuygun, N.; Gokce, S.; Dogu, F.; Ikinciogullari, A. Selective IgA deficiency: Clinical and laboratory features of 118 children in Turkey. J. Clin. Immunol. 2012, 32, 961–966. [Google Scholar]
- Todoric, K.; Koontz, J.B.; Mattox, D.; Tarrant, T.K. Autoimmunity in immunodeficiency. Curr. Allergy Asthma Rep. 2013, 13, 361–370. [Google Scholar]
- Price, P.; Witt, C.; Allcock, R.; Sayer, D.; Garlepp, M.; Kok, C.C.; French, M.; Mallal, S.; Christiansen, F. The genetic basis for the association of the 8.1 ancestral haplotype (A1, B8, DR3) with multiple immunopathological diseases. Immunol. Rev. 1999, 167, 257–274. [Google Scholar]
- Azizi, G.; Tavakol, M.; Rafiemanesh, H.; Kiaee, F.; Yazdani, R.; Heydari, A.; Abouhamzeh, K.; Anvari, P.; Mohammadikhajehdehi, S.; Sharifia, L.; et al. Autoimmunity in a cohort of 471 patients with primary antibody deficiencies. Expert Rev. Clin. Immunol. 2017, 13, 1099–1106. [Google Scholar] [PubMed]
- Nechvatalova, J.; Pikulova, Z.; Stikarovska, D.; Pesak, S.; Vlkova, M.; Litzman, J. B-lymphocyte subpopulations in patients with selective IgA deficiency. J. Clin. Immunol. 2012, 32, 441–448. [Google Scholar] [PubMed]
- Haimila, K.; Einarsdottir, E.; de Kauwe, A.; Koskinen, L.L.; Pan-Hammarström, Q.; Kaartinen, T.; Kurppa, K.; Ziberna, F.; Not, T.; Vatta, S.; et al. The shared CTLA4-ICOS risk locus in celiac disease, IgA deficiency and common variable immunodeficiency. Genes Immun. 2009, 10, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Odineal, D.D.; Gershwin, M.E. The Epidemiology and Clinical Manifestations of Autoimmunity in Selective IgA Deficiency. Clin. Rev. Allergy Immunol. 2020, 58, 107–133. [Google Scholar] [PubMed]
- Davies, E.G.; Thrasher, A.J. Update on the hyper immunoglobulin M syndromes. Br. J. Haematol. 2010, 149, 167–180. [Google Scholar]
- Notarangelo, L.D.; Hayward, A.R. X-linked immunodeficiency with hyper-IgM (XHIM). Clin. Exp. Immunol. 2000, 120, 399–405. [Google Scholar] [PubMed]
- Jesus, A.A.; Duarte, A.J.; Oliveira, J.B. Autoimmunity in hyper-IgM syndrome. J. Clin. Immunol. 2008, 28 (Suppl 1), S62–S66. [Google Scholar] [CrossRef]
- Revy, P.; Muto, T.; Levy, Y.; Geissmann, F.; Plebani, A.; Sanal, O.; Catalan, N.; Forveille, M.; Dufourcq-Lagelouse, R.; Gennery, A.; et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell 2000, 102, 565–575. [Google Scholar] [CrossRef]
- Imai, K.; Slupphaug, G.; Lee, W.I.; Revy, P.; Nonoyama, S.; Catalan, N.; Yel, L.; Forveille, M.; Kavli, B.; Krokan, H.E.; et al. Human uracil-DNA glycosylase deficiency associated with profoundly impaired immunoglobulin class-switch recombination. Nat. Immunol. 2003, 4, 1023–1028. [Google Scholar] [CrossRef]
- Ferrari, S.; Giliani, S.; Insalaco, A.; Al-Ghonaium, A.; Soresina, A.R.; Loubser, M.; Avanzini, M.A.; Marconi, M.; Badolato, R.; Ugazio, A.G.; et al. Mutations of CD40 gene cause an autosomal recessive form of immunodeficiency with hyper IgM. Proc. Natl. Acad. Sci. USA 2001, 98, 12614–12619. [Google Scholar] [CrossRef]
- Lehman, H.K. Autoimmunity and Immune Dysregulation in Primary Immune Deficiency Disorders. Curr. Allergy Asthma. Rep. 2015, 15, 53. [Google Scholar] [CrossRef]
- Winkelstein, J.A.; Marino, M.C.; Ochs, H.; Fuleihan, R.; Scholl, P.R.; Geha, R.; Stiehm, E.R.; Conley, M.E. The X-linked hyper-IgM syndrome: Clinical and immunologic features of 79 patients. Medicine 2003, 82, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.; Espanol-Boren, T.; Thomas, C.; Fischer, A.; Tovo, P.; Bordigoni, P.; Resnick, L.; Fasth, A.; Baer, M.; Gomez, L.; et al. Clinical spectrum of X-linked hyper-IgM syndrome. J. Pediatr. 1997, 131, 47–54. [Google Scholar] [CrossRef]
- Quartier, P.; Bustamante, J.; Sanal, O.; Plebani, A.; Debré, M.; Deville, A.; Litzmane, J.; Levy, J.; Fermand, J.-P.; Lane, P.; et al. Clinical, immunologic and genetic analysis of 29 patients with autosomal recessive hyper-IgM syndrome due to Activation-Induced Cytidine Deaminase deficiency. Clin. Immunol. 2004, 110, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Lacroix-Desmazes, S.; Resnick, I.; Stahl, D.; Mouthon, L.; Espanol, T.; Levy, J.; Kaveri, S.V.; Notarangelo, L.; Eibl, M.; Fischer, A.; et al. Defective self-reactive antibody repertoire of serum IgM in patients with hyper-IgM syndrome. J. Immunol. 1999, 162, 5601–5608. [Google Scholar] [PubMed]
- Lougaris, V.; Badolato, R.; Ferrari, S.; Plebani, A. Hyper immunoglobulin M syndrome due to CD40 deficiency: Clinical, molecular, and immunological features. Immunol. Rev. 2005, 203, 48–66. [Google Scholar] [CrossRef]
- Fleischer, D.M.; Sicherer, S.; Greenhawt, M.; Campbell, D.; Chan, E.; Muraro, A.; Halken, S.; Katz, Y.; Ebisawa, M.; Eichenfield, L.; et al. Consensus Communication on Early Peanut Introduction and Prevention of Peanut Allergy in High-Risk Infants. Pediatr. Dermatol. 2016, 33, 103–106. [Google Scholar] [CrossRef]
- Shillitoe, B.; Gennery, A. X-Linked Agammaglobulinaemia: Outcomes in the modern era. Clin. Immunol. 2017, 183, 54–62. [Google Scholar] [CrossRef]
- Pessach, I.M. The relationship of x-linked primary immune deficiencies and autoimmunity. Curr. Allergy Asthma. Rep. 2010, 10, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Winkelstein, J.A.; Marino, M.C.; Lederman, H.M.; Jones, S.M.; Sullivan, K.; Burks, A.W.; Conley, M.E.; Cunningham-Rundles, C.; Ochs, H.D. X-linked agammaglobulinemia: Report on a United States registry of 201 patients. Medicine 2006, 85, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Ochs, H.D.; Notarangelo, L.D. X-linked immunodeficiencies. Curr. Allergy Asthma. Rep. 2004, 4, 339–348. [Google Scholar] [CrossRef]
- Howard, V.; Greene, J.M.; Pahwa, S.; Winkelstein, J.A.; Boyle, J.M.; Kocak, M.; Conley, M.E. The health status and quality of life of adults with X-linked agammaglobulinemia. Clin. Immunol. 2006, 118, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Knight, A.K.; Cunningham-Rundles, C. Inflammatory and autoimmune complications of common variable immune deficiency. Autoimmun. Rev. 2006, 5, 156–159. [Google Scholar] [CrossRef]
- Lee, K.G.; Xu, S.; Wong, E.T.; Tergaonkar, V.; Lam, K.P. Bruton’s tyrosine kinase separately regulates NFkappaB p65RelA activation and cytokine interleukin (IL)-10/IL-12 production in TLR9-stimulated B Cells. J. Biol. Chem. 2008, 283, 11189–11198. [Google Scholar] [CrossRef]
- Kubo, T.; Uchida, Y.; Watanabe, Y.; Abe, M.; Nakamura, A.; Ono, M.; Akira, S.; Takai, T. Augmented TLR9-induced Btk activation in PIR-B-deficient B-1 cells provokes excessive autoantibody production and autoimmunity. J. Exp. Med. 2009, 206, 1971–1982. [Google Scholar] [CrossRef]
- Gobert, D.; Bussel, J.B.; Cunningham-Rundles, C.; Galicier, L.; Dechartres, A.; Berezne, A.; Bonnotte, B.; DeRevel, T.; Auzary, C.; Jaussaud, R.; et al. Efficacy and safety of rituximab in common variable immunodeficiency-associated immune cytopenias: A retrospective multicentre study on 33 patients. Br. J. Haematol. 2011, 155, 498–508. [Google Scholar] [CrossRef]
- Wang, J.; Cunningham-Rundles, C. Treatment and outcome of autoimmune hematologic disease in common variable immunodeficiency (CVID). J. Autoimmun. 2005, 25, 57–62. [Google Scholar] [CrossRef]
- Cirillo, E.; Giardino, G.; Gallo, V.; D’Assante, R.; Grasso, F.; Romano, R.; Lillo, C.D.; Galasso, G.; Pignata, C. Severe combined immunodeficiency—An update. Ann. N. Y. Acad. Sci. 2015, 1356, 90–106. [Google Scholar] [CrossRef] [PubMed]
- Costagliola, G.; Consolini, R. Lymphadenopathy at the crossroad between immunodeficiency and autoinflammation: An intriguing challenge. Clin. Exp. Immunol. 2021. Online ahead of print. [Google Scholar]
- Kato, M.; Kimura, H.; Seki, M.; Shimada, A.; Hayashi, Y.; Morio, T.; Kumaki, S.; Ishida, Y.; Kamachi, Y.; Yachie, A. Omenn syndrome--review of several phenotypes of Omenn syndrome and RAG1/RAG2 mutations in Japan. Allergol. Int. 2006, 55, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Aleman, K.; Noordzij, J.G.; de Groot, R.; van Dongen, J.J.; Hartwig, N.G. Reviewing Omenn syndrome. Eur. J. Pediatr. 2001, 160, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Giardino, G.; Gallo, V.; Prencipe, R.; Gaudino, G.; Romano, R.; De Cataldis, M. Unbalanced Immune System: Immunodeficiencies and Autoimmunity. Front. Pediatr. 2016, 4, 107. [Google Scholar] [CrossRef] [PubMed]
- Castagnoli, R.; Delmonte, O.M.; Calzoni, E.; Notarangelo, L.D. Hematopoietic Stem Cell Transplantation in Primary Immunodeficiency Diseases: Current Status and Future Perspectives. Front. Pediatr. 2019, 7, 295. [Google Scholar] [CrossRef]
- Villa, A.; Notarangelo, L.D. RAG gene defects at the verge of immunodeficiency and immune dysregulation. Immunol. Rev. 2019, 287, 73–90. [Google Scholar] [CrossRef]
- Notarangelo, L.D.; Kim, M.S.; Walter, J.E.; Lee, Y.N. Human RAG mutations: Biochemistry and clinical implications. Nat. Rev. Immunol. 2016, 16, 234–246. [Google Scholar] [CrossRef]
- Klein, L.; Robey, E.A.; Hsieh, C.S. Central CD4(+) T cell tolerance: Deletion versus regulatory T cell differentiation. Nat. Rev. Immunol. 2019, 19, 7–18. [Google Scholar] [CrossRef]
- Husebye, E.S.; Anderson, M.S.; Kämpe, O. Autoimmune Polyendocrine Syndromes. N. Engl. J. Med. 2018, 378, 1132–1141. [Google Scholar] [CrossRef]
- Guo, C.J.; Leung, P.S.C.; Zhang, W.; Ma, X.; Gershwin, M.E. The immunobiology and clinical features of type 1 autoimmune polyglandular syndrome (APS-1). Autoimmun. Rev. 2018, 17, 78–85. [Google Scholar] [CrossRef]
- Husebye, E.S.; Perheentupa, J.; Rautemaa, R.; Kämpe, O. Clinical manifestations and management of patients with autoimmune polyendocrine syndrome type I. J. Intern. Med. 2009, 265, 514–529. [Google Scholar] [CrossRef] [PubMed]
- McDonald-McGinn, D.M.; Sullivan, K.E.; Marino, B.; Philip, N.; Swillen, A.; Vorstman, J.A.; Zackai, E.H.; Emanuel, B.S.; Vermeesch, J.R.; Morrow, B.E.; et al. 22q11.2 deletion syndrome. Nat. Rev. Dis. Primers. 2015, 1, 15071. [Google Scholar] [CrossRef]
- Gennery, A.R. Immunological aspects of 22q11.2 deletion syndrome. Cell Mol. Life Sci. 2012, 69, 17–27. [Google Scholar] [CrossRef]
- McLean-Tooke, A.; Spickett, G.P.; Gennery, A.R. Immunodeficiency and autoimmunity in 22q11.2 deletion syndrome. Scand. J. Immunol. 2007, 66, 1–7. [Google Scholar] [CrossRef]
- Di Cesare, S.; Puliafito, P.; Ariganello, P.; Marcovecchio, G.E.; Mandolesi, M.; Capolino, R.; Digilio, M.C.; Aiuti, A.; Rossi, P.; Cancrini, C. Autoimmunity and regulatory T cells in 22q11.2 deletion syndrome patients. Pediatr. Allergy Immunol. 2015, 26, 591–594. [Google Scholar] [CrossRef]
- Marcovecchio, G.E.; Bortolomai, I.; Ferrua, F.; Fontana, E.; Imberti, L.; Conforti, E.; Amodio, D.; Bergante, S.; Macchiarulo, G.; D’Oria, V.; et al. Thymic Epithelium Abnormalities in DiGeorge and Down Syndrome Patients Contribute to Dysregulation in T Cell Development. Front Immunol. 2019, 10, 447. [Google Scholar] [CrossRef]
- Morsheimer, M.; Brown Whitehorn, T.F.; Heimall, J.; Sullivan, K.E. The immune deficiency of chromosome 22q11.2 deletion syndrome. Am. J. Med Genet. Part A 2017, 173, 2366–2372. [Google Scholar] [CrossRef]
- Legitimo, A.; Bertini, V.; Costagliola, G.; Baroncelli, G.I.; Morganti, R.; Valetto, A.; Consolini, R. Vitamin D status and the immune assessment in 22q11.2 deletion syndrome. Clin. Exp. Immunol. 2020, 200, 272–286. [Google Scholar] [CrossRef] [PubMed]
- McDonald-McGinn, D.M.; Sullivan, K.E. Chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Medicine 2011, 90, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Cepika, A.M.; Sato, Y.; Liu, J.M.; Uyeda, M.J.; Bacchetta, R.; Roncarolo, M.G. Tregopathies: Monogenic diseases resulting in regulatory T-cell deficiency. J. Allergy Clin. Immunol. 2018, 142, 1679–1695. [Google Scholar] [CrossRef]
- Gambineri, E.; Ciullini Mannurita, S.; Hagin, D.; Vignoli, M.; Anover-Sombke, S.; DeBoer, S. Clinical, Immunological, and Molecular Heterogeneity of 173 Patients With the Phenotype of Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked (IPEX) Syndrome. Front Immunol. 2018, 9, 2411. [Google Scholar] [CrossRef] [PubMed]
- Verma, N.; Burns, S.O.; Walker, L.S.K.; Sansom, D.M. Immune deficiency and autoimmunity in patients with CTLA-4 (CD152) mutations. Clin. Exp. Immunol. 2017, 190, 1–7. [Google Scholar] [CrossRef]
- Schwab, C.; Gabrysch, A.; Olbrich, P.; Patiño, V.; Warnatz, K.; Wolff, D.; Hoshino, A.; Kobayashi, M.; Imai, K.; Takag, M.; et al. Phenotype, penetrance, and treatment of 133 cytotoxic T-lymphocyte antigen 4-insufficient subjects. J. Allergy Clin. Immunol. 2018, 142, 1932–1946. [Google Scholar] [CrossRef] [PubMed]
- Delmonte, O.M.; Castagnoli, R.; Calzoni, E.; Notarangelo, L.D. Inborn Errors of Immunity With Immune Dysregulation: From Bench to Bedside. Front Pediatr. 2019, 7, 353. [Google Scholar] [CrossRef]
- Lo, B.; Abdel-Motal, U.M. Lessons from CTLA-4 deficiency and checkpoint inhibition. Curr. Opin. Immunol. 2017, 49, 14–19. [Google Scholar] [CrossRef]
- Gámez-Díaz, L.; August, D.; Stepensky, P.; Revel-Vilk, S.; Seidel, M.G.; Noriko, M.; Morio, T.; Worth, A.J.J.; Blessing, J.; Veerdonk, F.V.; et al. The extended phenotype of LPS-responsive beige-like anchor protein (LRBA) deficiency. J. Allergy Clin. Immunol. 2016, 137, 223–230. [Google Scholar] [CrossRef]
- Habibi, S.; Zaki-Dizaji, M.; Rafiemanesh, H.; Lo, B.; Jamee, M.; Gámez-Díaz, L.; Salami, F.; Kamali, A.N.; Mohammadi, H.; Abolhassani, H.; et al. Clinical, Immunologic, and Molecular Spectrum of Patients with LPS-Responsive Beige-Like Anchor Protein Deficiency: A Systematic Review. J. Allergy Clin. Immunol. Pract. 2019, 7, 2379–2386.e5. [Google Scholar] [CrossRef]
- Alkhairy, O.K.; Abolhassani, H.; Rezaei, N.; Fang, M.; Andersen, K.K.; Chavoshzadeh, Z.; Mohammadzadeh, I.; El-Rajab, M.A.; Massaad, M.; Chou, J.; et al. Spectrum of Phenotypes Associated with Mutations in LRBA. J. Clin. Immunol. 2016, 36, 33–45. [Google Scholar] [CrossRef]
- Lo, B.; Zhang, K.; Lu, W.; Zheng, L.; Zhang, Q.; Kanellopoulou, C.; Zhang, Y.; Liu, Z.; Fritz, J.M.; Marsh, R.; et al. AUTOIMMUNE DISEASE. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science 2015, 349, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Kiykim, A.; Ogulur, I.; Dursun, E.; Charbonnier, L.M.; Nain, E.; Cekic, S.; Dogruel, D.; Karaca, N.E.; Cogurlu, M.T.; Bilir, O.A.; et al. Abatacept as a Long-Term Targeted Therapy for LRBA Deficiency. J. Allergy Clin. Immunol. Pract. 2019, 7, 2790–2800.e15. [Google Scholar] [CrossRef] [PubMed]
- Lorenzini, T.; Dotta, L.; Giacomelli, M.; Vairo, D.; Badolato, R. STAT mutations as program switchers: Turning primary immunodeficiencies into autoimmune diseases. J. Leukoc. Biol. 2017, 101, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Asano, T.; Moriya, K.; Boisson-Dupuis, S.; Kobayashi, M.; Casanova, J.L.; Puel, A. Human STAT1 Gain-of-Function Heterozygous Mutations: Chronic Mucocutaneous Candidiasis and Type I Interferonopathy. J. Clin. Immunol. 2020, 40, 1065–1081. [Google Scholar] [CrossRef]
- Fabre, A.; Marchal, S.; Barlogis, V.; Mari, B.; Barbry, P.; Rohrlich, P.S.; Forbes, L.R.; Vogel, T.P.; Giovannini-Chami, L. Clinical Aspects of STAT3 Gain-of-Function Germline Mutations: A Systematic Review. J. Allergy Clin. Immunol. Pract. 2019, 7, 1958–1969.e9. [Google Scholar] [CrossRef]
- Vogel, T.P.; Milner, J.D.; Cooper, M.A. The Ying and Yang of STAT3 in Human Disease. J. Clin. Immunol. 2015, 35, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Hwa, V. STAT5B deficiency: Impacts on human growth and immunity. Growth Horm. IGF Res. 2016, 28, 16–20. [Google Scholar] [CrossRef]
- Khoury, T.; Molho-Pessach, V.; Ramot, Y.; Ayman, A.R.; Elpeleg, O.; Berkman, N.; Zlotogorski, A.; Ilan, Y. Tocilizumab Promotes Regulatory T-cell Alleviation in STAT3 Gain-of-function-associated Multi-organ Autoimmune Syndrome. Clin. Ther. 2017, 39, 444–449. [Google Scholar] [CrossRef]
- Afzali, B.; Grönholm, J.; Vandrovcova, J.; O’Brien, C.; Sun, H.W.; Vanderleyden, I.; Davis, F.P.; Khoder, A.; Zhang, Y.; Hegazy, A.N.; et al. BACH2 immunodeficiency illustrates an association between super-enhancers and haploinsufficiency. Nat. Immunol. 2017, 18, 813–823. [Google Scholar] [CrossRef]
- Tessarin, G.; Rossi, S.; Baronio, M.; Gazzurelli, L.; Colpani, M.; Benvenuto, A.; Zunica, F.; Cardinale, F.; Martire, B.; Brescia, L.; et al. Activated Phosphoinositide 3-Kinase Delta Syndrome 1, Clinical and Immunological Data from an Italian Cohort of Patients. J. Clin. Med. 2020, 9, 3335. [Google Scholar] [CrossRef]
- Coulter, T.I.; Chandra, A.; Bacon, C.M.; Babar, J.; Curtis, J.; Screaton, N.; Goodlad, J.R.; Farmer, G.; Steele, C.L.; Leahy, T.R.; et al. Clinical spectrum and features of activated phosphoinositide 3-kinase δ syndrome: A large patient cohort study. J. Allergy Clin. Immunol. 2017, 139, 597–606.e4. [Google Scholar] [CrossRef] [PubMed]
- Nunes-Santos, C.J.; Uzel, G.; Rosenzweig, S.D. PI3K pathway defects leading to immunodeficiency and immune dysregulation. J. Allergy Clin. Immunol. 2019, 143, 1676–1687. [Google Scholar] [CrossRef]
- Lucas, C.L.; Chandra, A.; Nejentsev, S.; Condliffe, A.M.; Okkenhaug, K. PI3Kδ and primary immunodeficiencies. Nat. Rev. Immunol. 2016, 16, 702–714. [Google Scholar] [CrossRef]
- Dimitrova, D.; Nademi, Z.; Maccari, M.E.; Ehl, S.; Uzel, G.; Tomoda, T.; Okano, T.; Imai, K.; Carpenter, B.; Ip, W.; et al. International retrospective study of allogeneic hematopoietic cell transplantation for activated PI3K-delta syndrome. J. Allergy Clin. Immunol. 2021, in press. [Google Scholar]
- Salzer, E.; Santos-Valente, E.; Keller, B.; Warnatz, K.; Boztug, K. Protein Kinase C δ: A Gatekeeper of Immune Homeostasis. J. Clin. Immunol. 2016, 36, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Duquesnes, N.; Lezoualc’h, F.; Crozatier, B. PKC-delta and PKC-epsilon: Foes of the same family or strangers? J. Mol. Cell Cardiol. 2011, 51, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, S.F. Distinctive activation mechanisms and functions for protein kinase Cdelta. Biochem. J. 2004, 384 Pt 3, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Kuehn, H.S.; Niemela, J.E.; Rangel-Santos, A.; Zhang, M.; Pittaluga, S.; Stoddard, J.L.; Hussey, A.A.; Evbuomwan, M.O.; Long Priel, D.A.; Kuhns, D.B.; et al. Loss-of-function of the protein kinase C δ (PKCδ) causes a B-cell lymphoproliferative syndrome in humans. Blood 2013, 121, 3117–3125. [Google Scholar] [CrossRef] [PubMed]
- Calvo, K.R.; Price, S.; Braylan, R.C.; Oliveira, J.B.; Lenardo, M.; Fleisher, T.A.; Koneti Rao, V. JMML and RALD (Ras-associated autoimmune leukoproliferative disorder): Common genetic etiology yet clinically distinct entities. Blood 2015, 125, 2753–2758. [Google Scholar] [CrossRef] [PubMed]
- Rivers, E.; Thrasher, A.J. Wiskott-Aldrich syndrome protein: Emerging mechanisms in immunity. Eur. J. Immunol. 2017, 47, 1857–1866. [Google Scholar] [CrossRef] [PubMed]
- Massaad, M.J.; Ramesh, N.; Geha, R.S. Wiskott-Aldrich syndrome: A comprehensive review. Ann. N. Y. Acad. Sci. 2013, 1285, 26–43. [Google Scholar] [CrossRef]
- Albert, M.H.; Freeman, A.F. Wiskott-Aldrich Syndrome (WAS) and Dedicator of Cytokinesis 8-(DOCK8) Deficiency. Front Pediatr. 2019, 7, 451. [Google Scholar] [CrossRef]
- Biggs, C.M.; Keles, S.; Chatila, T.A. DOCK8 deficiency: Insights into pathophysiology, clinical features and management. Clin. Immunol. 2017, 181, 75–82. [Google Scholar] [CrossRef]
- Schröder-Braunstein, J.; Kirschfink, M. Complement deficiencies and dysregulation: Pathophysiological consequences, modern analysis, and clinical management. Mol. Immunol. 2019, 114, 299–311. [Google Scholar] [CrossRef]
- Conigliaro, P.; Triggianese, P.; Ballanti, E.; Perricone, C.; Perricone, R.; Chimenti, M.S. Complement, infection, and autoimmunity. Curr. Opin. Rheumatol. 2019, 31, 532–541. [Google Scholar] [CrossRef]
- Vignesh, P.; Rawat, A.; Sharma, M.; Singh, S. Complement in autoimmune diseases. Clin. Chim. Acta. 2017, 465, 123–130. [Google Scholar] [CrossRef]
- Henrickson, S.E.; Jongco, A.M.; Thomsen, K.F.; Garabedian, E.K.; Thomsen, I.P. Noninfectious Manifestations and Complications of Chronic Granulomatous Disease. J. Pediatric Infect. Dis. Soc. 2018, 7 (Suppl. 1), S18–S24. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.H.; Yang, Y.H.; Chiang, B.L. Chronic Granulomatous Disease: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2020, 61, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Azizi, G.; Ziaee, V.; Tavakol, M.; Alinia, T.; Yazdai, R.; Mohammadi, H.; Abolhassani, H.; Aghamohammadi, A. Approach to the Management of Autoimmunity in Primary Immunodeficiency. Scand. J. Immunol. 2017, 85, 13–29. [Google Scholar] [CrossRef]
- Walter, J.E.; Farmer, J.R.; Foldvari, Z.; Torgerson, T.R.; Cooper, M.A. Mechanism-Based Strategies for the Management of Autoimmunity and Immune Dysregulation in Primary Immunodeficiencies. J. Allergy Clin. Immunol. Pract. 2016, 4, 1089–1100. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.S. How to evaluate for immunodeficiency in patients with autoimmune cytopenias: Laboratory evaluation for the diagnosis of inborn errors of immunity associated with immune dysregulation. Hematol. Am. Soc. Hematol. Educ. Program 2020, 2020, 661–672. [Google Scholar] [CrossRef]
- Consolini, R.; Costagliola, G.; Spatafora, D. The Centenary of Immune Thrombocytopenia-Part 2: Revising Diagnostic and Therapeutic Approach. Front Pediatr. 2017, 5, 179. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Provot, J.; Jais, J.P.; Alcais, A.; Mahlaoui, N. Autoimmune and inflammatory manifestations occur frequently in patients with primary immunodeficiencies. J. Allergy Clin. Immunol. 2017, 140, 1388–1393.e8. [Google Scholar] [CrossRef]
- Besnard, C.; Levy, E.; Aladjidi, N.; Stolzenberg, M.C.; Magerus-Chatinet, A.; Alibeu, O.; Nitschke, P.; Blanche, S.; Hermine, O.; Jeziorski, E.; et al. Pediatric-onset Evans syndrome: Heterogeneous presentation and high frequency of monogenic disorders including LRBA and CTLA4 mutations. Clin. Immunol. 2018, 188, 52–57. [Google Scholar] [CrossRef]
- Hadjadj, J.; Aladjidi, N.; Fernandes, H.; Leverger, G.; Magérus-Chatinet, A.; Mazerolles, F.; Hussey, A.A.; Evbuomwan, M.O.; Long Priel, D.A.; Kuhns, D.B.; et al. Pediatric Evans syndrome is associated with a high frequency of potentially damaging variants in immune genes. J. Am. Soc. Hematol. 2019, 134, 9–21. [Google Scholar]
- Costagliola, G.; Marco, S.D.; Comberiati, P.; D’Elios, S.; Petashvili, N.; Di Cicco, M.E.; Peroni, D. Practical Approach to Children Presenting with Eosinophila and Hypereosinophilia. Curr. Pediatr. Rev. 2020, 16, 81–88. [Google Scholar] [CrossRef]
- Shamriz, O.; Tal, Y.; Talmon, A.; Nahum, A. Chronic Mucocutaneous Candidiasis in Early Life: Insights Into Immune Mechanisms and Novel Targeted Therapies. Front Immunol. 2020, 11, 593289. [Google Scholar] [CrossRef] [PubMed]
- Leffler, J.; Bengtsson, A.A.; Blom, A.M. The complement system in systemic lupus erythematosus: An update. Ann. Rheum. Dis. 2014, 73, 1601–1606. [Google Scholar] [CrossRef] [PubMed]
- Boileau, J.; Mouillot, G.; Gerard, L.; Carmagnat, M.; Rabian, C.; Oksenhendler, E.; Pasquali, J.-L.; Korganow, A.-S.; DEFI Study Group. Autoimmunity in common variable immunodeficiency: Correlation with lymphocyte phenotype in the French DEFI study. J. Autoimmun. 2011, 36, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Ramón, S.; Radigan, L.; Yu, J.E.; Bard, S.; Cunningham-Rundles, C. Memory B cells in common variable immunodeficiency: Clinical associations and sex differences. Clin. Immunol. 2008, 128, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Cunningham-Rundles, C. Role of B cells in common variable immune deficiency. Expert Rev. Clin. Immunol. 2009, 5, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Arumugakani, G.; Wood, P.M.; Carter, C.R. Frequency of Treg cells is reduced in CVID patients with autoimmunity and splenomegaly and is associated with expanded CD21lo B lymphocytes. J. Clin. Immunol. 2010, 30, 292–300. [Google Scholar] [CrossRef]
- Picchianti Diamanti, A.; Rosado, M.M.; Scarsella, M.; Ceccarelli, S.; Laganà, B.; D’Amelio, R.; Carsetti, R. Increased serum IgM, immunodeficiency, and autoimmunity: A clinical series. Int. J. Immunopathol. Pharmacol. 2015, 28, 547–556. [Google Scholar] [CrossRef]
- Montin, D.; Marolda, A.; Licciardi, F.; Robasto, F.; Di Cesare, S.; Ricotti, E.; Ferro, F.; Scaioli, G.; Giancotta, C.; Amodio, D.; et al. Immunophenotype Anomalies Predict the Development of Autoimmune Cytopenia in 22q11.2 Deletion Syndrome. J. Allergy Clin. Immunol. Pract. 2019, 7, 2369–2376. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Disease | % of Patients with Autoimmunity | Autoimmune/Inflammatory Manifestations |
|---|---|---|
| CVID Specific genetic associations TACI defect BAFF-R defect ICOS deficiency NF-kB1 deficiency NF-kB2 deficiency | 20–30% | Autoimmune cytopenias (ITP, AIHA, neutropenia), organ specific autoimmune diseases (e.g., thyroiditis, T1D, ILD, IBD), systemic autoimmune diseases (RA, SLE), lymphoproliferation, lymphoma Variable autoimmune manifestations Variable autoimmune manifestations Autoimmune cytopenias, enteropathy, RA, SLE Autoimmune cytopenias, enteropathy, lymphoproliferation, lymphoma Autoimmunity affecting skin, hair and nails, pituitary hormone deficiencies, autoimmune cytopenias |
| sIgAD | 5–30% | Celiac disease, autoimmune cytopenias (ITP, AIHA), hypothyroidism, Graves’ disease, T1D, RA, SLE. |
| Hyper IgM syndromes XHIM AID deficiency NEMO | 10–20% 21% | AIHA, ITP, autoimmune hepatitis, T1D, Chron’s disease and uveitis, seronegative arthritis, hypothyroidism, SLE, sclerosing cholangitis AIHA, ITP, autoimmune hepatitis, T1D, Chron’s disease and uveitis, lymphoproliferation IBD, arthritis, AIHA |
| X-linked agammaglobulinemia | 15% | Arthritis, DM, IBD, AIHA, scleroderma, alopecia, T1D, glomerulonephritis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costagliola, G.; Cappelli, S.; Consolini, R. Autoimmunity in Primary Immunodeficiency Disorders: An Updated Review on Pathogenic and Clinical Implications. J. Clin. Med. 2021, 10, 4729. https://doi.org/10.3390/jcm10204729
Costagliola G, Cappelli S, Consolini R. Autoimmunity in Primary Immunodeficiency Disorders: An Updated Review on Pathogenic and Clinical Implications. Journal of Clinical Medicine. 2021; 10(20):4729. https://doi.org/10.3390/jcm10204729
Chicago/Turabian StyleCostagliola, Giorgio, Susanna Cappelli, and Rita Consolini. 2021. "Autoimmunity in Primary Immunodeficiency Disorders: An Updated Review on Pathogenic and Clinical Implications" Journal of Clinical Medicine 10, no. 20: 4729. https://doi.org/10.3390/jcm10204729
APA StyleCostagliola, G., Cappelli, S., & Consolini, R. (2021). Autoimmunity in Primary Immunodeficiency Disorders: An Updated Review on Pathogenic and Clinical Implications. Journal of Clinical Medicine, 10(20), 4729. https://doi.org/10.3390/jcm10204729