
Blood Biomarkers for Alzheimer’s Disease in Down Syndrome

 ,
,
Abstract
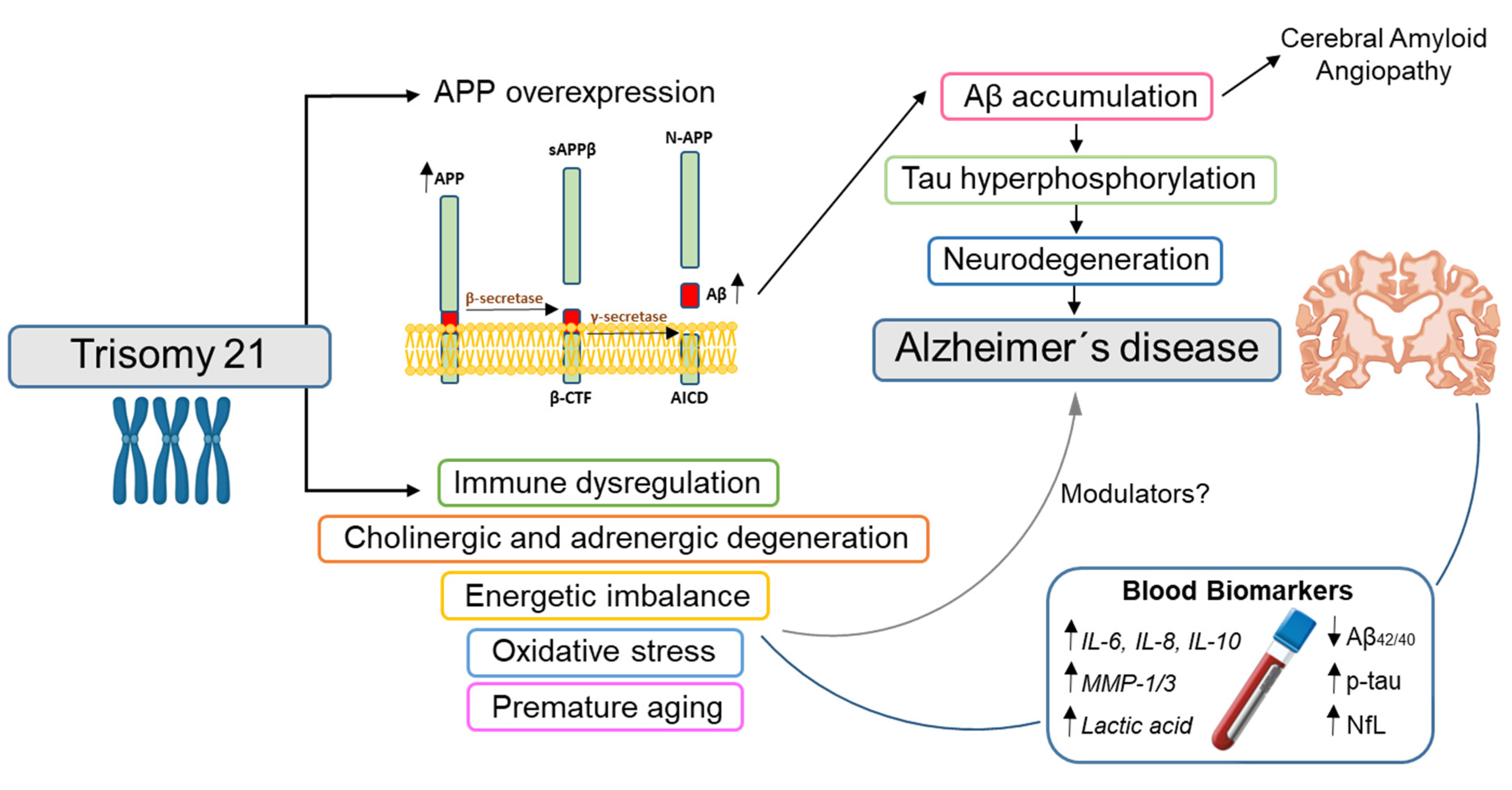
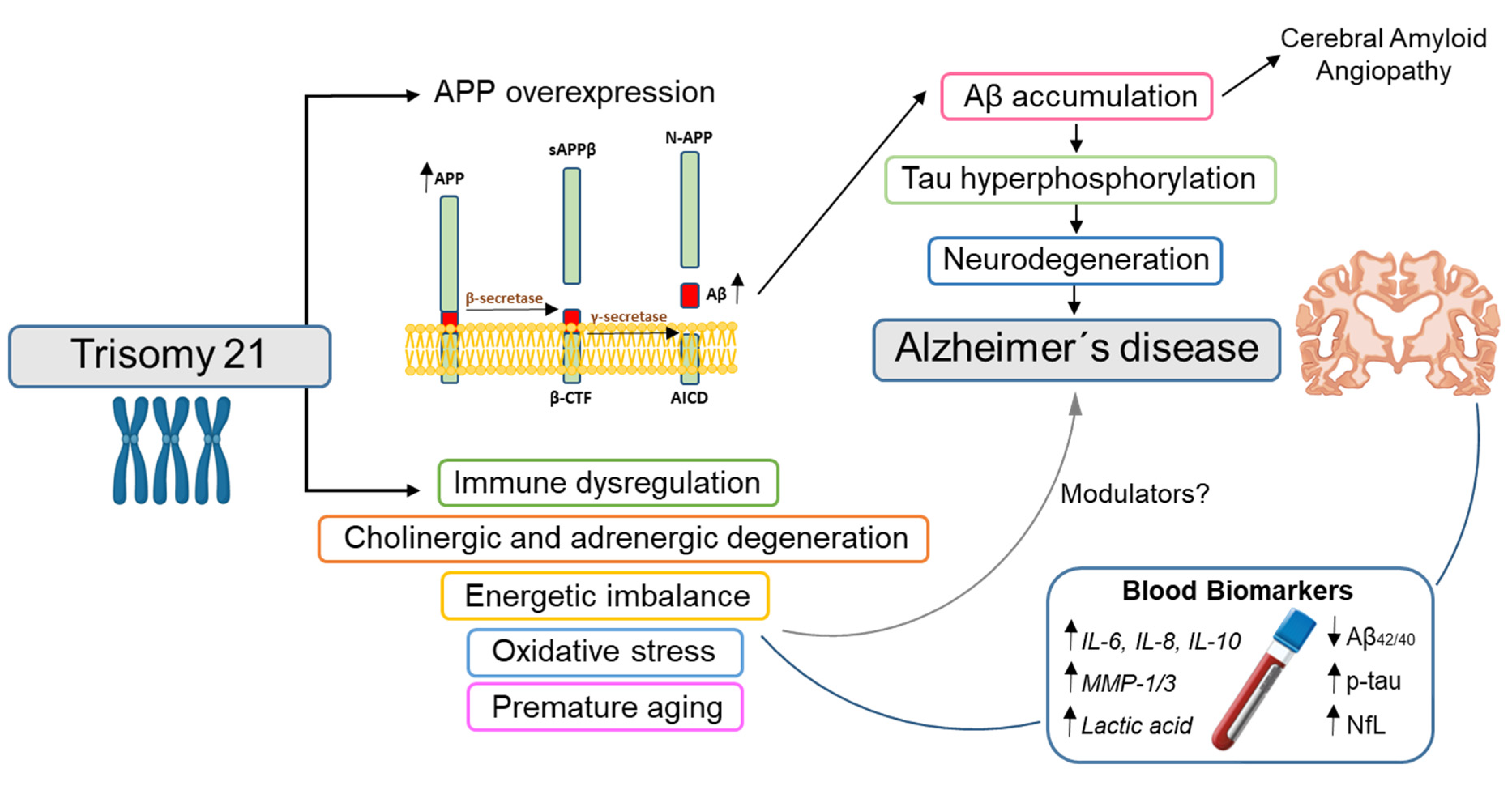
:1. Introduction
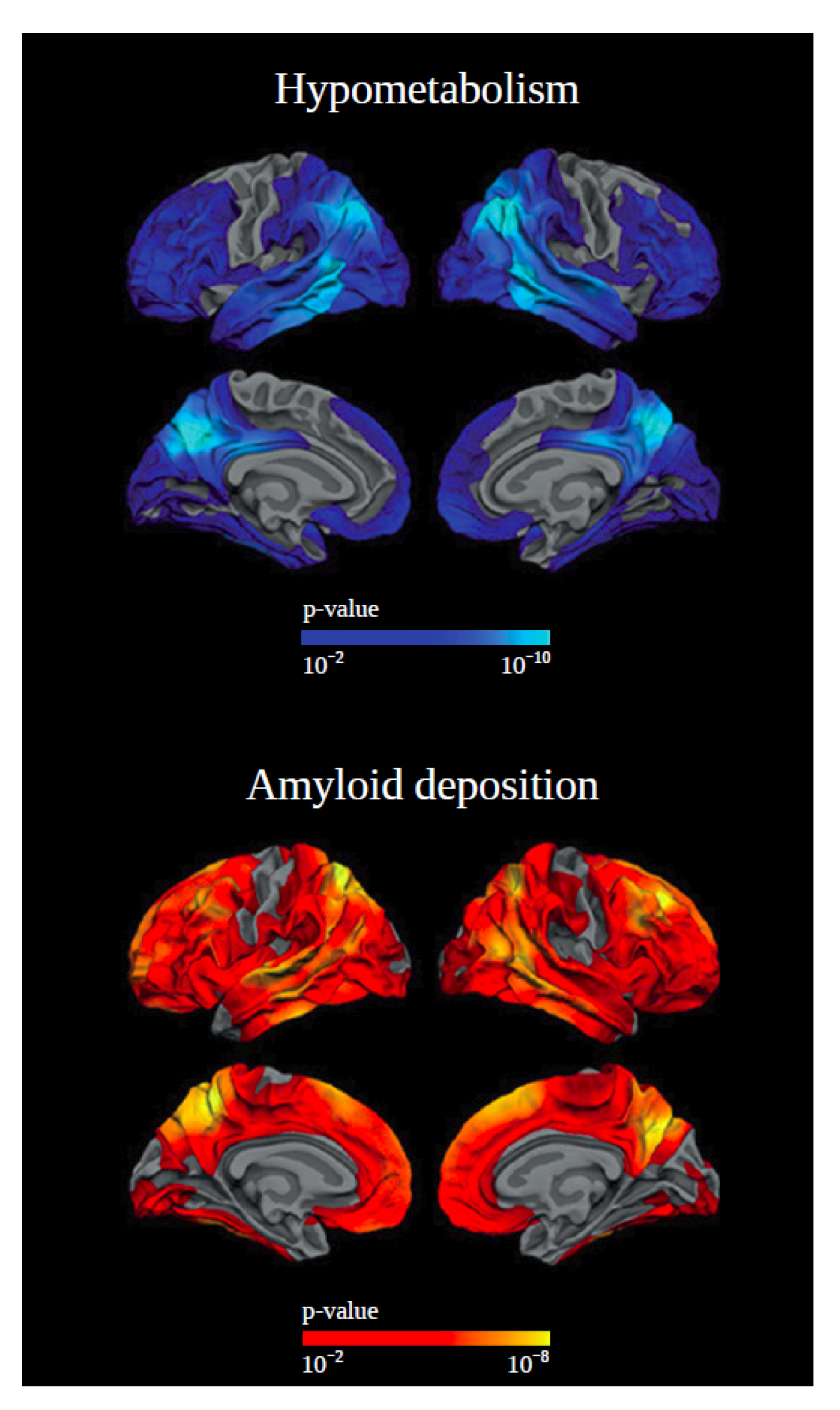
2. PET Biomarkers
3. CSF Biomarkers
4. Blood Biomarkers
4.1. Blood Biomarkers Also Used in Sporadic AD
4.1.1. Amyloid: Plasma Aβ
4.1.2. Tau: Plasma Total and Phosphorylated Tau
4.1.3. Neurodegeneration: Plasma/Serum NfL
4.2. Blood Biomarkers for Non-ATN Processes in DS
4.2.1. Inflammatory Biomarkers
4.2.2. Cholinergic and Adrenergic Biomarkers
4.2.3. Energy Metabolism and Oxidative Stress Biomarkers
4.2.4. DNA Biomarkers

{kind=link}
{kind=link}
| Affected Mechanism | Biomarker | Matrix | Technique | Platform | Results in DS Dementia | Refs |
|---|---|---|---|---|---|---|
| Inflammatory response | IFN-γ | Plasma | Mesoscale | Multi-Spot V-Plex Pro-inflammatory Panel (MesoScale Discovery) | DS ~ controls DS-AD > controls | [62] |
| TNF-α | DS > controls DS-AD > controls | |||||
| IL-6 | DS > controls DS-AD > controls | |||||
| IL-8 | DS ~ controls DS-AD > controls | |||||
| IL-10 | DS > controls DS-AD > controls | |||||
| NGAL | Serum | ELISA | ELISA kit (R&D systems) ELISA reader (Asys UVM 340 Biochrom) | Increased in DS. Association with species of Aβ depending on dementia progression | [108] | |
| NGF metabolic pathway | proNGF | Plasma | WB | Semi-dry transfer (Bio-Rad) | DS > controls DS-AD > controls | [116] |
| neuroserpin | WB | Semi-dry transfer (Bio-Rad) | Similar levels in all groups | |||
| tPA | ELISA | tPA ELISA kit (Abcam) | DS < controls DS-AD < controls | |||
| MMP-1 | Mesoscale | Multi-Spot MMP 3-Plex Ultra-Sensitive kit SECTOR Imager 2400 (MesoScale Discovery) | DS > controls DS-AD > controls | |||
| MMP-3 | DS > controls DS-AD > controls | |||||
| MMP-9 | Similar levels in all groups | |||||
| Biogenic amines | (nor)adrenergic (NA/A, MHPG) | Serum | RP-HPLC | AlexysTM Dual Monoamines Analyzer | Lower levels MHPG in DS dementia vs. non-demented DS and heathy controls | [118] |
| serotonergic (5-HT, 5-HIAA) | ||||||
| dopaminergic (DA, HVA, DOPAC) | ||||||
| Carbon metabolism | Lactic acid | Plasma | Targeted LC-MS/MS | Triple Quadrupole MS (Xevo-TQ-S, Waters Corporation) | DS-AD > non-AD-DS | [120] |
| Pyruvic acid | DS-AD > non-AD-DS | |||||
| Methyladipic acid | DS-AD > non-AD-DS | |||||
| Uridine | DS-AD < non-AD-DS | |||||
| Oxidative stress | SOD | Cytosolic and intracellular fractions from neutrophils | Spectro- photometry | Hitachi U-2010 spectrophotometer | SOD activity correlates to memory functioning in DS | [122] |
| DNA alterations | Telomere length | DNA extracted from T-lymphocytes | FISH | MetaSystems ISIS Image analyzer | Shortening of telomeres changes with AD progression in DS | [123] |
| DNA mehylation | DNA extracted from whole blood | Sodium bisulfite conversion | Infinium MethylationEPIC BeadChips and Illumina iScan | 2716 differentially methylated sites in DS, 9 related to dementia. | [124] |
5. Past Limitations and Future Perspectives
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Baird, P.A.; Sadovnick, A.D. Life expectancy in Down syndrome adults. Lancet 1988, 2, 1354–1356. [Google Scholar] [CrossRef]
- Rafii, M.S.; Santoro, S.L. Prevalence and Severity of Alzheimer Disease in Individuals with Down Syndrome. JAMA Neurol. 2019, 76, 142–143. [Google Scholar] [CrossRef] [PubMed]
- Ballard, C.; Mobley, W.; Hardy, J.; Williams, G.; Corbett, A. Dementia in Down’s syndrome. Lancet Neurol. 2016, 15, 622–636. [Google Scholar] [CrossRef]
- Dekker, A.D.; Fortea, J.; Blesa, R.; De Deyn, P.P. Cerebrospinal fluid biomarkers for Alzheimer’s disease in Down syndrome. Alzheimers Dement. 2017, 8, 1–10. [Google Scholar] [CrossRef]
- Lemere, C.A.; Blusztajn, J.K.; Yamaguchi, H.; Wisniewski, T.; Saido, T.C.; Selkoe, D.J. Sequence of deposition of heterogeneous amyloid beta-peptides and APO E in Down syndrome: Implications for initial events in amyloid plaque formation. Neurobiol. Dis. 1996, 3, 16–32. [Google Scholar] [CrossRef] [Green Version]
- Davidson, Y.S.; Robinson, A.; Prasher, V.P.; Mann, D.M.A. The age of onset and evolution of Braak tangle stage and Thal amyloid pathology of Alzheimer’s disease in individuals with Down syndrome. Acta Neuropathol. Commun. 2018, 6, 56. [Google Scholar] [CrossRef]
- Mann, D.M.A.; Davidson, Y.S.; Robinson, A.C.; Allen, N.; Hashimoto, T.; Richardson, A.; Jones, M.; Snowden, J.S.; Pendleton, N.; Potier, M.C.; et al. Patterns and severity of vascular amyloid in Alzheimer’s disease associated with duplications and missense mutations in APP gene, Down syndrome and sporadic Alzheimer’s disease. Acta Neuropathol. 2018, 136, 569–587. [Google Scholar] [CrossRef]
- Carmona-Iragui, M.; Videla, L.; Lleó, A.; Fortea, J. Down syndrome, Alzheimer disease, and cerebral amyloid angiopathy: The complex triangle of brain amyloidosis. Dev. Neurobiol. 2019, 79, 716–737. [Google Scholar] [CrossRef]
- Doran, E.; Keator, D.; Head, E.; Phelan, M.J.; Kim, R.; Totoiu, M.; Barrio, J.R.; Small, G.W.; Potkin, S.G.; Lott, I.T. Down Syndrome, Partial Trisomy 21, and Absence of Alzheimer’s Disease: The Role of APP. J. Alzheimers Dis. 2017, 56, 459–470. [Google Scholar] [CrossRef] [Green Version]
- Wiseman, F.K.; Pulford, L.J.; Barkus, C.; Liao, F.; Portelius, E.; Webb, R.; Chávez-Gutiérrez, L.; Cleverley, K.; Noy, S.; Sheppard, O.; et al. Trisomy of human chromosome 21 enhances amyloid-β deposition independently of an extra copy of APP. Brain 2018, 141, 2457–2474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, H.M.; Bain, L.J.; Brickman, A.M.; Carrillo, M.C.; Esbensen, A.J.; Espinosa, J.M.; Fernandez, F.; Fortea, J.; Hartley, S.L.; Head, E.; et al. Further understanding the connection between Alzheimer’s disease and Down syndrome. Alzheimers Dement. 2020, 16, 1065–1077. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.J.; Aisen, P.S.; De Strooper, B.; Fox, N.C.; Lemere, C.A.; Ringman, J.M.; Salloway, S.; Sperling, R.A.; Windisch, M.; Xiong, C. Autosomal-dominant Alzheimer’s disease: A review and proposal for the prevention of Alzheimer’s disease. Alzheimers Res. Ther. 2011, 3, 1. [Google Scholar] [CrossRef] [Green Version]
- Fortea, J.; Vilaplana, E.; Carmona-Iragui, M.; Benejam, B.; Videla, L.; Barroeta, I.; Fernández, S.; Altuna, M.; Pegueroles, J.; Montal, V.; et al. Clinical and biomarker changes of Alzheimer’s disease in adults with Down syndrome: A cross-sectional study. Lancet 2020, 395, 1988–1997. [Google Scholar] [CrossRef]
- Wisniewski, K.E.; Wisniewski, H.M.; Wen, G.Y. Occurrence of neuropathological changes and dementia of Alzheimer’s disease in Down’s syndrome. Ann Neurol 1985, 17, 278–282. [Google Scholar] [CrossRef]
- Fortea, J.; Carmona-Iragui, M.; Benejam, B.; Fernández, S.; Videla, L.; Barroeta, I.; Alcolea, D.; Pegueroles, J.; Muñoz, L.; Belbin, O.; et al. Plasma and CSF biomarkers for the diagnosis of Alzheimer’s disease in adults with Down syndrome: A cross-sectional study. Lancet Neurol. 2018, 17, 860–869. [Google Scholar] [CrossRef]
- Sabbagh, M.; Edgin, J. Clinical Assessment of Cognitive Decline in Adults with Down Syndrome. Curr. Alzheimer Res. 2016, 13, 30–34. [Google Scholar] [CrossRef]
- Folstein, M.F.; Folstein, S.E.; McHugh, P.R. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res. 1975, 12, 189–198. [Google Scholar] [CrossRef]
- Evenhuis, H.M. Evaluation of a screening instrument for dementia in ageing mentally retarded persons. J. Intellect. Disabil. Res. 1992, 36 Pt 4, 337–347. [Google Scholar] [CrossRef]
- Moran, J.A.; Rafii, M.S.; Keller, S.M.; Singh, B.K.; Janicki, M.P. The National Task Group on Intellectual Disabilities and Dementia Practices consensus recommendations for the evaluation and management of dementia in adults with intellectual disabilities. Mayo Clin. Proc. 2013, 88, 831–840. [Google Scholar] [CrossRef] [Green Version]
- Tsou, A.Y.; Bulova, P.; Capone, G.; Chicoine, B.; Gelaro, B.; Harville, T.O.; Martin, B.A.; McGuire, D.E.; McKelvey, K.D.; Peterson, M.; et al. Medical Care of Adults with Down Syndrome: A Clinical Guideline. JAMA 2020, 324, 1543–1556. [Google Scholar] [CrossRef] [PubMed]
- Haxby, J.V. Neuropsychological evaluation of adults with Down’s syndrome: Patterns of selective impairment in non-demented old adults. J. Ment. Defic. Res. 1989, 33 Pt 3, 193–210. [Google Scholar] [CrossRef]
- Hon, J.; Huppert, F.A.; Holland, A.J.; Watson, P. Neuropsychological assessment of older adults with Down’s syndrome: An epidemiological study using the Cambridge Cognitive Examination (CAMCOG). Br. J. Clin. Psychol. 1999, 38, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Ball, S.L.; Holland, A.J.; Huppert, F.A.; Treppner, P.; Watson, P.; Hon, J. The modified CAMDEX informant interview is a valid and reliable tool for use in the diagnosis of dementia in adults with Down’s syndrome. J. Intellect. Disabil. Res. 2004, 48, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Startin, C.M.; Lowe, B.; Hamburg, S.; Hithersay, R.; Strydom, A. Validating the Cognitive Scale for Down Syndrome (CS-DS) to Detect Longitudinal Cognitive Decline in Adults with Down Syndrome. Front. Psychiatry 2019, 10, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, D.M.; Yates, P.O.; Marcyniuk, B. Alzheimer’s presenile dementia, senile dementia of Alzheimer type and Down’s syndrome in middle age form an age related continuum of pathological changes. Neuropathol. Appl. Neurobiol. 1984, 10, 185–207. [Google Scholar] [CrossRef] [PubMed]
- Annus, T.; Wilson, L.R.; Hong, Y.T.; Acosta-Cabronero, J.; Fryer, T.D.; Cardenas-Blanco, A.; Smith, R.; Boros, I.; Coles, J.P.; Aigbirhio, F.I.; et al. The pattern of amyloid accumulation in the brains of adults with Down syndrome. Alzheimers Dement. 2016, 12, 538–545. [Google Scholar] [CrossRef] [Green Version]
- Sabbagh, M.N.; Chen, K.; Rogers, J.; Fleisher, A.S.; Liebsack, C.; Bandy, D.; Belden, C.; Protas, H.; Thiyyagura, P.; Liu, X.; et al. Florbetapir PET, FDG PET, and MRI in Down syndrome individuals with and without Alzheimer’s dementia. Alzheimers Dement. 2015, 11, 994–1004. [Google Scholar] [CrossRef] [Green Version]
- Cole, J.H.; Annus, T.; Wilson, L.R.; Remtulla, R.; Hong, Y.T.; Fryer, T.D.; Acosta-Cabronero, J.; Cardenas-Blanco, A.; Smith, R.; Menon, D.K.; et al. Brain-predicted age in Down syndrome is associated with beta amyloid deposition and cognitive decline. Neurobiol. Aging 2017, 56, 41–49. [Google Scholar] [CrossRef]
- Rafii, M.S.; Wishnek, H.; Brewer, J.B.; Donohue, M.C.; Ness, S.; Mobley, W.C.; Aisen, P.S.; Rissman, R.A. The down syndrome biomarker initiative (DSBI) pilot: Proof of concept for deep phenotyping of Alzheimer’s disease biomarkers in down syndrome. Front. Behav. Neurosci. 2015, 9, 239. [Google Scholar] [CrossRef] [Green Version]
- Matthews, D.C.; Lukic, A.S.; Andrews, R.D.; Marendic, B.; Brewer, J.; Rissman, R.A.; Mosconi, L.; Strother, S.C.; Wernick, M.N.; Mobley, W.C.; et al. Dissociation of Down syndrome and Alzheimer’s disease effects with imaging. Alzheimers Dement. 2016, 2, 69–81. [Google Scholar] [CrossRef] [Green Version]
- Lao, P.J.; Betthauser, T.J.; Hillmer, A.T.; Price, J.C.; Klunk, W.E.; Mihaila, I.; Higgins, A.T.; Bulova, P.D.; Hartley, S.L.; Hardison, R.; et al. The effects of normal aging on amyloid-β deposition in nondemented adults with Down syndrome as imaged by carbon 11-labeled Pittsburgh compound B. Alzheimers Dement. 2016, 12, 380–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeVine, H., 3rd; Spielmann, H.P.; Matveev, S.; Cauvi, F.M.; Murphy, M.P.; Beckett, T.L.; McCarty, K.; Lott, I.T.; Doran, E.; Schmitt, F.; et al. Down syndrome: Age-dependence of PiB binding in postmortem frontal cortex across the lifespan. Neurobiol. Aging 2017, 54, 163–169. [Google Scholar] [CrossRef] [Green Version]
- Jennings, D.; Seibyl, J.; Sabbagh, M.; Lai, F.; Hopkins, W.; Bullich, S.; Gimenez, M.; Reininger, C.; Putz, B.; Stephens, A.; et al. Age dependence of brain β-amyloid deposition in Down syndrome: An [18F] florbetaben PET study. Neurology 2015, 84, 500–507. [Google Scholar] [CrossRef]
- Handen, B.L.; Cohen, A.D.; Channamalappa, U.; Bulova, P.; Cannon, S.A.; Cohen, W.I.; Mathis, C.A.; Price, J.C.; Klunk, W.E. Imaging brain amyloid in nondemented young adults with Down syndrome using Pittsburgh compound B. Alzheimers Dement. 2012, 8, 496–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klunk, W.E.; Price, J.C.; Mathis, C.A.; Tsopelas, N.D.; Lopresti, B.J.; Ziolko, S.K.; Bi, W.; Hoge, J.A.; Cohen, A.D.; Ikonomovic, M.D.; et al. Amyloid deposition begins in the striatum of presenilin-1 mutation carriers from two unrelated pedigrees. J. Neurosci. 2007, 27, 6174–6184. [Google Scholar] [CrossRef] [Green Version]
- Schöll, M.; Maass, A.; Mattsson, N.; Ashton, N.J.; Blennow, K.; Zetterberg, H.; Jagust, W. Biomarkers for tau pathology. Mol. Cell Neurosci. 2019, 97, 18–33. [Google Scholar] [CrossRef]
- Rafii, M.S.; Lukic, A.S.; Andrews, R.D.; Brewer, J.; Rissman, R.A.; Strother, S.C.; Wernick, M.N.; Pennington, C.; Mobley, W.C.; Ness, S.; et al. PET Imaging of Tau Pathology and Relationship to Amyloid, Longitudinal MRI, and Cognitive Change in Down Syndrome: Results from the Down Syndrome Biomarker Initiative (DSBI). J. Alzheimers Dis. 2017, 60, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Haier, R.J.; Head, K.; Head, E.; Lott, I.T. Neuroimaging of individuals with Down’s syndrome at-risk for dementia: Evidence for possible compensatory events. Neuroimage 2008, 39, 1324–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Leuzy, A.; Ashton, N.J.; Mattsson-Carlgren, N.; Dodich, A.; Boccardi, M.; Corre, J.; Drzezga, A.; Nordberg, A.; Ossenkoppele, R.; Zetterberg, H.; et al. 2020 update on the clinical validity of cerebrospinal fluid amyloid, tau, and phospho-tau as biomarkers for Alzheimer’s disease in the context of a structured 5-phase development framework. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 2121–2139. [Google Scholar] [CrossRef]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Hölttä, M.; Rosén, C.; Olsson, C.; Strobel, G.; et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef]
- Portelius, E.; Hölttä, M.; Soininen, H.; Bjerke, M.; Zetterberg, H.; Westerlund, A.; Herukka, S.K.; Blennow, K.; Mattsson, N. Altered cerebrospinal fluid levels of amyloid β and amyloid precursor-like protein 1 peptides in Down’s syndrome. Neuromol. Med. 2014, 16, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Henson, R.L.; Doran, E.; Christian, B.T.; Handen, B.L.; Klunk, W.E.; Lai, F.; Lee, J.H.; Rosas, H.D.; Schupf, N.; Zaman, S.H.; et al. Cerebrospinal fluid biomarkers of Alzheimer’s disease in a cohort of adults with Down syndrome. Alzheimers Dement. 2020, 12, e12057. [Google Scholar] [CrossRef]
- Tapiola, T.; Alafuzoff, I.; Herukka, S.K.; Parkkinen, L.; Hartikainen, P.; Soininen, H.; Pirttilä, T. Cerebrospinal fluid {beta}-amyloid 42 and tau proteins as biomarkers of Alzheimer-type pathologic changes in the brain. Arch. Neurol. 2009, 66, 382–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bejanin, A.; Iulita, M.F.; Vilaplana, E.; Carmona-Iragui, M.; Benejam, B.; Videla, L.; Barroeta, I.; Fernandez, S.; Altuna, M.; Pegueroles, J.; et al. Association of Apolipoprotein E ɛ4 Allele with Clinical and Multimodal Biomarker Changes of Alzheimer Disease in Adults with Down Syndrome. JAMA Neurol. 2021, 78, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Portelius, E.; Soininen, H.; Andreasson, U.; Zetterberg, H.; Persson, R.; Karlsson, G.; Blennow, K.; Herukka, S.K.; Mattsson, N. Exploring Alzheimer molecular pathology in Down’s syndrome cerebrospinal fluid. Neurodegener Dis. 2014, 14, 98–106. [Google Scholar] [CrossRef]
- Delaby, C.; Alcolea, D.; Carmona-Iragui, M.; Illán-Gala, I.; Morenas-Rodríguez, E.; Barroeta, I.; Altuna, M.; Estellés, T.; Santos-Santos, M.; Turon-Sans, J.; et al. Differential levels of Neurofilament Light protein in cerebrospinal fluid in patients with a wide range of neurodegenerative disorders. Sci. Rep. 2020, 10, 9161. [Google Scholar] [CrossRef] [PubMed]
- Galasko, D.; Xiao, M.; Xu, D.; Smirnov, D.; Salmon, D.P.; Dewit, N.; Vanbrabant, J.; Jacobs, D.; Vanderstichele, H.; Vanmechelen, E.; et al. Synaptic biomarkers in CSF aid in diagnosis, correlate with cognition and predict progression in MCI and Alzheimer’s disease. Alzheimers Dement. 2019, 5, 871–882. [Google Scholar] [CrossRef]
- Shao, K.; Shan, S.; Ru, W.; Ma, C. Association between serum NPTX2 and cognitive function in patients with vascular dementia. Brain Behav. 2020, 10, e01779. [Google Scholar] [CrossRef] [PubMed]
- van der Ende, E.L.; Xiao, M.; Xu, D.; Poos, J.M.; Panman, J.L.; Jiskoot, L.C.; Meeter, L.H.; Dopper, E.G.; Papma, J.M.; Heller, C.; et al. Neuronal pentraxin 2: A synapse-derived CSF biomarker in genetic frontotemporal dementia. J. Neurol. Neurosurg. Psychiatry 2020, 91, 612–621. [Google Scholar] [CrossRef] [Green Version]
- Belbin, O.; Xiao, M.F.; Xu, D.; Carmona-Iragui, M.; Pegueroles, J.; Benejam, B.; Videla, L.; Fernández, S.; Barroeta, I.; Nuñez-Llaves, R.; et al. Cerebrospinal fluid profile of NPTX2 supports role of Alzheimer’s disease-related inhibitory circuit dysfunction in adults with Down syndrome. Mol. Neurodegener 2020, 15, 46. [Google Scholar] [CrossRef] [PubMed]
- Lleó, A.; Carmona-Iragui, M.; Videla, L.; Fernández, S.; Benejam, B.; Pegueroles, J.; Barroeta, I.; Altuna, M.; Valldeneu, S.; Xiao, M.F.; et al. VAMP-2 is a surrogate cerebrospinal fluid marker of Alzheimer-related cognitive impairment in adults with Down syndrome. Alzheimers Res. Ther. 2021, 13, 119. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Feldman, H.H.; Frisoni, G.B.; Hampel, H.; Jagust, W.J.; Johnson, K.A.; Knopman, D.S.; et al. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016, 87, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Koychev, I.; Jansen, K.; Dette, A.; Shi, L.; Holling, H. Blood-Based ATN Biomarkers of Alzheimer’s Disease: A Meta-Analysis. J. Alzheimers Dis. 2021, 79, 177–195. [Google Scholar] [CrossRef]
- Rafii, M.S.; Ances, B.M.; Schupf, N.; Krinsky-McHale, S.J.; Mapstone, M.; Silverman, W.; Lott, I.; Klunk, W.; Head, E.; Christian, B.; et al. The AT(N) framework for Alzheimer’s disease in adults with Down syndrome. Alzheimers Dement. 2020, 12, e12062. [Google Scholar] [CrossRef]
- Mehta, P.D.; Capone, G.; Jewell, A.; Freedland, R.L. Increased amyloid beta protein levels in children and adolescents with Down syndrome. J. Neurol. Sci. 2007, 254, 22–27. [Google Scholar] [CrossRef]
- Mehta, P.D.; Dalton, A.J.; Mehta, S.P.; Kim, K.S.; Sersen, E.A.; Wisniewski, H.M. Increased plasma amyloid beta protein 1-42 levels in Down syndrome. Neurosci. Lett. 1998, 241, 13–16. [Google Scholar] [CrossRef]
- Mehta, P.D.; Mehta, S.P.; Fedor, B.; Patrick, B.A.; Emmerling, M.; Dalton, A.J. Plasma amyloid beta protein 1-42 levels are increased in old Down Syndrome but not in young Down Syndrome. Neurosci. Lett. 2003, 342, 155–158. [Google Scholar] [CrossRef]
- Mehta, P.D.; Pirttila, T.; Patrick, B.A.; Barshatzky, M.; Mehta, S.P. Amyloid beta protein 1-40 and 1-42 levels in matched cerebrospinal fluid and plasma from patients with Alzheimer disease. Neurosci. Lett. 2001, 304, 102–106. [Google Scholar] [CrossRef]
- Schupf, N.; Patel, B.; Pang, D.; Zigman, W.B.; Silverman, W.; Mehta, P.D.; Mayeux, R. Elevated plasma beta-amyloid peptide Abeta(42) levels, incident dementia, and mortality in Down syndrome. Arch. Neurol. 2007, 64, 1007–1013. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, Y.; Andrews, H.F.; Becker, A.G.; Gray, A.J.; Mehta, P.D.; Sano, M.C.; Dalton, A.J.; Aisen, P.S. The relationship of plasma Abeta levels to dementia in aging individuals with Down syndrome. Alzheimer Dis. Assoc. Disord. 2009, 23, 315–318. [Google Scholar] [CrossRef]
- Iulita, M.F.; Ower, A.; Barone, C.; Pentz, R.; Gubert, P.; Romano, C.; Cantarella, R.A.; Elia, F.; Buono, S.; Recupero, M.; et al. An inflammatory and trophic disconnect biomarker profile revealed in Down syndrome plasma: Relation to cognitive decline and longitudinal evaluation. Alzheimers Dement. 2016, 12, 1132–1148. [Google Scholar] [CrossRef]
- Head, E.; Doran, E.; Nistor, M.; Hill, M.; Schmitt, F.A.; Haier, R.J.; Lott, I.T. Plasma amyloid-β as a function of age, level of intellectual disability, and presence of dementia in Down syndrome. J. Alzheimers Dis. 2011, 23, 399–409. [Google Scholar] [CrossRef]
- Schupf, N.; Zigman, W.B.; Tang, M.X.; Pang, D.; Mayeux, R.; Mehta, P.; Silverman, W. Change in plasma Aß peptides and onset of dementia in adults with Down syndrome. Neurology 2010, 75, 1639–1644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schupf, N.; Patel, B.; Silverman, W.; Zigman, W.B.; Zhong, N.; Tycko, B.; Mehta, P.D.; Mayeux, R. Elevated plasma amyloid beta-peptide 1-42 and onset of dementia in adults with Down syndrome. Neurosci. Lett. 2001, 301, 199–203. [Google Scholar] [CrossRef]
- Jones, E.L.; Hanney, M.; Francis, P.T.; Ballard, C.G. Amyloid beta concentrations in older people with Down syndrome and dementia. Neurosci Lett 2009, 451, 162–164. [Google Scholar] [CrossRef] [PubMed]
- Prasher, V.P.; Sajith, S.G.; Mehta, P.; Zigman, W.B.; Schupf, N. Plasma beta-amyloid and duration of Alzheimer’s disease in adults with Down syndrome. Int. J. Geriatr. Psychiatry 2010, 25, 202–207. [Google Scholar] [CrossRef] [Green Version]
- Lee, N.C.; Yang, S.Y.; Chieh, J.J.; Huang, P.T.; Chang, L.M.; Chiu, Y.N.; Huang, A.C.; Chien, Y.H.; Hwu, W.L.; Chiu, M.J. Blood Beta-Amyloid and Tau in Down Syndrome: A Comparison with Alzheimer’s Disease. Front. Aging Neurosci. 2016, 8, 316. [Google Scholar] [CrossRef] [Green Version]
- Coppus, A.M.; Schuur, M.; Vergeer, J.; Janssens, A.C.; Oostra, B.A.; Verbeek, M.M.; van Duijn, C.M. Plasma β amyloid and the risk of Alzheimer’s disease in Down syndrome. Neurobiol. Aging 2012, 33, 1988–1994. [Google Scholar] [CrossRef] [Green Version]
- Alhajraf, F.; Ness, D.; Hye, A.; Strydom, A. Plasma amyloid and tau as dementia biomarkers in Down syndrome: Systematic review and meta-analyses. Dev. Neurobiol. 2019, 79, 684–698. [Google Scholar] [CrossRef]
- Alawode, D.O.; Heslegrave, A.J.; Ashton, N.J.; Karikari, T.K.; Simrén, J.; Montoliu-Gaya, L.; Pannee, J.; O’Connor, A.; Weston, P.S.; Lantero-Rodriguez, J.; et al. Transitioning from cerebrospinal fluid to blood tests to facilitate diagnosis and disease monitoring in Alzheimer’s disease. J. Intern Med. 2021. [Google Scholar] [CrossRef]
- Mengel, D.; Liu, W.; Glynn, R.J.; Selkoe, D.J.; Strydom, A.; Lai, F.; Rosas, H.D.; Torres, A.; Patsiogiannis, V.; Skotko, B.; et al. Dynamics of plasma biomarkers in Down syndrome: The relative levels of Aβ42 decrease with age, whereas NT1 tau and NfL increase. Alzheimers Res. Ther. 2020, 12, 27. [Google Scholar] [CrossRef] [Green Version]
- Startin, C.M.; Ashton, N.J.; Hamburg, S.; Hithersay, R.; Wiseman, F.K.; Mok, K.Y.; Hardy, J.; Lleó, A.; Lovestone, S.; Parnetti, L.; et al. Plasma biomarkers for amyloid, tau, and cytokines in Down syndrome and sporadic Alzheimer’s disease. Alzheimers Res. Ther. 2019, 11, 26. [Google Scholar] [CrossRef]
- Nakamura, A.; Kaneko, N.; Villemagne, V.L.; Kato, T.; Doecke, J.; Doré, V.; Fowler, C.; Li, Q.X.; Martins, R.; Rowe, C.; et al. High performance plasma amyloid-β biomarkers for Alzheimer’s disease. Nature 2018, 554, 249–254. [Google Scholar] [CrossRef]
- Schindler, S.E.; Bollinger, J.G.; Ovod, V.; Mawuenyega, K.G.; Li, Y.; Gordon, B.A.; Holtzman, D.M.; Morris, J.C.; Benzinger, T.L.S.; Xiong, C.; et al. High-precision plasma β-amyloid 42/40 predicts current and future brain amyloidosis. Neurology 2019, 93, e1647–e1659. [Google Scholar] [CrossRef]
- Palmqvist, S.; Janelidze, S.; Stomrud, E.; Zetterberg, H.; Karl, J.; Zink, K.; Bittner, T.; Mattsson, N.; Eichenlaub, U.; Blennow, K.; et al. Performance of Fully Automated Plasma Assays as Screening Tests for Alzheimer Disease-Related β-Amyloid Status. JAMA Neurol. 2019, 76, 1060–1069. [Google Scholar] [CrossRef] [PubMed]
- Keshavan, A.; Pannee, J.; Karikari, T.K.; Rodriguez, J.L.; Ashton, N.J.; Nicholas, J.M.; Cash, D.M.; Coath, W.; Lane, C.A.; Parker, T.D.; et al. Population-based blood screening for preclinical Alzheimer’s disease in a British birth cohort at age 70. Brain 2021. [Google Scholar] [CrossRef]
- Kasai, T.; Tatebe, H.; Kondo, M.; Ishii, R.; Ohmichi, T.; Yeung, W.T.E.; Morimoto, M.; Chiyonobu, T.; Terada, N.; Allsop, D.; et al. Increased levels of plasma total tau in adult Down syndrome. PLoS ONE 2017, 12, e0188802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, M.E.; Rafii, M.S.; Zhang, F.; Hall, J.; Julovich, D.; Ances, B.M.; Schupf, N.; Krinsky-McHale, S.J.; Mapstone, M.; Silverman, W.; et al. Plasma Total-Tau and Neurofilament Light Chain as Diagnostic Biomarkers of Alzheimer’s Disease Dementia and Mild Cognitive Impairment in Adults with Down Syndrome. J. Alzheimers Dis. 2021, 79, 671–681. [Google Scholar] [CrossRef]
- Simrén, J.; Leuzy, A.; Karikari, T.K.; Hye, A.; Benedet, A.L.; Lantero-Rodriguez, J.; Mattsson-Carlgren, N.; Schöll, M.; Mecocci, P.; Vellas, B.; et al. The diagnostic and prognostic capabilities of plasma biomarkers in Alzheimer’s disease. Alzheimers Dement. 2021. [Google Scholar] [CrossRef] [PubMed]
- Mattsson, N.; Zetterberg, H.; Janelidze, S.; Insel, P.S.; Andreasson, U.; Stomrud, E.; Palmqvist, S.; Baker, D.; Tan Hehir, C.A.; Jeromin, A.; et al. Plasma tau in Alzheimer disease. Neurology 2016, 87, 1827–1835. [Google Scholar] [CrossRef] [Green Version]
- Karikari, T.K.; Benedet, A.L.; Ashton, N.J.; Lantero Rodriguez, J.; Snellman, A.; Suárez-Calvet, M.; Saha-Chaudhuri, P.; Lussier, F.; Kvartsberg, H.; Rial, A.M.; et al. Diagnostic performance and prediction of clinical progression of plasma phospho-tau181 in the Alzheimer’s Disease Neuroimaging Initiative. Mol. Psychiatry 2020. [Google Scholar] [CrossRef]
- O’Connor, A.; Karikari, T.K.; Poole, T.; Ashton, N.J.; Lantero Rodriguez, J.; Khatun, A.; Swift, I.; Heslegrave, A.J.; Abel, E.; Chung, E.; et al. Plasma phospho-tau181 in presymptomatic and symptomatic familial Alzheimer’s disease: A longitudinal cohort study. Mol Psychiatry 2020. [Google Scholar] [CrossRef]
- Karikari, T.K.; Pascoal, T.A.; Ashton, N.J.; Janelidze, S.; Benedet, A.L.; Rodriguez, J.L.; Chamoun, M.; Savard, M.; Kang, M.S.; Therriault, J.; et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer’s disease: A diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. 2020, 19, 422–433. [Google Scholar] [CrossRef]
- Lantero Rodriguez, J.; Karikari, T.K.; Suárez-Calvet, M.; Troakes, C.; King, A.; Emersic, A.; Aarsland, D.; Hye, A.; Zetterberg, H.; Blennow, K.; et al. Plasma p-tau181 accurately predicts Alzheimer’s disease pathology at least 8 years prior to post-mortem and improves the clinical characterisation of cognitive decline. Acta Neuropathol. 2020, 140, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Mattsson-Carlgren, N.; Janelidze, S.; Palmqvist, S.; Cullen, N.; Svenningsson, A.L.; Strandberg, O.; Mengel, D.; Walsh, D.M.; Stomrud, E.; Dage, J.L.; et al. Longitudinal plasma p-tau217 is increased in early stages of Alzheimer’s disease. Brain 2020, 143, 3234–3241. [Google Scholar] [CrossRef]
- Ashton, N.J.; Pascoal, T.A.; Karikari, T.K.; Benedet, A.L.; Lantero-Rodriguez, J.; Brinkmalm, G.; Snellman, A.; Schöll, M.; Troakes, C.; Hye, A.; et al. Plasma p-tau231: A new biomarker for incipient Alzheimer’s disease pathology. Acta Neuropathol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Tatebe, H.; Kasai, T.; Ohmichi, T.; Kishi, Y.; Kakeya, T.; Waragai, M.; Kondo, M.; Allsop, D.; Tokuda, T. Quantification of plasma phosphorylated tau to use as a biomarker for brain Alzheimer pathology: Pilot case-control studies including patients with Alzheimer’s disease and down syndrome. Mol. Neurodegener 2017, 12, 63. [Google Scholar] [CrossRef] [Green Version]
- Hamlett, E.D.; Goetzl, E.J.; Ledreux, A.; Vasilevko, V.; Boger, H.A.; LaRosa, A.; Clark, D.; Carroll, S.L.; Carmona-Iragui, M.; Fortea, J.; et al. Neuronal exosomes reveal Alzheimer’s disease biomarkers in Down syndrome. Alzheimers Dement. 2017, 13, 541–549. [Google Scholar] [CrossRef] [Green Version]
- Lleó, A.; Zetterberg, H.; Pegueroles, J.; Karikari, T.K.; Carmona-Iragui, M.; Ashton, N.J.; Montal, V.; Barroeta, I.; Lantero-Rodríguez, J.; Videla, L.; et al. Phosphorylated tau 181 in plasma as a biomarker for Alzheimer’s disease in adults with Down syndrome: A cross-sectional study. Nat. Commun. 2021, 12, 4304. [Google Scholar] [CrossRef] [PubMed]
- Gafson, A.R.; Barthélemy, N.R.; Bomont, P.; Carare, R.O.; Durham, H.D.; Julien, J.P.; Kuhle, J.; Leppert, D.; Nixon, R.A.; Weller, R.O.; et al. Neurofilaments: Neurobiological foundations for biomarker applications. Brain 2020, 143, 1975–1998. [Google Scholar] [CrossRef]
- Benedet, A.L.; Ashton, N.J.; Pascoal, T.A.; Leuzy, A.; Mathotaarachchi, S.; Kang, M.S.; Therriault, J.; Savard, M.; Chamoun, M.; Schöll, M.; et al. Plasma neurofilament light associates with Alzheimer’s disease metabolic decline in amyloid-positive individuals. Alzheimers Dement. 2019, 11, 679–689. [Google Scholar] [CrossRef]
- Benedet, A.L.; Leuzy, A.; Pascoal, T.A.; Ashton, N.J.; Mathotaarachchi, S.; Savard, M.; Therriault, J.; Kang, M.S.; Chamoun, M.; Schöll, M.; et al. Stage-specific links between plasma neurofilament light and imaging biomarkers of Alzheimer’s disease. Brain 2020, 143, 3793–3804. [Google Scholar] [CrossRef]
- Blennow, K.; Zetterberg, H. Fluid biomarker-based molecular phenotyping of Alzheimer’s disease patients in research and clinical settings. Prog. Mol. Biol. Transl. Sci. 2019, 168, 3–23. [Google Scholar] [CrossRef]
- Strydom, A.; Heslegrave, A.; Startin, C.M.; Mok, K.Y.; Hardy, J.; Groet, J.; Nizetic, D.; Zetterberg, H. Neurofilament light as a blood biomarker for neurodegeneration in Down syndrome. Alzheimers Res. Ther. 2018, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- Shinomoto, M.; Kasai, T.; Tatebe, H.; Kondo, M.; Ohmichi, T.; Morimoto, M.; Chiyonobu, T.; Terada, N.; Allsop, D.; Yokota, I.; et al. Plasma neurofilament light chain: A potential prognostic biomarker of dementia in adult Down syndrome patients. PLoS ONE 2019, 14, e0211575. [Google Scholar] [CrossRef] [PubMed]
- Ashton, N.J.; Janelidze, S.; Al Khleifat, A.; Leuzy, A.; van der Ende, E.L.; Karikari, T.K.; Benedet, A.L.; Pascoal, T.A.; Lleó, A.; Parnetti, L.; et al. A multicentre validation study of the diagnostic value of plasma neurofilament light. Nat. Commun. 2021, 12, 3400. [Google Scholar] [CrossRef] [PubMed]
- Rafii, M.S.; Donohue, M.C.; Matthews, D.C.; Muranevici, G.; Ness, S.; O’Bryant, S.E.; Rissman, R.A. Plasma Neurofilament Light and Alzheimer’s Disease Biomarkers in Down Syndrome: Results from the Down Syndrome Biomarker Initiative (DSBI). J Alzheimers Dis. 2019, 70, 131–138. [Google Scholar] [CrossRef]
- Pape, S.E.; Al Janabi, T.; Ashton, N.J.; Hye, A.; Sheehan, R.; Gallagher, P.; Knight, B.; Prins, A.M.; Courtenay, K.; Jordanova, V.; et al. The reliability and validity of DSM 5 diagnostic criteria for neurocognitive disorder and relationship with plasma neurofilament light in a down syndrome population. Sci. Rep. 2021, 11, 13438. [Google Scholar] [CrossRef]
- Illouz, T.; Biragyn, A.; Iulita, M.F.; Flores-Aguilar, L.; Dierssen, M.; De Toma, I.; Antonarakis, S.E.; Yu, E.; Herault, Y.; Potier, M.C.; et al. Immune Dysregulation and the Increased Risk of Complications and Mortality Following Respiratory Tract Infections in Adults With Down Syndrome. Front. Immunol. 2021, 12, 621440. [Google Scholar] [CrossRef]
- Wilcock, D.M.; Griffin, W.S. Down’s syndrome, neuroinflammation, and Alzheimer neuropathogenesis. J. Neuroinflamm. 2013, 10, 84. [Google Scholar] [CrossRef] [Green Version]
- Cetiner, S.; Demirhan, O.; Inal, T.C.; Tastemir, D.; Sertdemir, Y. Analysis of peripheral blood T-cell subsets, natural killer cells and serum levels of cytokines in children with Down syndrome. Int. J. Immunogenet. 2010, 37, 233–237. [Google Scholar] [CrossRef]
- Zaki, M.E.; El-Bassyouni, H.T.; Tosson, A.M.; Youness, E.; Hussein, J. Coenzyme Q10 and pro-inflammatory markers in children with Down syndrome: Clinical and biochemical aspects. J. Pediatr. 2017, 93, 100–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corsi, M.M.; Dogliotti, G.; Pedroni, F.; Palazzi, E.; Magni, P.; Chiappelli, M.; Licastro, F. Plasma nerve growth factor (NGF) and inflammatory cytokines (IL-6 and MCP-1) in young and adult subjects with Down syndrome: An interesting pathway. Neuro Endocrinol. Lett. 2006, 27, 773–778. [Google Scholar] [PubMed]
- Nateghi Rostami, M.; Douraghi, M.; Miramin Mohammadi, A.; Nikmanesh, B. Altered serum pro-inflammatory cytokines in children with Down’s syndrome. Eur. Cytokine Netw. 2012, 23, 64–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Che, M.; Yuan, J.; Yu, Y.; Cao, C.; Qin, X.Y.; Cheng, Y. Aberrations in circulating inflammatory cytokine levels in patients with Down syndrome: A meta-analysis. Oncotarget 2017, 8, 84489–84496. [Google Scholar] [CrossRef] [Green Version]
- Naudé, P.J.; Nyakas, C.; Eiden, L.E.; Ait-Ali, D.; van der Heide, R.; Engelborghs, S.; Luiten, P.G.; De Deyn, P.P.; den Boer, J.A.; Eisel, U.L. Lipocalin 2: Novel component of proinflammatory signaling in Alzheimer’s disease. FASEB J. 2012, 26, 2811–2823. [Google Scholar] [CrossRef] [Green Version]
- Naudé, P.J.; Dekker, A.D.; Coppus, A.M.; Vermeiren, Y.; Eisel, U.L.; van Duijn, C.M.; Van Dam, D.; De Deyn, P.P. Serum NGAL is Associated with Distinct Plasma Amyloid-β Peptides According to the Clinical Diagnosis of Dementia in Down Syndrome. J. Alzheimers Dis. 2015, 45, 733–743. [Google Scholar] [CrossRef]
- Wilcock, D.M.; Hurban, J.; Helman, A.M.; Sudduth, T.L.; McCarty, K.L.; Beckett, T.L.; Ferrell, J.C.; Murphy, M.P.; Abner, E.L.; Schmitt, F.A.; et al. Down syndrome individuals with Alzheimer’s disease have a distinct neuroinflammatory phenotype compared to sporadic Alzheimer’s disease. Neurobiol. Aging 2015, 36, 2468–2474. [Google Scholar] [CrossRef] [Green Version]
- Petersen, M.E.; Zhang, F.; Schupf, N.; Krinsky-McHale, S.J.; Hall, J.; Mapstone, M.; Cheema, A.; Silverman, W.; Lott, I.; Rafii, M.S.; et al. Proteomic profiles for Alzheimer’s disease and mild cognitive impairment among adults with Down syndrome spanning serum and plasma: An Alzheimer’s Biomarker Consortium-Down Syndrome (ABC-DS) study. Alzheimers Dement. 2020, 12, e12039. [Google Scholar] [CrossRef]
- Casanova, M.F.; Walker, L.C.; Whitehouse, P.J.; Price, D.L. Abnormalities of the nucleus basalis in Down’s syndrome. Ann. Neurol. 1985, 18, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Etienne, P.; Robitaille, Y.; Wood, P.; Gauthier, S.; Nair, N.P.; Quirion, R. Nucleus basalis neuronal loss, neuritic plaques and choline acetyltransferase activity in advanced Alzheimer’s disease. Neuroscience 1986, 19, 1279–1291. [Google Scholar] [CrossRef]
- Cuello, A.C. Effects of trophic factors on the CNS cholinergic phenotype. Prog. Brain Res. 1996, 109, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Bruno, M.A.; Leon, W.C.; Fragoso, G.; Mushynski, W.E.; Almazan, G.; Cuello, A.C. Amyloid beta-induced nerve growth factor dysmetabolism in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2009, 68, 857–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iulita, M.F.; Do Carmo, S.; Ower, A.K.; Fortress, A.M.; Flores Aguilar, L.; Hanna, M.; Wisniewski, T.; Granholm, A.C.; Buhusi, M.; Busciglio, J.; et al. Nerve growth factor metabolic dysfunction in Down’s syndrome brains. Brain 2014, 137, 860–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pentz, R.; Iulita, M.F.; Ducatenzeiler, A.; Videla, L.; Benejam, B.; Iragui, M.C.; Blesa, R.; Lleó, A.; Fortea, J.; Cuello, A.C. Nerve growth factor (NGF) pathway biomarkers in Down syndrome prior to and after the onset of clinical Alzheimer’s disease: A paired CSF and plasma study. Alzheimers Dement. 2021, 17, 605–617. [Google Scholar] [CrossRef]
- Lanari, A.; Amenta, F.; Silvestrelli, G.; Tomassoni, D.; Parnetti, L. Neurotransmitter deficits in behavioural and psychological symptoms of Alzheimer’s disease. Mech. Ageing Dev. 2006, 127, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Dekker, A.D.; Coppus, A.M.; Vermeiren, Y.; Aerts, T.; van Duijn, C.M.; Kremer, B.P.; Naudé, P.J.; Van Dam, D.; De Deyn, P.P. Serum MHPG strongly predicts conversion to Alzheimer’s disease in behaviorally characterized subjects with Down syndrome. J. Alzheimers Dis. 2015, 43, 871–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosconi, L.; Pupi, A.; De Leon, M.J. Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2008, 1147, 180–195. [Google Scholar] [CrossRef]
- Gross, T.J.; Doran, E.; Cheema, A.K.; Head, E.; Lott, I.T.; Mapstone, M. Plasma metabolites related to cellular energy metabolism are altered in adults with Down syndrome and Alzheimer’s disease. Dev. Neurobiol. 2019, 79, 622–638. [Google Scholar] [CrossRef]
- Coskun, P.; Helguera, P.; Nemati, Z.; Bohannan, R.C.; Thomas, J.; Samuel, S.E.; Argueta, J.; Doran, E.; Wallace, D.C.; Lott, I.T.; et al. Metabolic and Growth Rate Alterations in Lymphoblastic Cell Lines Discriminate Between Down Syndrome and Alzheimer’s Disease. J. Alzheimers Dis. 2017, 55, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Zis, P.; Dickinson, M.; Shende, S.; Walker, Z.; Strydom, A. Oxidative stress and memory decline in adults with Down syndrome: Longitudinal study. J. Alzheimers Dis. 2012, 31, 277–283. [Google Scholar] [CrossRef]
- Jenkins, E.C.; Ye, L.; Krinsky-McHale, S.J.; Zigman, W.B.; Schupf, N.; Silverman, W.P. Telomere longitudinal shortening as a biomarker for dementia status of adults with Down syndrome. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2016, 171b, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Haertle, L.; Müller, T.; Lardenoije, R.; Maierhofer, A.; Dittrich, M.; Riemens, R.J.M.; Stora, S.; Roche, M.; Leber, M.; Riedel-Heller, S.; et al. Methylomic profiling in trisomy 21 identifies cognition- and Alzheimer’s disease-related dysregulation. Clin. Epigenet. 2019, 11, 195. [Google Scholar] [CrossRef] [Green Version]
- Rafii, M.S.; Zaman, S.; Handen, B.L. Integrating Biomarker Outcomes into Clinical Trials for Alzheimer’s Disease in Down Syndrome. J. Prev. Alzheimers Dis. 2021, 8, 48–51. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.B.; Janelidze, S.; Smith, R.; Mattsson-Carlgren, N.; Palmqvist, S.; Teunissen, C.E.; Zetterberg, H.; Stomrud, E.; Ashton, N.J.; Blennow, K.; et al. Plasma GFAP is an early marker of amyloid-β but not tau pathology in Alzheimer’s disease. Brain 2021. [Google Scholar] [CrossRef] [PubMed]
- Simrén, J.; Ashton, N.J.; Blennow, K.; Zetterberg, H. Blood neurofilament light in remote settings: Alternative protocols to support sample collection in challenging pre-analytical conditions. Alzheimers Dement. 2021, 13, e12145. [Google Scholar] [CrossRef]
- Altmann, P.; Ponleitner, M.; Rommer, P.S.; Haslacher, H.; Mucher, P.; Leutmezer, F.; Petzold, A.; Wotawa, C.; Lanzenberger, R.; Berger, T.; et al. Seven day pre-analytical stability of serum and plasma neurofilament light chain. Sci. Rep. 2021, 11, 11034. [Google Scholar] [CrossRef]
- Ashton, N.J.; Suárez-Calvet, M.; Karikari, T.K.; Lantero-Rodriguez, J.; Snellman, A.; Sauer, M.; Simrén, J.; Minguillon, C.; Fauria, K.; Blennow, K.; et al. Effects of pre-analytical procedures on blood biomarkers for Alzheimer’s pathophysiology, glial activation, and neurodegeneration. Alzheimers Dement. 2021, 13, e12168. [Google Scholar] [CrossRef]
- Lott, I.T.; Head, E. Dementia in Down syndrome: Unique insights for Alzheimer disease research. Nat. Rev. Neurol. 2019, 15, 135–147. [Google Scholar] [CrossRef]
- Flores-Aguilar, L.; Iulita, M.F.; Kovecses, O.; Torres, M.D.; Levi, S.M.; Zhang, Y.; Askenazi, M.; Wisniewski, T.; Busciglio, J.; Cuello, A.C. Evolution of neuroinflammation across the lifespan of individuals with Down syndrome. Brain 2020, 143, 3653–3671. [Google Scholar] [CrossRef] [PubMed]
- Handen, B.L.; Lott, I.T.; Christian, B.T.; Schupf, N.; O’Bryant, S.; Mapstone, M.; Fagan, A.M.; Lee, J.H.; Tudorascu, D.; Wang, M.C.; et al. The Alzheimer’s Biomarker Consortium-Down Syndrome: Rationale and methodology. Alzheimers Dement. 2020, 12, e12065. [Google Scholar] [CrossRef]
- Handen, B.L. The Search for Biomarkers of Alzheimer’s Disease in Down Syndrome. Am. J. Intellect. Dev. Disabil. 2020, 125, 97–99. [Google Scholar] [CrossRef] [PubMed]
- FDA. Available online: https://www.fda.gov/drugs/news-events-human-drugs/fdas-decision-approve-new-treatment-alzheimers-disease (accessed on 10 June 2021).
| Biomarker | DS Compared with Healthy Controls | DS-AD Compared with Cognitively Stable DS | Diagnostic Application | Future Challenges |
|---|---|---|---|---|
| Aβ (A) | Increased levels of Aβ40 and Aβ42. Contradictory results for Aβ42/Aβ40 ratio and for association between Aβ42 and age. | Higher plasma Aβ40 and lower Aβ42/Aβ40 ratio in demented DS. No association between Aβ42 and dementia status. | Low diagnostic performance. Overlap between groups. | Quantification by sensitive IP-MS. Analysis of other Aβ species other than Aβ40 and Aβ42 (Aβ37, Aβ38, Aβ41) |
| p-tau181 (T) | Increased from early 30s. P-tau396 also found increased in neuronal exosomes. | Increased levels in both prodromal AD and AD dementia. Earlier increases in APOE ε4 allele carriers. | High diagnostic accuracy to differentiate AD dementia, low for prodromal AD. | Study of other tau phosphorylations: p-tau205, p-tau217, p-tau231. |
| T-tau (N) | Increased. Positive correlation between age and plasma t-tau in DS. | Significantly higher levels in DS-AD but weak increase in prodromal AD. | Low diagnostic performance. Overlap between groups. | New sensitive assays that target CNS specific tau. |
| NfL (N) | Increased. Positive correlation between age and plasma NfL. | Increased levels in both prodromal AD and AD dementia. | High diagnostic performance for both prodromal AD and AD dementia. | Use as diagnostic tool in clinical routine. Combination with cognitive assessment. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montoliu-Gaya, L.; Strydom, A.; Blennow, K.; Zetterberg, H.; Ashton, N.J. Blood Biomarkers for Alzheimer’s Disease in Down Syndrome. J. Clin. Med. 2021, 10, 3639. https://doi.org/10.3390/jcm10163639
Montoliu-Gaya L, Strydom A, Blennow K, Zetterberg H, Ashton NJ. Blood Biomarkers for Alzheimer’s Disease in Down Syndrome. Journal of Clinical Medicine. 2021; 10(16):3639. https://doi.org/10.3390/jcm10163639
Chicago/Turabian StyleMontoliu-Gaya, Laia, Andre Strydom, Kaj Blennow, Henrik Zetterberg, and Nicholas James Ashton. 2021. "Blood Biomarkers for Alzheimer’s Disease in Down Syndrome" Journal of Clinical Medicine 10, no. 16: 3639. https://doi.org/10.3390/jcm10163639
APA StyleMontoliu-Gaya, L., Strydom, A., Blennow, K., Zetterberg, H., & Ashton, N. J. (2021). Blood Biomarkers for Alzheimer’s Disease in Down Syndrome. Journal of Clinical Medicine, 10(16), 3639. https://doi.org/10.3390/jcm10163639