Proposal of the Definition for COVID-19-Associated Coagulopathy

 ,
,  and
and 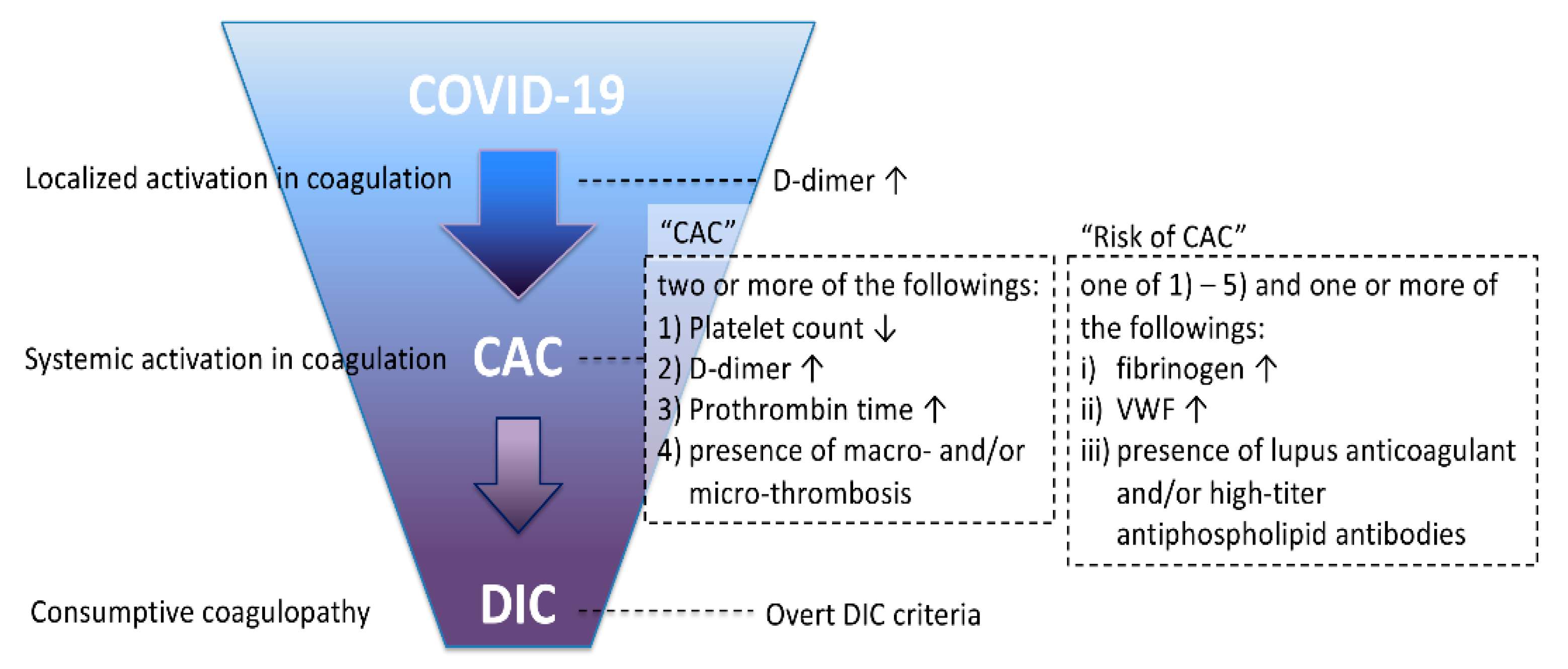{kind=link}
{kind=link}
Abstract
1. Introduction
2. Characteristic Differences between COVID-19-Associated Coagulopathy and Sepsis-Associated DIC
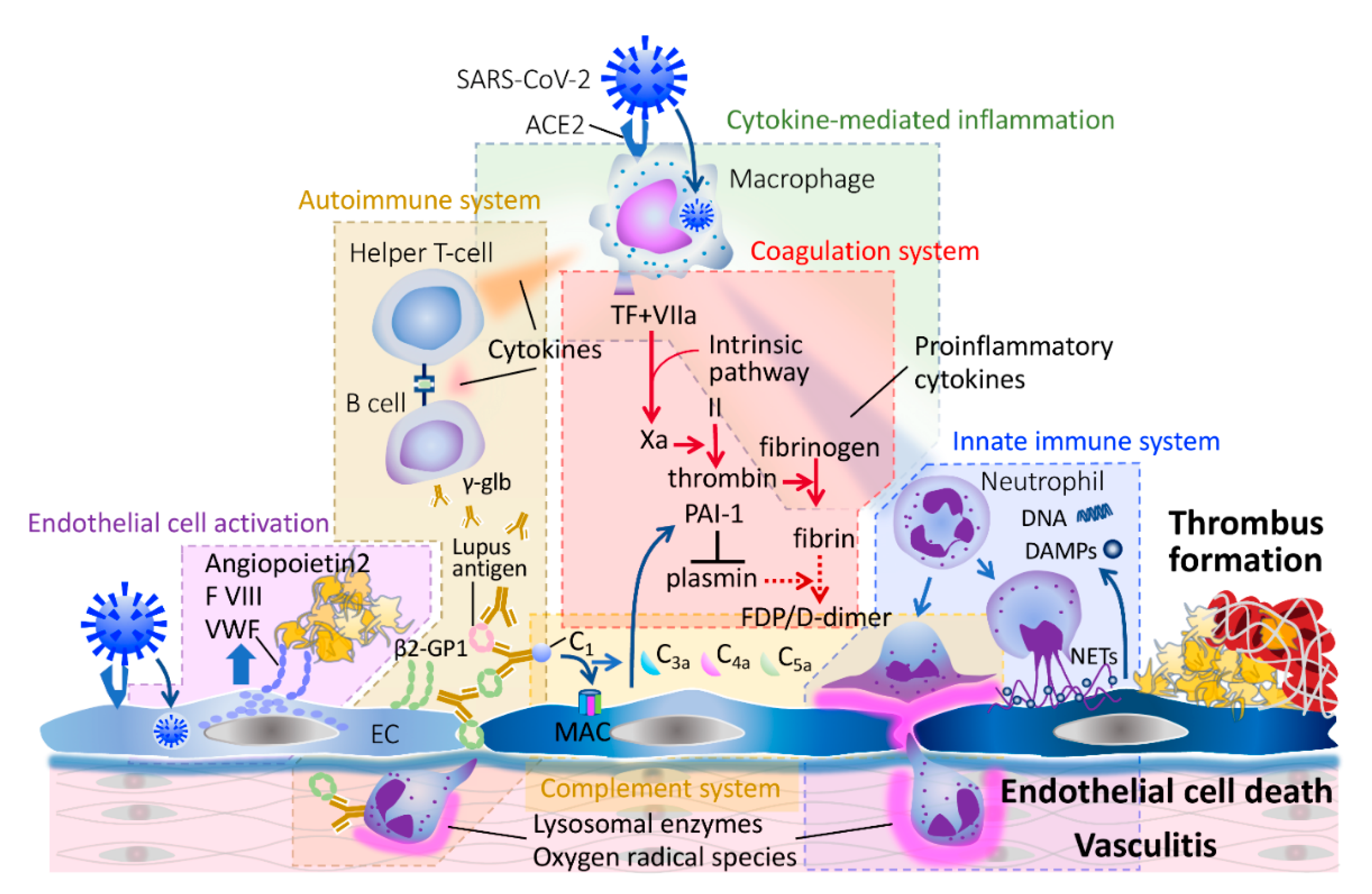
3. Pathophysiology of COVID-19-Associated Coagulopathy and Bacterial Sepsis-Associated DIC
3.1. Endothelial Activation and Damage
3.2. Coagulation and Fibrinolytic Systems in Infection
3.3. Innate Immune System
3.4. Adaptive Immune System
3.5. Complement System
4. Diagnosis of CAC and DIC
5. Skin and Acral Lesions
6. Anticoagulant Therapy Using Heparin for CAC and DIC
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Taylor, F.B., Jr.; Toh, C.H.; Hoots, W.K.; Wada, H.; Levi, M. Scientific Subcommittee on Disseminated Intravascular Coagulation (DIC) of the International Society on Thrombosis and Haemostasis (ISTH). Towards definition, clinical and laboratory criteria, and a scoring system for disseminated intravascular coagulation. Thromb. Haemost. 2001, 86, 1327–1330. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.; Li, D.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Iba, T.; Levy, J.H.; Connors, J.M.; Warkentin, T.E.; Thachil, J.; Levi, M. The unique characteristics of COVID-19 coagulopathy. Crit. Care 2020, 24, 1–8. [Google Scholar] [CrossRef]
- Dolhnikoff, M.; Duarte-Neto, A.N.; Monteiro, R.A.D.A.; Da Silva, L.F.F.; De Oliveira, E.P.; Saldiva, P.H.N.; Mauad, T.; Negri, E.M. Pathological evidence of pulmonary thrombotic phenomena in severe COVID-19. J. Thromb. Haemost. 2020, 18, 1517–1519. [Google Scholar] [CrossRef]
- Asakura, H. Classifying types of disseminated intravascular coagulation: Clinical and animal models. J. Intensive Care 2014, 2, 1–7. [Google Scholar] [CrossRef]
- Gando, S.; Levi, M.; Toh, C.-H. Disseminated intravascular coagulation. Nat. Rev. Dis. Prim. 2016, 2, 16037. [Google Scholar] [CrossRef]
- Al-Samkari, H.; Leaf, R.S.K.; Dzik, W.H.; Carlson, J.C.; Fogerty, A.E.; Waheed, A.; Goodarzi, K.; Bendapudi, P.K.; Bornikova, L.; Gupta, S.; et al. COVID-19 and coagulation: Bleeding and thrombotic manifestations of SARS-CoV-2 infection. Blood 2020, 136, 489–500. [Google Scholar] [CrossRef]
- Konopka, K.E.; Nguyen, T.; Jentzen, J.M.; Rayes, O.; Schmidt, C.J.; Wilson, A.M.; Farver, C.F.; Myers, J.L. Diffuse alveolar damage (DAD) resulting from coronavirus disease 2019 Infection is Morphologically Indistinguishable from Other Causes of DAD. Histopathology 2020, 77, 570–578. [Google Scholar] [CrossRef]
- Goshua, G.; Pine, A.B.; Meizlish, M.L.; Chang, C.-H.; Zhang, H.; Bahel, P.; Baluha, A.; Bar, N.; Bona, R.D.; Burns, A.J.; et al. Endotheliopathy in COVID-19-associated coagulopathy: Evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020, 7, e575–e582. [Google Scholar] [CrossRef]
- Brojakowska, A.; Narula, J.; Shimony, R.; Bander, J. Clinical implications of SARS CoV-2 interaction with renin angiotensin system: JACC review topic of the week. J. Am. Coll. Cardiol. 2020, 75, 3085–3095. [Google Scholar] [CrossRef] [PubMed]
- Schwameis, M.; Schörgenhofer, C.; Assinger, A.; Steiner, M.M.; Jilma, B. VWF excess and ADAMTS13 deficiency: A unifying pathomechanism linking inflammation to thrombosis in DIC, malaria, and TTP. Thromb. Haemost. 2015, 113, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, A.; Ball, C.; Nolasco, L.; Moake, J.F.; Dong, J.-F. Effects of inflammatory cytokines on the release and cleavage of the endothelial cell–derived ultralarge von Willebrand factor multimers under flow. Blood 2004, 104, 100–106. [Google Scholar] [CrossRef]
- Iba, T.; Levy, J.H.; Levi, M.; Connors, J.M.; Thachil, J. Coagulopathy of Coronavirus Disease 2019. Crit. Care Med. 2020, 48, 1358–1364. [Google Scholar] [CrossRef] [PubMed]
- Peetermans, M.; Meyers, S.; Liesenborghs, L.; Vanhoorelbeke, K.; De Meyer, S.F.; Vandenbriele, C.; Lox, M.; Hoylaerts, M.F.; Martinod, K.; Jacquemin, M.; et al. Von Willebrand factor and ADAMTS13 impact on the outcome of Staphylococcus aureus sepsis. J. Thromb. Haemost. 2020, 18, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Tomashefski, J.F., Jr.; Davies, P.; Boggis, C.; Greene, R.; Zapol, W.M.; Reid, L.M. The pulmonary vascular lesions of the adult respiratory distress syndrome. Am. J. Pathol. 1983, 112, 112–126. [Google Scholar] [PubMed]
- Smadja, D.M.; Guerin, C.L.; Chocron, R.; Yatim, N.; Boussier, J.; Gendron, N.; Khider, L.; Hadjadj, J.; Goudot, G.; DeBuc, B.; et al. Angiopoietin-2 as a marker of endothelial activation is a good predictor factor for intensive care unit admission of COVID-19 patients. Angiogenesis 2020, 23, 611–620. [Google Scholar] [CrossRef]
- Uchimido, R.; Schmidt, E.P.; Shapiro, N.I. The glycocalyx: A novel diagnostic and therapeutic target in sepsis. Crit. Care 2019, 23, 1–12. [Google Scholar] [CrossRef]
- Iba, T.; Levy, J.H. Inflammation and thrombosis: Roles of neutrophils, platelets and endothelial cells and their interactions in thrombus formation during sepsis. J. Thromb. Haemost. 2018, 16, 231–241. [Google Scholar] [CrossRef]
- Iba, T.; Ogura, H. Role of extracellular vesicles in the development of sepsis-induced coagulopathy. J. Intensive Care 2018, 6, 68. [Google Scholar] [CrossRef]
- D’Alonzo, D.; De Fenza, M.; Pavone, V. COVID-19 and pneumonia: A role for the uPA/uPAR system. Drug Discov. Today 2020, 25, 1528–1534. [Google Scholar] [CrossRef] [PubMed]
- McGonagle, D.; O’Donnell, J.S.; Sharif, K.; Emery, P.; Bridgewood, C. Immune mechanisms of pulmonary intravascular coag-ulopathy in COVID-19 pneumonia. Lancet Rheumatol. 2020, 2, e437–e445. [Google Scholar] [CrossRef]
- Karakike, E.; Giamarellos-Bourboulis, E.J. Macrophage Activation-Like Syndrome: A Distinct Entity Leading to Early Death in Sepsis. Front. Immunol. 2019, 10, 55. [Google Scholar] [CrossRef]
- Middleton, E.A.; He, X.-Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef]
- Bautista-Vargas, M.; Bonilla-Abadía, F.; Cañas, C.A. Potential role for tissue factor in the pathogenesis of hypercoagulability associated with in COVID-19. J. Thromb. Thrombolysis 2020, 50, 479–483. [Google Scholar] [CrossRef]
- Rodríguez, Y.; Novelli, L.; Rojas, M.; De Santis, M.; Acosta-Ampudia, Y.; Monsalve, D.M.; Ramírez-Santana, C.; Costanzo, A.; Ridgway, W.M.; Ansari, A.A.; et al. Autoinflammatory and autoimmune conditions at the crossroad of COVID-19. J. Autoimmun. 2020, 114, 102506. [Google Scholar] [CrossRef]
- Wiedermann, F.J.; Lederer, W.; Mayr, A.J.; Sepp, N.; Herold, M.; Schobersberger, W. Prospective observational study of antiphospholipid antibodies in acute lung injury and acute respiratory distress syndrome: Comparison with catastrophic antiphospholipid syndrome. Lupus 2003, 12, 462–467. [Google Scholar] [CrossRef]
- Chen, M.; Daha, M.R.; Kallenberg, C.G.M. The complement system in systemic autoimmune disease. J. Autoimmun. 2010, 34, J276–J286. [Google Scholar] [CrossRef]
- Magro, C.; Mulvey, J.J.; Berlin, D.; Nuovo, G.; Salvatore, S.; Harp, J.; Baxter-Stoltzfus, A.; Laurence, J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases. Transl. Res. 2020, 220, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.J.; Wiffen, L.J.; Brown, T.P. COVID-19: A collision of complement, coagulation and inflammatory pathways. J. Thromb. Haemost. 2020, 18, 2110–2117. [Google Scholar] [CrossRef]
- Levi, M. COVID-19 coagulopathy vs disseminated intravascular coagulation. Blood Adv. 2020, 4, 2850. [Google Scholar] [CrossRef] [PubMed]
- Ranucci, M.; Ballotta, A.; Di Dedda, U.; Bayshnikova, E.; Poli, M.D.; Resta, M.; Falco, M.; Albano, G.; Menicanti, L. The procoagulant pattern of patients with COVID-19 acute respiratory distress syndrome. J. Thromb. Haemost. 2020, 18, 1747–1751. [Google Scholar] [CrossRef]
- Matsubara, T.; Yamakawa, K.; Umemura, Y.; Gando, S.; Ogura, H.; Shiraishi, A.; Kushimoto, S.; Abe, T.; Tarui, T.; Hagiwara, A.; et al. Significance of plasma fibrinogen level and antithrombin activity in sepsis: A multicenter cohort study using a cubic spline model. Thromb. Res. 2019, 181, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Iba, T.; Levy, J.H.; Warkentin, T.E.; Thachil, J.; Van Der Poll, T.; Levi, M. Diagnosis and management of sepsis-induced coagulopathy and disseminated intravascular coagulation. J. Thromb. Haemost. 2019, 17, 1989–1994. [Google Scholar] [CrossRef] [PubMed]
- Semeraro, F.; Ammollo, C.T.; Caironi, P.; Masson, S.; Latini, R.; Panigada, M.; Semeraro, N.; Gattinoni, L.; Colucci, M. Low D-dimer levels in sepsis: Good or bad? Thromb. Res. 2019, 174, 13–15. [Google Scholar] [CrossRef]
- Kidokoro, A.; Iba, T.; Fukunaga, M.; Yagi, Y. Alterations in coagulation and fibrinolysis during sepsis. Shock 1996, 5, 223–228. [Google Scholar] [CrossRef]
- Iba, T.; Levy, J.H.; Thachil, J.; Wada, H.; Levi, M. The progression from coagulopathy to disseminated intravascular coagulation in representative underlying diseases. Thromb. Res. 2019, 179, 11–14. [Google Scholar] [CrossRef]
- Levi, M.; Thachil, J. Coronavirus Disease 2019 Coagulopathy: Disseminated Intravascular Coagulation and Thrombotic Microangiopathy—Either, Neither, or Both. Semin. Thromb. Hemost. 2020, 46, 781–784. [Google Scholar] [CrossRef]
- Hernandez, C.; Bruckner, A.L. Focus on “COVID Toes. ” JAMA Dermatol. 2020, 156, 1003. [Google Scholar] [CrossRef]
- Llamas-Velasco, M.; Muñoz-Hernández, P.; Lázaro-González, J.; Reolid-Pérez, A.; Abad-Santamaría, B.; Fraga, J.; Daudén-Tello, E. Thrombotic occlusive vasculopathy in a skin biopsy from a livedoid lesion of a patient with COVID -19. Br. J. Dermatol. 2020, 183, 591–593. [Google Scholar] [CrossRef]
- Levy, J.H.; Ghadimi, K.; Faraoni, D.; Van Diepen, S.; Levy, B.; Hotchkiss, R.; Connors, J.M.; Iba, T.; Warkentin, T.E. Ischemic limb necrosis in septic shock: What is the role of high-dose vasopressor therapy? J. Thromb. Haemost. 2019, 17, 1973–1978. [Google Scholar] [CrossRef] [PubMed]
- Thachil, J.; Tang, N.; Gando, S.; Falanga, A.; Cattaneo, M.; Levi, M.; Clark, C.; Iba, T. ISTH interim guidance on recognition and management of coagulopathy in COVID-19. J. Thromb. Haemost. 2020, 18, 1023–1026. [Google Scholar] [CrossRef] [PubMed]
- Murao, S.; Yamakawa, K. A Systematic Summary of Systematic Reviews on Anticoagulant Therapy in Sepsis. J. Clin. Med. 2019, 8, 1869. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iba, T.; Warkentin, T.E.; Thachil, J.; Levi, M.; Levy, J.H. Proposal of the Definition for COVID-19-Associated Coagulopathy. J. Clin. Med. 2021, 10, 191. https://doi.org/10.3390/jcm10020191
Iba T, Warkentin TE, Thachil J, Levi M, Levy JH. Proposal of the Definition for COVID-19-Associated Coagulopathy. Journal of Clinical Medicine. 2021; 10(2):191. https://doi.org/10.3390/jcm10020191
Chicago/Turabian StyleIba, Toshiaki, Theodore E. Warkentin, Jecko Thachil, Marcel Levi, and Jerrold H. Levy. 2021. "Proposal of the Definition for COVID-19-Associated Coagulopathy" Journal of Clinical Medicine 10, no. 2: 191. https://doi.org/10.3390/jcm10020191
APA StyleIba, T., Warkentin, T. E., Thachil, J., Levi, M., & Levy, J. H. (2021). Proposal of the Definition for COVID-19-Associated Coagulopathy. Journal of Clinical Medicine, 10(2), 191. https://doi.org/10.3390/jcm10020191