
Aberrantly Glycosylated IgA1 in IgA Nephropathy: What We Know and What We Don’t Know

Abstract
1. Introduction
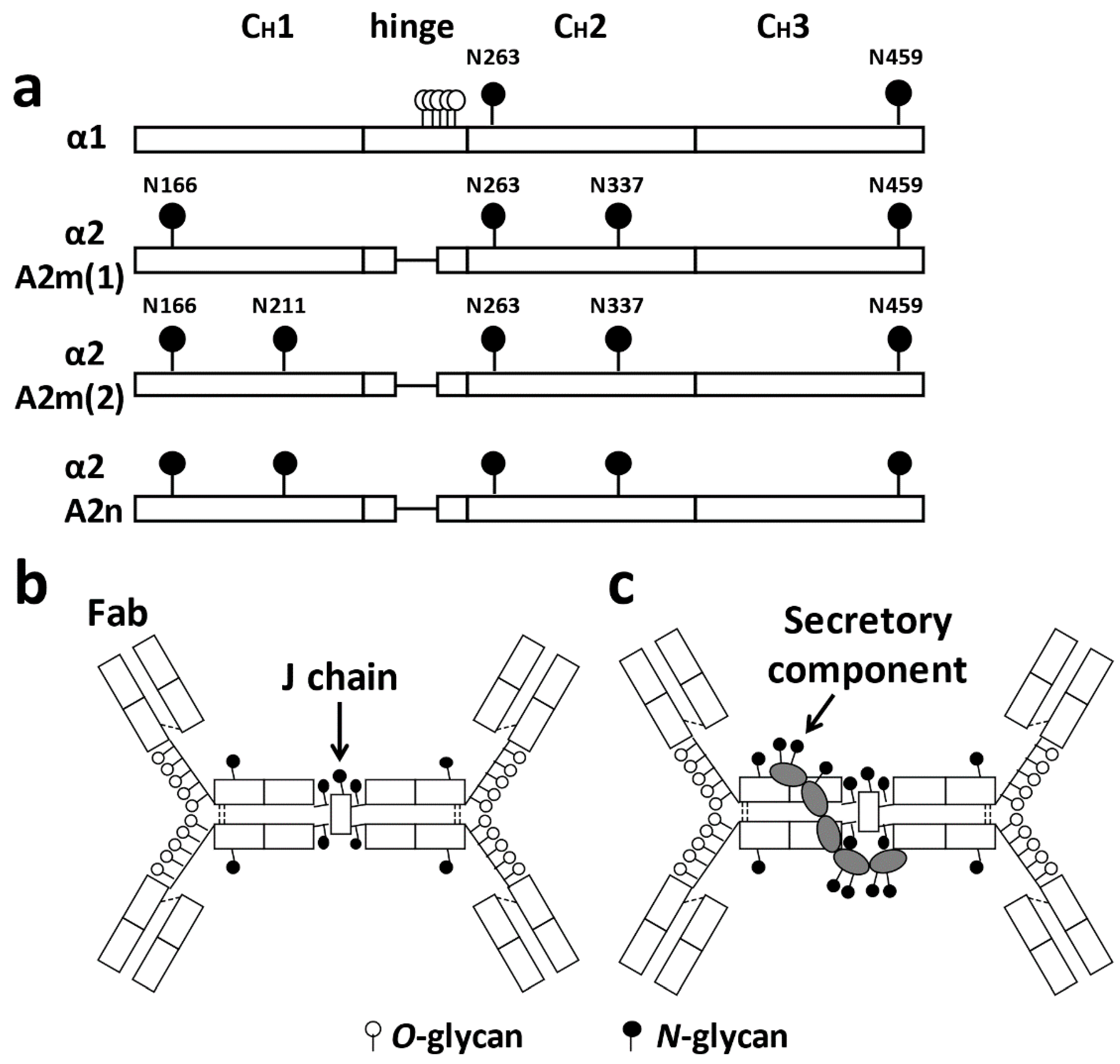
2. Structure of IgA
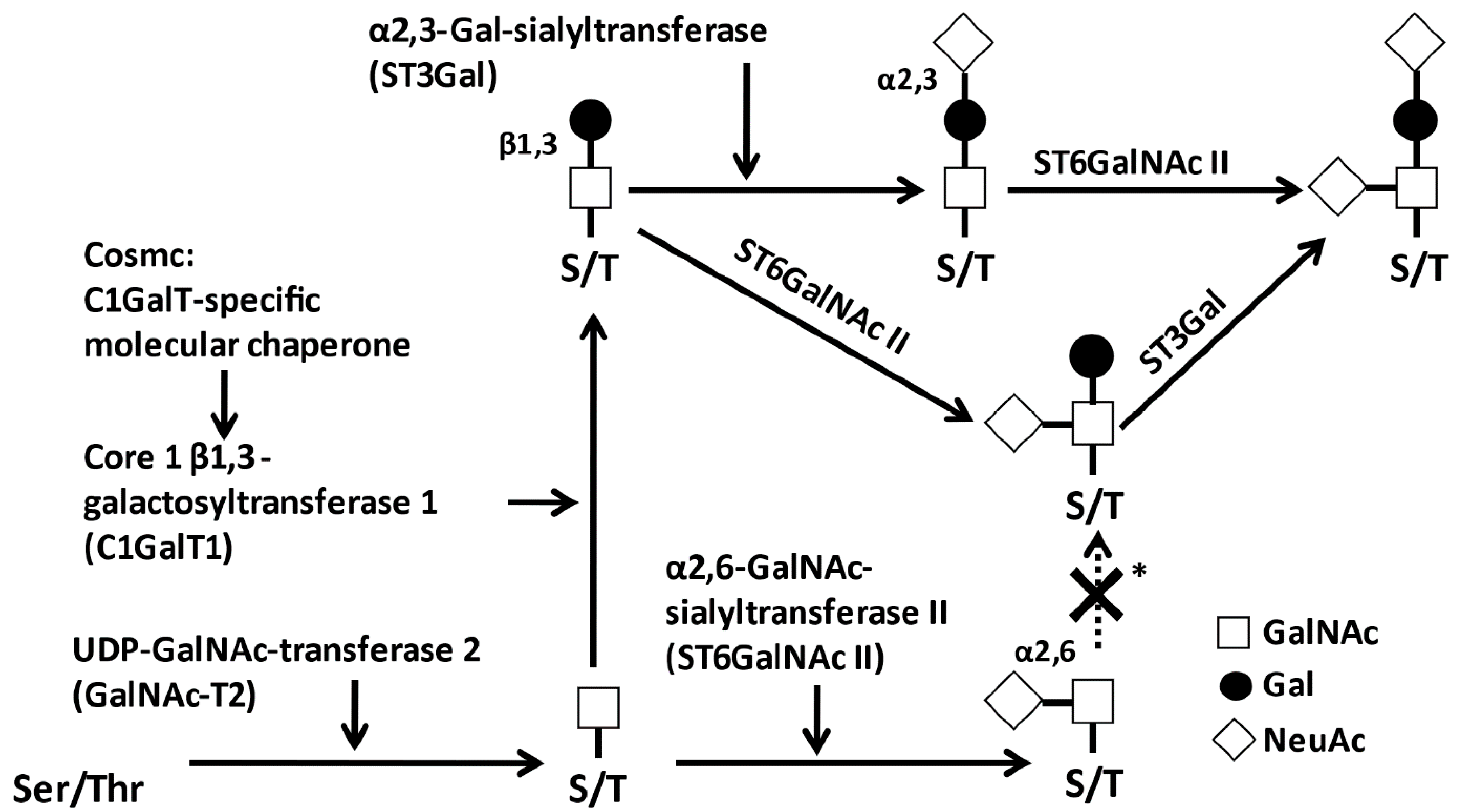
2.1. Glycosylation of IgA1
2.2. Biological Roles of O-Glycosylation in IgA1 HR
2.3. Aberrantly Glycosylated IgA1
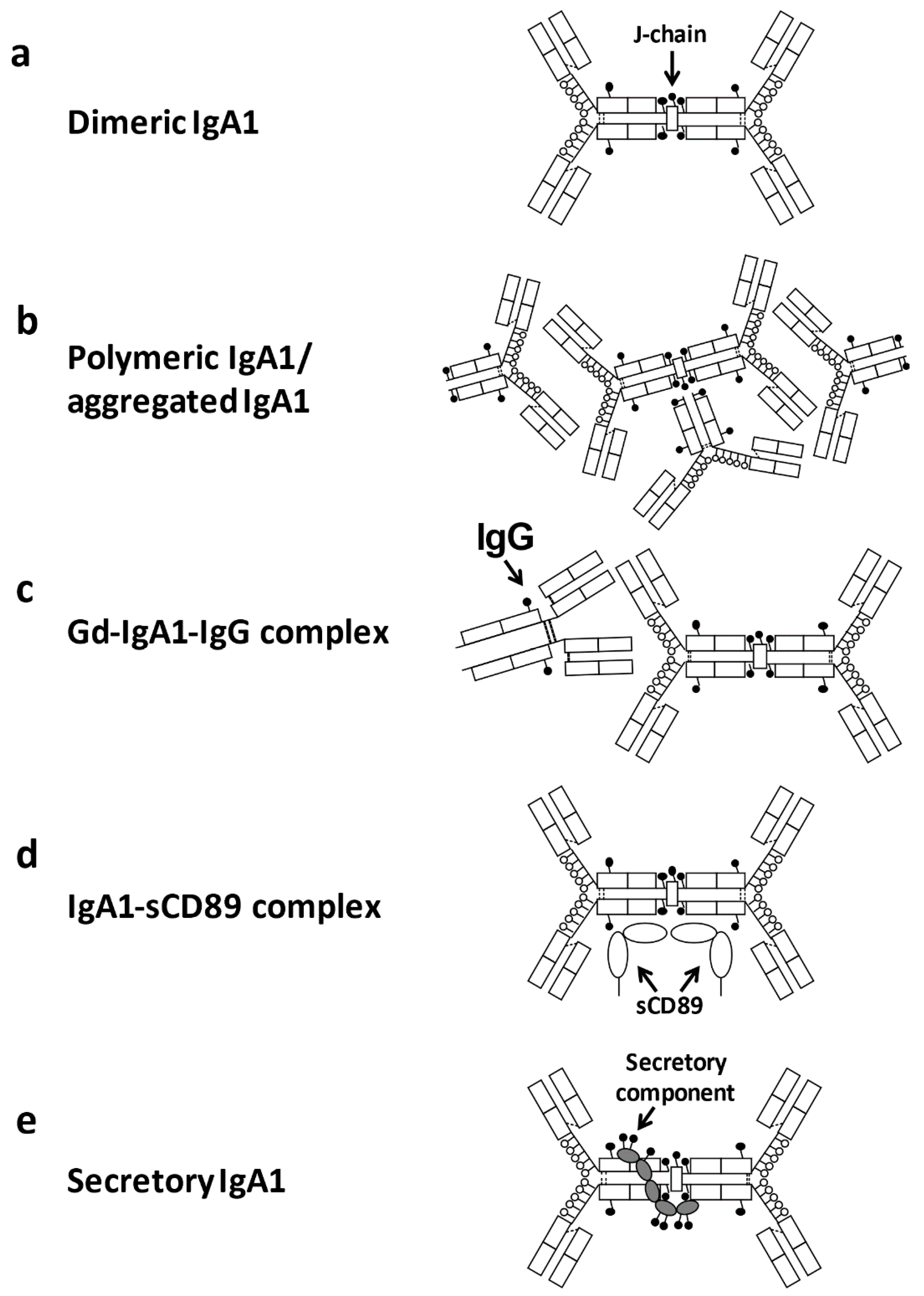
3. Pathogenic Significance of Aberrantly Glycosylated IgA1
3.1. Genetic Factors Associated to Gd-IgA1 Production
3.2. Alteration of Mucosal Immunity Associated with Gd-IgA1 Production
3.3. Antigenicity of Gd-IgA1 Related to Autoantibody Production
3.4. Impact of IgA1 O-Glycosylation of Interactions with IgA Receptors
3.5. Complement Activation
4. Clinical Significance of Aberrantly Glycosylated IgA1
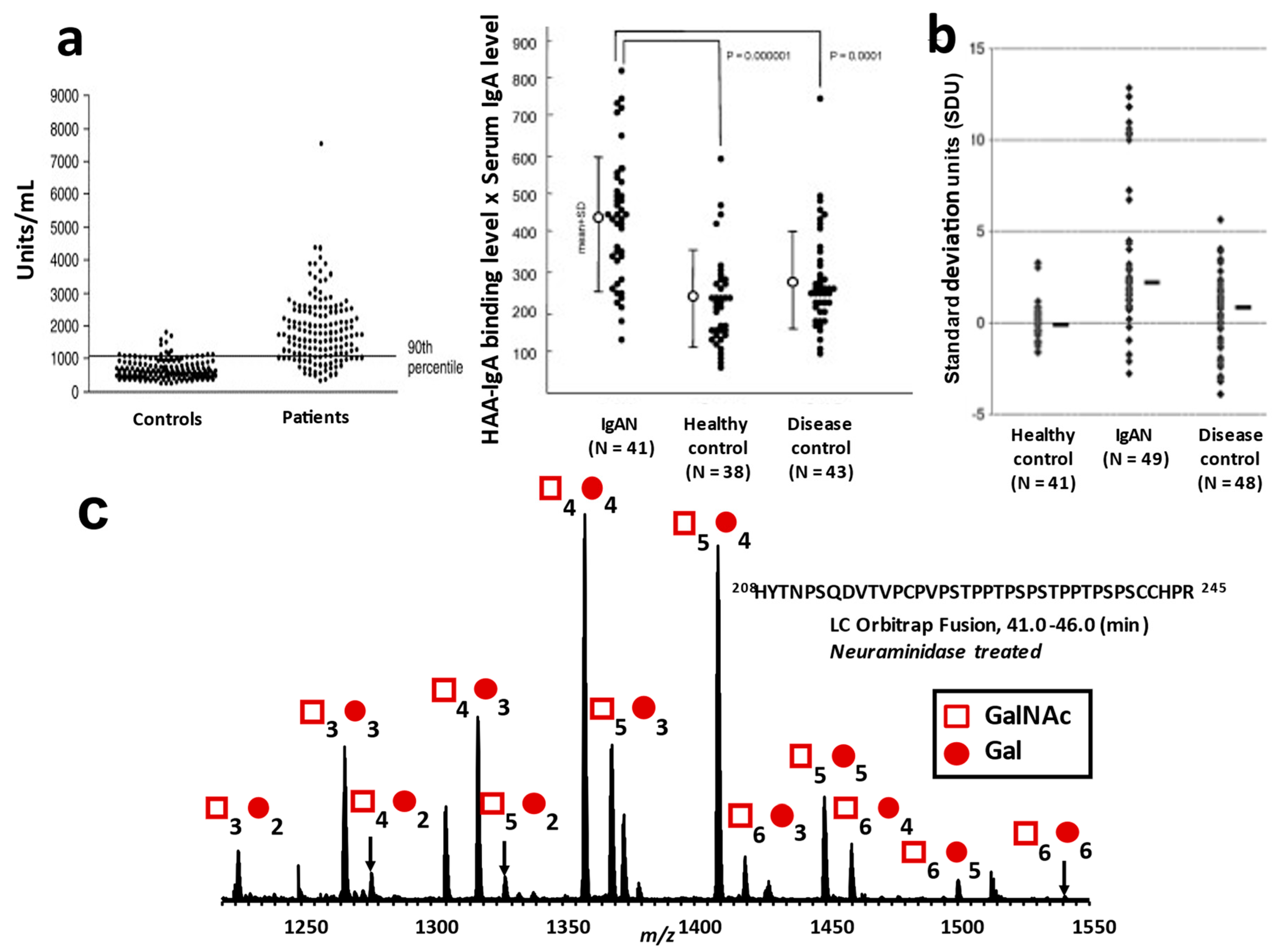
4.1. Approaches for Detection of Aberrantly Glycosylated IgA1
4.2. Reliability of Gd-IgA1 as a Diagnostic and Prognostic Biomarker
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Berger, J.; Hinglais, N. Intercapillary deposits of IgA-IgG. J. Urol. Nephrol. 1968, 74, 694–695. [Google Scholar]
- Conley, M.E.; Cooper, M.D.; Michael, A.F. Selective deposition of immunoglobulin A1 in immunoglobulin A nephropathy, anaphylactoid purpura nephritis, and systemic lupus erythematosus. J. Clin. Investig. 1980, 66, 1432–1436. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.; Yaneva, H.; Nabarra, B.; Barbanel, C. Recurrence of mesangial deposition of IgA after renal transplantation. Kidney Int. 1975, 7, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Ponticelli, C.; Traversi, L.; Feliciani, A.; Cesana, B.M.; Banfi, G.; Tarantino, A. Kidney transplantation in patients with IgA mesangial glomerulonephritis. Kidney Int. 2001, 60, 1948–1954. [Google Scholar] [CrossRef] [PubMed]
- Berger, J. Recurrence of IgA nephropathy in renal allografts. Am. J. Kidney Dis. 1988, 12, 371–372. [Google Scholar] [CrossRef]
- Floege, J. Recurrent IgA nephropathy after renal transplantation. Semin. Nephrol. 2004, 24, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Sanfilippo, F.; Croker, B.P.; Bollinger, R.R. Fate of four cadaveric donor renal allografts with mesangial IgA deposits. Transplantation 1982, 33, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Silva, F.G.; Chander, P.; Pirani, C.L.; Hardy, M.A. Disappearance of glomerular mesangial IgA deposits after renal allograft transplantation. Transplantation 1982, 33, 241–246. [Google Scholar]
- Cuevas, X.; Lloveras, J.; Mir, M.; Aubia, J.; Masramon, J. Disappearance of mesangial IgA deposits from the kidneys of two donors after transplantation. Transplant. Proc. 1987, 19, 2208–2209. [Google Scholar]
- Iwata, Y.; Wada, T.; Uchiyama, A.; Miwa, A.; Nakaya, I.; Tohyama, T.; Yamada, Y.; Kurokawa, T.; Yoshida, T.; Ohta, S.; et al. Remission of IgA nephropathy after allogeneic peripheral blood stem cell transplantation followed by immunosuppression for acute lymphocytic leukemia. Intern. Med. 2006, 45, 1291–1295. [Google Scholar] [CrossRef][Green Version]
- Hu, S.L.; Colvin, G.A.; Rifai, A.; Suzuki, H.; Novak, J.; Esparza, A.; Farooqi, S.; Julian, B.A. Glomerulonephritis after hematopoietic cell transplantation: IgA nephropathy with increased excretion of galactose-deficient IgA1. Nephrol. Dial. Transplant. 2010, 25, 1708–1713. [Google Scholar] [CrossRef] [PubMed]
- Zickerman, A.M.; Allen, A.C.; Talwar, V.; Olczak, S.A.; Brownlee, A.; Holland, M.; Furness, P.N.; Brunskill, N.J.; Feehally, J. IgA myeloma presenting as Henoch-Schönlein purpura with nephritis. Am. J. Kidney Dis. 2000, 36, E19. [Google Scholar] [CrossRef] [PubMed]
- Van Der Helm-Van Mil, A.H.; Smith, A.C.; Pouria, S.; Tarelli, E.; Brunskill, N.J.; Eikenboom, H.C. Immunoglobulin A multiple myeloma presenting with Henoch-Schönlein purpura associated with reduced sialylation of IgA1. Br. J. Haematol. 2003, 122, 915–917. [Google Scholar] [CrossRef] [PubMed]
- Floege, J. The pathogenesis of IgA nephropathy: What is new and how does it change therapeutic approaches? Am. J. Kidney Dis. 2011, 58, 992–1004. [Google Scholar] [CrossRef] [PubMed]
- Donadio, J.V.; Grande, J.P. IgA nephropathy. N. Engl. J. Med. 2002, 347, 738–748. [Google Scholar] [CrossRef]
- Pouria, S.; Barratt, J. Secondary IgA nephropathy. Semin. Nephrol. 2008, 28, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Glassock, R.J. The pathogenesis of IgA nephropathy. Curr. Opin. Nephrol. Hypertens. 2011, 20, 153–160. [Google Scholar] [CrossRef]
- Boyd, J.K.; Cheung, C.K.; Molyneux, K.; Feehally, J.; Barratt, J. An update on the pathogenesis and treatment of IgA nephropathy. Kidney Int. 2012, 81, 833–843. [Google Scholar] [CrossRef]
- Mestecky, J. Immunobiology of IgA. Am. J. Kidney Dis. 1988, 12, 378–383. [Google Scholar] [CrossRef]
- Tomino, Y.; Sakai, H.; Miura, M.; Endoh, M.; Nomoto, Y. Detection of polymeric IgA in glomeruli from patients with IgA nephropathy. Clin. Exp. Immunol. 1982, 49, 419–425. [Google Scholar]
- Bene, M.C.; Faure, G.; Duheille, J. IgA nephropathy: Characterization of the polymeric nature of mesangial deposits by in vitro binding of free secretory component. Clin. Exp. Immunol. 1982, 47, 527–534. [Google Scholar] [PubMed]
- Suzuki, K.; Honda, K.; Tanabe, K.; Toma, H.; Nihei, H.; Yamaguchi, Y. Incidence of latent mesangial IgA deposition in renal allograft donors in Japan. Kidney Int. 2003, 63, 2286–2294. [Google Scholar] [CrossRef] [PubMed]
- Van Der Boog, P.J.M.; Van Kooten, C.; De Fijter, J.W.; Daha, M.R. Role of macromolecular IgA in IgA nephropathy. Kidney Int. 2005, 67, 813–821. [Google Scholar] [CrossRef]
- Narita, I.; Gejyo, F. Pathogenetic significance of aberrant glycosylation of IgA1 in IgA nephropathy. Clin. Exp. Immunol. 2008, 12, 332–338. [Google Scholar] [CrossRef]
- Suzuki, H.; Kiryluk, K.; Novak, J.; Moldoveanu, Z.; Herr, A.B.; Renfrow, M.B.; Wyatt, R.J.; Scolari, F.; Mestecky, J.; Gharavi, A.G.; et al. The pathophysiology of IgA nephropathy. J. Am. Soc. Nephrol. 2011, 22, 1795–1803. [Google Scholar] [CrossRef] [PubMed]
- Mestecky, J.; Raska, M.; Julian, B.A.; Gharavi, A.G.; Renfrow, M.B.; Moldoveanu, Z.; Novak, L.; Matousovic, K.; Novak, J. IgA nephropathy: Molecular mechanisms of the disease. Annu. Rev. Pathol. 2013, 8, 217–240. [Google Scholar] [CrossRef]
- Novak, J.; Julian, B.A.; Mestecky, J.; Renfrow, M.B. Glycosylation of IgA1 and pathogenesis of IgA nephropathy. Semin. Immunopathol. 2012, 34, 365–382. [Google Scholar] [CrossRef]
- Coppo, R.; Amore, A.; Peruzzi, L.; Vergano, L.; Camilla, R. Innate immunity and IgA nephropathy. J. Nephrol. 2010, 23, 626–632. [Google Scholar]
- Allen, A.C.; Harper, S.J.; Feehally, J. Galactosylation of N- and O-linked carbohydrate moieties of IgA1 and IgG in IgA nephropathy. Clin. Exp. Immunol. 1995, 100, 470–474. [Google Scholar] [CrossRef]
- Allen, A.C.; Bailey, E.M.; Brenchley, P.E.; Buck, K.S.; Barratt, J.; Feehally, J. Mesangial IgA1 in IgA nephropathy exhibits aberrant O-glycosylation: Observations in three patients. Kidney Int. 2001, 60, 969–973. [Google Scholar] [CrossRef]
- Hiki, Y.; Odani, H.; Takahashi, M.; Yasuda, Y.; Nishimoto, A.; Iwase, H.; Shinzato, T.; Kobayashi, Y.; Maeda, K. Mass spectrometry proves under-O-glycosylation of glomerular IgA1 in IgA nephropathy. Kidney Int. 2001, 59, 1077–1085. [Google Scholar] [CrossRef] [PubMed]
- Moldoveanu, Z.; Wyatt, R.J.; Lee, J.Y.; Tomana, M.; Julian, B.A.; Mestecky, J.; Huang, W.Q.; Anreddy, S.R.; Hall, S.; Hastings, M.C.; et al. Patients with IgA nephropathy have increased serum galactose-deficient IgA1 levels. Kidney Int. 2007, 71, 1148–1154. [Google Scholar] [CrossRef] [PubMed]
- Shimozato, S.; Hiki, Y.; Odani, H.; Takahashi, K.; Yamamoto, K.; Sugiyama, S. Serum under-galactosylated IgA1 is increased in Japanese patients with IgA nephropathy. Nephrol. Dial. Transplant. 2008, 23, 1931–1939. [Google Scholar] [CrossRef] [PubMed]
- Moura, I.C.; Benhamou, M.; Launay, P.; Vrtovsnik, F.; Blank, U.; Monteiro, R.C. The glomerular response to IgA deposition in IgA nephropathy. Semin. Nephrol. 2008, 28, 88–95. [Google Scholar] [CrossRef]
- Hiki, Y. O-linked oligosaccharides of the IgA1 hinge region: Roles of its aberrant structure in the occurrence and/or progression of IgA nephropathy. Clin. Exp. Immunol. 2009, 13, 415–423. [Google Scholar] [CrossRef]
- Lai, K.N. Pathogenesis of IgA nephropathy. Nat. Rev. Nephrol. 2012, 8, 275–283. [Google Scholar] [CrossRef]
- Rizk, D.V.; Maillard, N.; Julian, B.A.; Knoppova, B.; Green, T.J.; Novak, J.; Wyatt, R.J. The emerging role of complement proteins as a target for therapy of IgA nephropathy. Front. Immunol. 2019, 10, 504. [Google Scholar] [CrossRef]
- Knoppova, B.; Reily, C.; Maillard, N.; Rizk, D.V.; Moldoveanu, Z.; Mestecky, J.; Raska, M.; Renfrow, M.B.; Julian, B.A.; Novak, J. The origin and activities of IgA1-containing immune complexes in IgA nephropathy. Front. Immunol. 2016, 7, 117. [Google Scholar] [CrossRef]
- Maillard, N.; Wyatt, R.J.; Julian, B.A.; Kiryluk, K.; Gharavi, A.; Fremeaux-Bacchi, V.; Novak, J. Current understanding of the role of complement in IgA nephropathy. J. Am. Soc. Nephrol. 2015, 26, 1503–1512. [Google Scholar] [CrossRef]
- Yagame, M.; Tomino, Y.; Miura, M.; Tanigaki, T.; Suga, T.; Nomoto, Y.; Sakai, H. Detection of IgA-class circulating immune complexes (CIC) in sera from patients with IgA nephropathy using a solid-phase anti-C3 Facb enzyme immunoassay (EIA). Clin. Exp. Immunol. 1987, 67, 270–276. [Google Scholar]
- Valentijn, R.M.; van Es, L.A.; Daha, M.R. The specific detection of IgG, IgA and the complement components C3 and C4 in circulating immune complexes. J. Clin. Lab. Immunol. 1984, 14, 81–86. [Google Scholar] [PubMed]
- Czerkinsky, C.; Koopman, W.J.; Jackson, S.; Collins, J.E.; Crago, S.S.; Schrohenloher, R.E.; Julian, B.A.; Galla, J.H.; Mestecky, J. Circulating immune complexes and immunoglobulin A rheumatoid factor in patients with mesangial immunoglobulin A nephropathies. J. Clin. Investig. 1986, 77, 1931–1938. [Google Scholar] [CrossRef] [PubMed]
- Papista, C.; Berthelot, L.; Monteiro, R.C. Dysfunctions of the Iga system: A common link between intestinal and renal diseases. Cell. Mol. Immunol. 2011, 8, 126–134. [Google Scholar] [CrossRef]
- Arnold, J.N.; Wormald, M.R.; Sim, R.B.; Rudd, P.M.; Dwek, R.A. The impact of glycosylation on the biological function and structure of human immunoglobulins. Annu. Rev. Immunol. 2007, 25, 21–50. [Google Scholar] [CrossRef] [PubMed]
- Tsuzukida, Y.; Wang, C.C.; Putnam, F.W. Structure of the A2m(1) allotype of human IgA—A recombinant molecule. Proc. Natl. Acad. Sci. USA 1979, 76, 1104–1108. [Google Scholar] [CrossRef] [PubMed]
- Mattu, T.S.; Pleass, R.J.; Willis, A.C.; Kilian, M.; Wormald, M.R.; Lellouch, A.C.; Rudd, P.M.; Woof, J.M.; Dwek, R.A. The glycosylation and structure of human serum IgA1, Fab, and Fc regions and the role of N-glycosylation on Fcα receptor interactions. J. Biol. Chem. 1998, 273, 2260–2272. [Google Scholar] [CrossRef] [PubMed]
- Woof, J.M.; Mestecky, J. Mucosal immunoglobulins. Immunol. Rev. 2005, 206, 64–82. [Google Scholar] [CrossRef]
- Woof, J.M.; Mestecky, J. Mucosal Immunoglobulins. In Mucosal Immunology, 3rd ed.; Academic Press: New York, NY, USA, 2005; Volume 1, pp. 153–181. [Google Scholar]
- Peppard, J.V.; Kaetzel, C.S.; Russell, M.W. Phylogeny and Comparative Physiology of IgA. In Mucosal Immunology, 3rd ed.; Academic Press: New York, NY, USA, 2005; pp. 195–210. [Google Scholar]
- Renfrow, M.B.; Cooper, H.J.; Tomana, M.; Kulhavy, R.; Hiki, Y.; Toma, K.; Emmett, M.R.; Mestecky, J.; Marshall, A.G.; Novak, J. Determination of aberrant O-glycosylation in the IgA1 hinge region by electron capture dissociation fourier transform-ion cyclotron resonance mass spectrometry. J. Biol. Chem. 2005, 280, 19136–19145. [Google Scholar] [CrossRef]
- Iwasaki, H.; Zhang, Y.; Tachibana, K.; Gotoh, M.; Kikuchi, N.; Kwon, Y.D.; Togayachi, A.; Kudo, T.; Kubota, T.; Narimatsu, H. Initiation of O-glycan synthesis in IgA1 hinge region is determined by a single enzyme, UDP-N-acetyl-α-D-galactosamine:polypeptide N-acetylgalactosaminyltransferase 2. J. Biol. Chem. 2003, 278, 5613–5621. [Google Scholar] [CrossRef]
- Takahashi, K.; Wall, S.B.; Suzuki, H.; Smith, A.D.t.; Hall, S.; Poulsen, K.; Kilian, M.; Mobley, J.A.; Julian, B.A.; Mestecky, J.; et al. Clustered O-glycans of IgA1: Defining macro- and microheterogeneity by use of electron capture/transfer dissociation. Mol. Cell. Proteom. 2010, 9, 2545–2557. [Google Scholar] [CrossRef]
- Takahashi, K.; Smith, A.D.; Poulsen, K.; Kilian, M.; Julian, B.A.; Mestecky, J.; Novak, J.; Renfrow, M.B. Naturally occurring structural isomers in serum IgA1 O-glycosylation. J. Proteome Res. 2012, 11, 692–702. [Google Scholar] [CrossRef]
- Tarelli, E.; Smith, A.C.; Hendry, B.M.; Challacombe, S.J.; Pouria, S. Human serum IgA1 is substituted with up to six O-glycans as shown by matrix assisted laser desorption ionisation time-of-flight mass spectrometry. Carbohydr. Res. 2004, 339, 2329–2335. [Google Scholar] [CrossRef]
- Ohyama, Y.; Yamaguchi, H.; Nakajima, K.; Mizuno, T.; Fukamachi, Y.; Yokoi, Y.; Tsuboi, N.; Inaguma, D.; Hasegawa, M.; Renfrow, M.B.; et al. Analysis of O-glycoforms of the IgA1 hinge region by sequential deglycosylation. Sci. Rep. 2020, 10, 671. [Google Scholar] [CrossRef]
- Novak, J.; Julian, B.A.; Tomana, M.; Mestecky, J. IgA glycosylation and IgA immune complexes in the pathogenesis of IgA nephropathy. Semin. Nephrol. 2008, 28, 78–87. [Google Scholar] [CrossRef]
- Wandall, H.H.; Irazoqui, F.; Tarp, M.A.; Bennett, E.P.; Mandel, U.; Takeuchi, H.; Kato, K.; Irimura, T.; Suryanarayanan, G.; Hollingsworth, M.A.; et al. The lectin domains of polypeptide GalNAc-transferases exhibit carbohydrate-binding specificity for GalNAc: Lectin binding to GalNAc-glycopeptide substrates is required for high density GalNAc-O-glycosylation. Glycobiology 2007, 17, 374–387. [Google Scholar] [CrossRef]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef] [PubMed]
- Stewart, T.J.; Takahashi, K.; Whitaker, R.H.; Raska, M.; Placzek, W.J.; Novak, J.; Renfrow, M.B. IgA1 hinge-region clustered glycan fidelity is established early during semi-ordered glycosylation by GalNAc-T2. Glycobiology 2019, 29, 543–556. [Google Scholar] [CrossRef]
- Stewart, T.J.; Takahashi, K.; Xu, N.; Prakash, A.; Brown, R.; Raska, M.; Renfrow, M.B.; Novak, J. Quantitative assessment of successive carbohydrate additions to the clustered O-glycosylation sites of IgA1 by glycosyltransferases. Glycobiology 2021, 31, 540–556. [Google Scholar] [CrossRef] [PubMed]
- Ju, T.; Cummings, R.D. Protein glycosylation: Chaperone mutation in Tn syndrome. Nature 2005, 437, 1252. [Google Scholar] [CrossRef] [PubMed]
- Schachter, H.; McGuire, E.J.; Roseman, S. Sialic acids. 13. A uridine diphosphate D-galactose: Mucin galactosyltransferase from porcine submaxillary gland. J. Biol. Chem. 1971, 246, 5321–5328. [Google Scholar] [CrossRef]
- Novak, J.; Julian, B.A.; Tomana, M.; Mesteck, J. Progress in molecular and genetic studies of IgA nephropathy. J. Clin. Immunol. 2001, 21, 310–327. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Raska, M.; Yamada, K.; Moldoveanu, Z.; Julian, B.A.; Wyatt, R.J.; Tomino, Y.; Gharavi, A.G.; Novak, J. Cytokines alter IgA1 O-glycosylation by dysregulating C1GalT1 and ST6GalNAc-II enzymes. J. Biol. Chem. 2014, 289, 5330–5339. [Google Scholar] [CrossRef] [PubMed]
- Varki, A. Biological roles of glycans. Glycobiology 2017, 27, 3–49. [Google Scholar] [CrossRef] [PubMed]
- Furtado, P.B.; Whitty, P.W.; Robertson, A.; Eaton, J.T.; Almogren, A.; Kerr, M.A.; Woof, J.M.; Perkins, S.J. Solution structure determination of monomeric human IgA2 by X-ray and neutron scattering, analytical ultracentrifugation and constrained modelling: A comparison with monomeric human IgA1. J. Mol. Biol. 2004, 338, 921–941. [Google Scholar] [CrossRef] [PubMed]
- Narimatsu, Y.; Joshi, H.J.; Schjoldager, K.T.; Hintze, J.; Halim, A.; Steentoft, C.; Nason, R.; Mandel, U.; Bennett, E.P.; Clausen, H.; et al. Exploring Regulation of Protein O-Glycosylation in Isogenic Human HEK293 Cells by Differential O-Glycoproteomics. Mol. Cell. Proteom. 2019, 18, 1396–1409. [Google Scholar] [CrossRef] [PubMed]
- Royle, L.; Roos, A.; Harvey, D.J.; Wormald, M.R.; van Gijlswijk-Janssen, D.; Redwan, E.-R.M.; Wilson, I.A.; Daha, M.R.; Dwek, R.A.; Rudd, P.M. Secretory IgA N- and O-glycans provide a link between the innate and adaptive immune systems. J. Biol. Chem. 2003, 278, 20140–20153. [Google Scholar] [CrossRef]
- Tomana, M.; Novak, J.; Julian, B.A.; Matousovic, K.; Konecny, K.; Mestecky, J. Circulating immune complexes in IgA nephropathy consist of IgA1 with galactose-deficient hinge region and antiglycan antibodies. J. Clin. Investig. 1999, 104, 73–81. [Google Scholar] [CrossRef]
- Moore, J.S.; Kulhavy, R.; Tomana, M.; Moldoveanu, Z.; Suzuki, H.; Brown, R.; Hall, S.; Kilian, M.; Poulsen, K.; Mestecky, J.; et al. Reactivities of N-acetylgalactosamine-specific lectins with human IgA1 proteins. Mol. Immunol. 2007, 44, 2598–2604. [Google Scholar] [CrossRef]
- Reily, C.; Rizk, D.V.; Julian, B.A.; Novak, J. Assay for galactose-deficient IgA1 enables mechanistic studies with primary cells from IgA nephropathy patients. Biotechniques 2018, 65, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Gomes, M.M.; Suzuki, H.; Brooks, M.T.; Tomana, M.; Moldoveanu, Z.; Mestecky, J.; Julian, B.A.; Novak, J.; Herr, A.B. Recognition of galactose-deficient O-glycans in the hinge region of IgA1 by N-acetylgalactosamine-specific snail lectins: A comparative binding study. Biochemistry 2010, 49, 5671–5682. [Google Scholar] [CrossRef]
- Leung, J.C.; Poon, P.Y.; Lai, K.N. Increased sialylation of polymeric immunoglobulin A1: Mechanism of selective glomerular deposition in immunoglobulin A nephropathy? J. Lab. Clin. Med. 1999, 133, 152–160. [Google Scholar] [CrossRef]
- Amore, A.; Cirina, P.; Conti, G.; Brusa, P.; Peruzzi, L.; Coppo, R. Glycosylation of circulating IgA in patients with IgA nephropathy modulates proliferation and apoptosis of mesangial cells. J. Am. Soc. Nephrol. 2001, 12, 1862–1871. [Google Scholar] [CrossRef] [PubMed]
- Leung, J.C.; Tang, S.C.; Chan, D.T.; Lui, S.L.; Lai, K.N. Increased sialylation of polymeric lambda-IgA1 in patients with IgA nephropathy. J. Clin. Lab. Anal. 2002, 16, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Moldoveanu, Z.; Hall, S.; Brown, R.; Vu, H.L.; Novak, L.; Julian, B.A.; Tomana, M.; Wyatt, R.J.; Edberg, J.C.; et al. IgA1-secreting cell lines from patients with IgA nephropathy produce aberrantly glycosylated IgA1. J. Clin. Investig. 2008, 118, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Wada, Y.; Tajiri, M.; Ohshima, S. Quantitation of saccharide compositions of O-glycans by mass spectrometry of glycopeptides and its application to rheumatoid arthritis. J. Proteome Res. 2010, 9, 1367–1373. [Google Scholar] [CrossRef]
- Smith, A.C.; de Wolff, J.F.; Molyneux, K.; Feehally, J.; Barratt, J. O-glycosylation of serum IgD in IgA nephropathy. J. Am. Soc. Nephrol. 2006, 17, 1192–1199. [Google Scholar] [CrossRef]
- Gharavi, A.G.; Moldoveanu, Z.; Wyatt, R.J.; Barker, C.V.; Woodford, S.Y.; Lifton, R.P.; Mestecky, J.; Novak, J.; Julian, B.A. Aberrant IgA1 glycosylation is inherited in familial and sporadic IgA nephropathy. J. Am. Soc. Nephrol. 2008, 19, 1008–1014. [Google Scholar] [CrossRef]
- Rizk, D.V.; Saha, M.K.; Hall, S.; Novak, L.; Brown, R.; Huang, Z.Q.; Fatima, H.; Julian, B.A.; Novak, J. Glomerular immunodeposits of patients with IgA nephropathy are enriched for IgG autoantibodies specific for galactose-deficient IgA1. J. Am. Soc. Nephrol. 2019, 30, 2017–2026. [Google Scholar] [CrossRef]
- Moldoveanu, Z.; Suzuki, H.; Reily, C.; Satake, K.; Novak, L.; Xu, N.; Huang, Z.Q.; Knoppova, B.; Khan, A.; Hall, S.; et al. Experimental evidence of pathogenic role of IgG autoantibodies in IgA nephropathy. J. Autoimmun. 2021, 118, 102593. [Google Scholar] [CrossRef]
- Tam, K.Y.; Leung, J.C.K.; Chan, L.Y.Y.; Lam, M.F.; Tang, S.C.W.; Lai, K.N. Macromolecular IgA1 taken from patients with familial IgA nephropathy or their asymptomatic relatives have higher reactivity to mesangial cells in vitro. Kidney Int. 2009, 75, 1330–1339. [Google Scholar] [CrossRef] [PubMed]
- Kiryluk, K.; Moldoveanu, Z.; Sanders, J.T.; Eison, T.M.; Suzuki, H.; Julian, B.A.; Novak, J.; Gharavi, A.G.; Wyatt, R.J. Aberrant glycosylation of IgA1 is inherited in both pediatric IgA nephropathy and Henoch-Schönlein purpura nephritis. Kidney Int. 2011, 80, 79–87. [Google Scholar] [CrossRef]
- Lin, X.; Ding, J.; Zhu, L.; Shi, S.; Jiang, L.; Zhao, M.; Zhang, H. Aberrant galactosylation of IgA1 is involved in the genetic susceptibility of Chinese patients with IgA nephropathy. Nephrol. Dial. Transplant. 2009, 24, 3372–3375. [Google Scholar] [CrossRef] [PubMed]
- Hastings, M.C.; Moldoveanu, Z.; Julian, B.A.; Novak, J.; Sanders, J.T.; McGlothan, K.R.; Gharavi, A.G.; Wyatt, R.J. Galactose-deficient IgA1 in African Americans with IgA nephropathy: Serum levels and heritability. Clin. J. Am. Soc. Nephrol. 2010, 5, 2069–2074. [Google Scholar] [CrossRef] [PubMed]
- Li, G.S.; Zhang, H.; Lv, J.C.; Shen, Y.; Wang, H.Y. Variants of C1GALT1 gene are associated with the genetic susceptibility to IgA nephropathy. Kidney Int. 2007, 71, 448–453. [Google Scholar] [CrossRef]
- Pirulli, D.; Crovella, S.; Ulivi, S.; Zadro, C.; Bertok, S.; Rendine, S.; Scolari, F.; Foramitti, M.; Ravani, P.; Roccatello, D.; et al. Genetic variant of C1GalT1 contributes to the susceptibility to IgA nephropathy. J. Nephrol. 2009, 22, 152–159. [Google Scholar]
- Zhu, L.; Tang, W.; Li, G.; Lv, J.; Ding, J.; Yu, L.; Zhao, M.; Li, Y.; Zhang, X.; Shen, Y.; et al. Interaction between variants of two glycosyltransferase genes in IgA nephropathy. Kidney Int. 2009, 76, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Li, G.S.; Zhu, L.; Zhang, H.; Lv, J.C.; Ding, J.X.; Zhao, M.H.; Shen, Y.; Wang, H.Y. Variants of the ST6GALNAC2 promoter influence transcriptional activity and contribute to genetic susceptibility to IgA nephropathy. Hum. Mutat. 2007, 28, 950–957. [Google Scholar] [CrossRef] [PubMed]
- Malycha, F.; Eggermann, T.; Hristov, M.; Schena, F.P.; Mertens, P.R.; Zerres, K.; Floege, J.; Eitner, F. No evidence for a role of cosmc-chaperone mutations in European IgA nephropathy patients. Nephrol. Dial. Transplant. 2009, 24, 321–324. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yu, X.Q.; Li, M.; Zhang, H.; Low, H.Q.; Wei, X.; Wang, J.Q.; Sun, L.D.; Sim, K.S.; Li, Y.; Foo, J.N.; et al. A genome-wide association study in Han Chinese identifies multiple susceptibility loci for IgA nephropathy. Nat. Genet. 2011, 44, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Kiryluk, K.; Li, Y.; Scolari, F.; Sanna-Cherchi, S.; Choi, M.; Verbitsky, M.; Fasel, D.; Lata, S.; Prakash, S.; Shapiro, S.; et al. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat. Genet. 2014, 46, 1187–1196. [Google Scholar] [CrossRef]
- Qi, Y.Y.; Zhou, X.J.; Cheng, F.J.; Hou, P.; Zhu, L.; Shi, S.F.; Liu, L.J.; Lv, J.C.; Zhang, H. DEFA gene variants associated with IgA nephropathy in a Chinese population. Genes Immun. 2015, 16, 231–237. [Google Scholar] [CrossRef]
- Gharavi, A.G.; Kiryluk, K.; Choi, M.; Li, Y.; Hou, P.; Xie, J.; Sanna-Cherchi, S.; Men, C.J.; Julian, B.A.; Wyatt, R.J.; et al. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat. Genet. 2011, 43, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Gale, D.P.; Molyneux, K.; Wimbury, D.; Higgins, P.; Levine, A.P.; Caplin, B.; Ferlin, A.; Yin, P.; Nelson, C.P.; Stanescu, H.; et al. Galactosylation of IgA1 Is Associated with Common Variation in C1GALT1. J. Am. Soc. Nephrol. 2017, 28, 2158–2166. [Google Scholar] [CrossRef]
- Kiryluk, K.; Li, Y.; Moldoveanu, Z.; Suzuki, H.; Reily, C.; Hou, P.; Xie, J.; Mladkova, N.; Prakash, S.; Fischman, C.; et al. GWAS for serum galactose-deficient IgA1 implicates critical genes of the O-glycosylation pathway. PLoS Genet. 2017, 13, e1006609. [Google Scholar] [CrossRef]
- Thu, C.T.; Mahal, L.K. Sweet Control: MicroRNA regulation of the glycome. Biochemistry 2020, 59, 3098–3110. [Google Scholar] [CrossRef] [PubMed]
- Serino, G.; Sallustio, F.; Cox, S.N.; Pesce, F.; Schena, F.P. Abnormal miR-148b expression promotes aberrant glycosylation of IgA1 in IgA nephropathy. J. Am. Soc. Nephrol. 2012, 23, 814–824. [Google Scholar] [CrossRef] [PubMed]
- Serino, G.; Sallustio, F.; Curci, C.; Cox, S.N.; Pesce, F.; De Palma, G.; Schena, F.P. Role of let-7b in the regulation of N-acetylgalactosaminyltransferase 2 in IgA nephropathy. Nephrol. Dial. Transplant. 2015, 30, 1132–1139. [Google Scholar] [CrossRef] [PubMed]
- Serino, G.; Pesce, F.; Sallustio, F.; De Palma, G.; Cox, S.N.; Curci, C.; Zaza, G.; Lai, K.N.; Leung, J.C.; Tang, S.C.; et al. In a retrospective international study, circulating miR-148b and let-7b were found to be serum markers for detecting primary IgA nephropathy. Kidney Int. 2016, 89, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liu, D.; Liu, Y.; Xia, M.; Li, Y.; Li, M.; Liu, H. Comprehensive analysis of circRNA expression profiles and circRNA-associated competing endogenous RNA networks in IgA nephropathy. PeerJ 2020, 8, e10395. [Google Scholar] [CrossRef]
- Liu, C.; Ye, M.Y.; Yan, W.Z.; Peng, X.F.; He, L.Y.; Peng, Y.M. microRNA-630 regulates underglycosylated IgA1 production in the tonsils by targeting TLR4 in IgA nephropathy. Front. Immunol. 2020, 11, 563699. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liao, Y.; Wang, L.; Lin, Y.; Ye, Z.; Zeng, X.; Liu, X.; Wei, F.; Yang, N. Small RNA deep sequencing reveals novel miRNAs in peripheral blood mononuclear cells from patients with IgA nephropathy. Mol. Med. Rep. 2020, 22, 3378–3386. [Google Scholar] [CrossRef]
- Trimarchi, H.M.; Iotti, A.; Iotti, R.; Freixas, E.A.; Peters, R. Immunoglobulin A nephropathy and ulcerative colitis. A focus on their pathogenesis. Am. J. Nephrol. 2001, 21, 400–405. [Google Scholar] [CrossRef]
- Forshaw, M.J.; Guirguis, O.; Hennigan, T.W. IgA nephropathy in association with Crohn’s disease. Int. J. Colorectal. Dis. 2005, 20, 463–465. [Google Scholar] [CrossRef]
- de Moura, C.G.; de Moura, T.G.; de Souza, S.P.; Testagrossa, L. Inflammatory bowel disease, ankylosing spondylitis, and IgA nephropathy. J. Clin. Rheumatol. 2006, 12, 106–107. [Google Scholar] [CrossRef] [PubMed]
- Smerud, H.K.; Fellstrom, B.; Hallgren, R.; Osagie, S.; Venge, P.; Kristjansson, G. Gluten sensitivity in patients with IgA nephropathy. Nephrol. Dial. Transplant. 2009, 24, 2476–2481. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.J.; Xu, L.X.; Liu, G.; Zhao, M.H.; Wang, H.Y. The level of serum secretory IgA of patients with IgA nephropathy is elevated and associated with pathological phenotypes. Nephrol. Dial. Transplant. 2008, 23, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Oortwijn, B.D.; Rastaldi, M.P.; Roos, A.; Mattinzoli, D.; Daha, M.R.; van Kooten, C. Demonstration of secretory IgA in kidneys of patients with IgA nephropathy. Nephrol. Dial. Transplant. 2007, 22, 3191–3195. [Google Scholar] [CrossRef] [PubMed]
- Harper, S.J.; Allen, A.C.; Pringle, J.H.; Feehally, J. Increased dimeric IgA producing B cells in the bone marrow in IgA nephropathy determined by in situ hybridisation for J chain mRNA. J. Clin. Pathol. 1996, 49, 38–42. [Google Scholar] [CrossRef]
- Novak, J.; Barratt, J.; Julian, B.A.; Renfrow, M.B. Aberrant glycosylation of the IgA1 molecule in IgA nephropathy. Semin. Nephrol. 2018, 38, 461–476. [Google Scholar] [CrossRef]
- Hirano, K.; Matsuzaki, K.; Yasuda, T.; Nishikawa, M.; Yasuda, Y.; Koike, K.; Maruyama, S.; Yokoo, T.; Matsuo, S.; Kawamura, T.; et al. Association between tonsillectomy and outcomes in patients with immunoglobulin A nephropathy. JAMA Netw Open 2019, 2, e194772. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Chen, X.; Nishi, S.; Narita, I.; Gejyo, F. Relationship between tonsils and IgA nephropathy as well as indications of tonsillectomy. Kidney Int. 2004, 65, 1135–1144. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Nishi, S.; Ueno, M.; Imai, N.; Sakatsume, M.; Narita, I.; Suzuki, Y.; Akazawa, K.; Shimada, H.; Arakawa, M.; et al. The efficacy of tonsillectomy on long-term renal survival in patients with IgA nephropathy. Kidney Int. 2003, 63, 1861–1867. [Google Scholar] [CrossRef]
- Inoue, T.; Sugiyama, H.; Hiki, Y.; Takiue, K.; Morinaga, H.; Kitagawa, M.; Maeshima, Y.; Fukushima, K.; Nishizaki, K.; Akagi, H.; et al. Differential expression of glycogenes in tonsillar B lymphocytes in association with proteinuria and renal dysfunction in IgA nephropathy. Clin. Immunol. 2010, 136, 447–455. [Google Scholar] [CrossRef]
- He, L.; Peng, Y.; Liu, H.; Yin, W.; Chen, X.; Peng, X.; Shao, J.; Liu, Y.; Liu, F. Th1/Th2 polarization in tonsillar lymphocyte form patients with IgA nephropathy. Ren. Fail. 2014, 36, 407–412. [Google Scholar] [CrossRef]
- Yamada, K.; Kobayashi, N.; Ikeda, T.; Suzuki, Y.; Tsuge, T.; Horikoshi, S.; Emancipator, S.N.; Tomino, Y. Down-regulation of core 1 beta1,3-galactosyltransferase and Cosmc by Th2 cytokine alters O-glycosylation of IgA1. Nephrol. Dial. Transplant. 2010, 25, 3890–3897. [Google Scholar] [CrossRef] [PubMed]
- Muto, M.; Manfroi, B.; Suzuki, H.; Joh, K.; Nagai, M.; Wakai, S.; Righini, C.; Maiguma, M.; Izui, S.; Tomino, Y.; et al. Toll-Like Receptor 9 stimulation induces aberrant expression of a proliferation-inducing ligand by tonsillar germinal center B cells in IgA nephropathy. J. Am. Soc. Nephrol. 2017, 28, 1227–1238. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Huang, Z.Q.; Raska, M.; Reily, C.; Anderson, J.C.; Suzuki, H.; Kiryluk, K.; Gharavi, A.G.; Julian, B.A.; Willey, C.D.; et al. Leukemia inhibitory factor signaling enhances production of galactose-deficient IgA1 in IgA nephropathy. Kidney Dis. 2020, 6, 168–180. [Google Scholar] [CrossRef]
- Yamada, K.; Huang, Z.Q.; Raska, M.; Reily, C.; Anderson, J.C.; Suzuki, H.; Ueda, H.; Moldoveanu, Z.; Kiryluk, K.; Suzuki, Y.; et al. Inhibition of STAT3 signaling reduces IgA1 autoantigen production in IgA nephropathy. Kidney Int. Rep. 2017, 2, 1194–1207. [Google Scholar] [CrossRef]
- Novak, J.; Rizk, D.; Takahashi, K.; Zhang, X.; Bian, Q.; Ueda, H.; Ueda, Y.; Reily, C.; Lai, L.Y.; Hao, C.; et al. New insights into the pathogenesis of IgA nephropathy. Kidney Dis. 2015, 1, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Reily, C.; Ueda, H.; Huang, Z.Q.; Mestecky, J.; Julian, B.A.; Willey, C.D.; Novak, J. Cellular signaling and production of galactose-deficient IgA1 in IgA nephropathy, an autoimmune disease. J. Immunol. Res. 2014, 2014, 197548. [Google Scholar] [CrossRef] [PubMed]
- Makita, Y.; Suzuki, H.; Kano, T.; Takahata, A.; Julian, B.A.; Novak, J.; Suzuki, Y. TLR9 activation induces aberrant IgA glycosylation via APRIL- and IL-6-mediated pathways in IgA nephropathy. Kidney Int. 2019, 97, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Tomana, M.; Matousovic, K.; Julian, B.A.; Radl, J.; Konecny, K.; Mestecky, J. Galactose-deficient IgA1 in sera of IgA nephropathy patients is present in complexes with IgG. Kidney Int. 1997, 52, 509–516. [Google Scholar] [CrossRef]
- Suzuki, H.; Fan, R.; Zhang, Z.; Brown, R.; Hall, S.; Julian, B.A.; Chatham, W.W.; Suzuki, Y.; Wyatt, R.J.; Moldoveanu, Z.; et al. Aberrantly glycosylated IgA1 in IgA nephropathy patients is recognized by IgG antibodies with restricted heterogeneity. J. Clin. Investig. 2009, 119, 1668–1677. [Google Scholar] [CrossRef] [PubMed]
- Placzek, W.J.; Yanagawa, H.; Makita, Y.; Renfrow, M.B.; Julian, B.A.; Rizk, D.V.; Suzuki, Y.; Novak, J.; Suzuki, H. Serum galactose-deficient-IgA1 and IgG autoantibodies correlate in patients with IgA nephropathy. PLoS ONE 2018, 13, e0190967. [Google Scholar] [CrossRef] [PubMed]
- Kokubo, T.; Hiki, Y.; Iwase, H.; Tanaka, A.; Toma, K.; Hotta, K.; Kobayashi, Y. Protective role of IgA1 glycans against IgA1 self-aggregation and adhesion to extracellular matrix proteins. J. Am. Soc. Nephrol. 1998, 9, 2048–2054. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.L.; Fridovich, S.E.; Knowles, B.B.; Lodish, H.F. Characterization of the asialoglycoprotein receptor in a continuous hepatoma line. J. Biol. Chem. 1981, 256, 8878–8881. [Google Scholar] [CrossRef]
- Grossetete, B.; Launay, P.; Lehuen, A.; Jungers, P.; Bach, J.F.; Monteiro, R.C. Down-regulation of Fc alpha receptors on blood cells of IgA nephropathy patients: Evidence for a negative regulatory role of serum IgA. Kidney Int. 1998, 53, 1321–1335. [Google Scholar] [CrossRef] [PubMed]
- van Zandbergen, G.; van Kooten, C.; Mohamad, N.K.; Reterink, T.J.; de Fijter, J.W.; van de Winkel, J.G.; Daha, M.R. Reduced binding of immunoglobulin A (IgA) from patients with primary IgA nephropathy to the myeloid IgA Fc-receptor, CD89. Nephrol. Dial. Transplant. 1998, 13, 3058–3064. [Google Scholar] [CrossRef]
- Goritzer, K.; Turupcu, A.; Maresch, D.; Novak, J.; Altmann, F.; Oostenbrink, C.; Obinger, C.; Strasser, R. Distinct Fc α receptor N-glycans modulate the binding affinity to immunoglobulin A (IgA) antibodies. J. Biol. Chem. 2019, 294, 13995–14008. [Google Scholar] [CrossRef]
- Launay, P.; Grossetete, B.; Arcos-Fajardo, M.; Gaudin, E.; Torres, S.P.; Beaudoin, L.; Patey-Mariaud de Serre, N.; Lehuen, A.; Monteiro, R.C. Fcα receptor (CD89) mediates the development of immunoglobulin A (IgA) nephropathy (Berger’s disease). Evidence for pathogenic soluble receptor-IgA complexes in patients and CD89 transgenic mice. J. Exp. Med. 2000, 191, 1999–2009. [Google Scholar] [CrossRef] [PubMed]
- Berthelot, L.; Papista, C.; Maciel, T.T.; Biarnes-Pelicot, M.; Tissandie, E.; Wang, P.H.; Tamouza, H.; Jamin, A.; Bex-Coudrat, J.; Gestin, A.; et al. Transglutaminase is essential for IgA nephropathy development acting through IgA receptors. J. Exp. Med. 2012, 209, 793–806. [Google Scholar] [CrossRef]
- Berthelot, L.; Robert, T.; Vuiblet, V.; Tabary, T.; Braconnier, A.; Drame, M.; Toupance, O.; Rieu, P.; Monteiro, R.C.; Toure, F. Recurrent IgA nephropathy is predicted by altered glycosylated IgA, autoantibodies and soluble CD89 complexes. Kidney Int. 2015, 88, 815–822. [Google Scholar] [CrossRef]
- McDonald, K.J.; Cameron, A.J.; Allen, J.M.; Jardine, A.G. Expression of Fc alpha/mu receptor by human mesangial cells: A candidate receptor for immune complex deposition in IgA nephropathy. Biochem. Biophys. Res. Commun. 2002, 290, 438–442. [Google Scholar] [CrossRef]
- Moura, I.C.; Centelles, M.N.; Arcos-Fajardo, M.; Malheiros, D.M.; Collawn, J.F.; Cooper, M.D.; Monteiro, R.C. Identification of the transferrin receptor as a novel immunoglobulin (Ig)A1 receptor and its enhanced expression on mesangial cells in IgA nephropathy. J. Exp. Med. 2001, 194, 417–425. [Google Scholar] [CrossRef]
- Leung, J.C.; Tsang, A.W.; Chan, D.T.; Lai, K.N. Absence of CD89, polymeric immunoglobulin receptor, and asialoglycoprotein receptor on human mesangial cells. J. Am. Soc. Nephrol. 2000, 11, 241–249. [Google Scholar] [CrossRef]
- Haddad, E. Enhanced expression of the CD71 mesangial IgA1 receptor in Berger Disease and Henoch-Schönlein Nephritis: Association between CD71 expression and IgA deposits. J. Am. Soc. Nephrol. 2003, 14, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Moura, I.C. Glycosylation and size of IgA1 are essential for interaction with mesangial transferrin receptor in IgA nephropathy. J. Am. Soc. Nephrol. 2004, 15, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Molyneux, K.; Wimbury, D.; Pawluczyk, I.; Muto, M.; Bhachu, J.; Mertens, P.R.; Feehally, J.; Barratt, J. β1,4-galactosyltransferase 1 is a novel receptor for IgA in human mesangial cells. Kidney Int. 2017, 92, 1458–1468. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, Y.; Otsuka, T.; Tsuchida, Y.; Gejyo, F.; Narita, I. Integrin α1/β1 and α2/β1 as a receptor for IgA1 in human glomerular mesangial cells in IgA nephropathy. Int. Immunol. 2012, 24, 219–232. [Google Scholar] [CrossRef]
- Monteiro, R.C. Recent advances in the physiopathology of IgA nephropathy. Nephrol. Ther. 2018, 14 (Suppl. 1), S1–S8. [Google Scholar] [CrossRef] [PubMed]
- Tomana, M.; Zikan, J.; Moldoveanu, Z.; Kulhavy, R.; Bennett, J.C.; Mestecky, J. Interactions of cell-surface galactosyltransferase with immunoglobulins. Mol. Immunol. 1993, 30, 265–275. [Google Scholar] [CrossRef]
- Tortajada, A.; Gutierrez, E.; Pickering, M.C.; Praga Terente, M.; Medjeral-Thomas, N. The role of complement in IgA nephropathy. Mol. Immunol. 2019, 114, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Rauterberg, E.W.; Lieberknecht, H.M.; Wingen, A.M.; Ritz, E. Complement membrane attack (MAC) in idiopathic IgA-glomerulonephritis. Kidney Int. 1987, 31, 820–829. [Google Scholar] [CrossRef]
- Roos, A.; Rastaldi, M.P.; Calvaresi, N.; Oortwijn, B.D.; Schlagwein, N.; van Gijlswijk-Janssen, D.J.; Stahl, G.L.; Matsushita, M.; Fujita, T.; van Kooten, C.; et al. Glomerular activation of the lectin pathway of complement in IgA nephropathy is associated with more severe renal disease. J. Am. Soc. Nephrol. 2006, 17, 1724–1734. [Google Scholar] [CrossRef] [PubMed]
- Stad, R.K.; Bogers, W.M.; Thoomes-van der Sluys, M.E.; Van Es, L.A.; Daha, M.R. In vivo activation of complement by IgA in a rat model. Clin. Exp. Immunol. 1992, 87, 138–143. [Google Scholar] [CrossRef]
- Stad, R.K.; Bruijn, J.A.; van Gijlswijk-Janssen, D.J.; van Es, L.A.; Daha, M.R. An acute model for IgA-mediated glomerular inflammation in rats induced by monoclonal polymeric rat IgA antibodies. Clin. Exp. Immunol. 1993, 92, 514–521. [Google Scholar] [CrossRef]
- Oortwijn, B.D.; Roos, A.; Royle, L.; van Gijlswijk-Janssen, D.J.; Faber-Krol, M.C.; Eijgenraam, J.W.; Dwek, R.A.; Daha, M.R.; Rudd, P.M.; van Kooten, C. Differential glycosylation of polymeric and monomeric IgA: A possible role in glomerular inflammation in IgA nephropathy. J. Am. Soc. Nephrol. 2006, 17, 3529–3539. [Google Scholar] [CrossRef]
- Arnold, J.N.; Wormald, M.R.; Suter, D.M.; Radcliffe, C.M.; Harvey, D.J.; Dwek, R.A.; Rudd, P.M.; Sim, R.B. Human serum IgM glycosylation: Identification of glycoforms that can bind to mannan-binding lectin. J. Biol. Chem. 2005, 280, 29080–29087. [Google Scholar] [CrossRef]
- Malhotra, R.; Wormald, M.R.; Rudd, P.M.; Fischer, P.B.; Dwek, R.A.; Sim, R.B. Glycosylation changes of IgG associated with rheumatoid arthritis can activate complement via the mannose-binding protein. Nat. Med. 1995, 1, 237–243. [Google Scholar] [CrossRef]
- Yasutake, J.; Suzuki, Y.; Suzuki, H.; Hiura, N.; Yanagawa, H.; Makita, Y.; Kaneko, E.; Tomino, Y. Novel lectin-independent approach to detect galactose-deficient IgA1 in IgA nephropathy. Nephrol. Dial. Transplant. 2015, 30, 1315–1321. [Google Scholar] [CrossRef]
- Takahashi, K.; Raska, M.; Stuchlova Horynova, M.; Hall, S.D.; Poulsen, K.; Kilian, M.; Hiki, Y.; Yuzawa, Y.; Moldoveanu, Z.; Julian, B.A.; et al. Enzymatic sialylation of IgA1 O-glycans: Implications for studies of IgA nephropathy. PLoS ONE 2014, 9, e99026. [Google Scholar] [CrossRef]
- Hiki, Y.; Hori, H.; Yamamoto, K.; Yamamoto, Y.; Yuzawa, Y.; Kitaguchi, N.; Takahashi, K. Specificity of two monoclonal antibodies against a synthetic glycopeptide, an analogue to the hypo-galactosylated IgA1 hinge region. J. Nephrol. 2015, 28, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Zhang, Z.; Zhang, H.; Liu, X. Aberrant IgA1 glycosylation in IgA nephropathy: A Systematic Review. PLoS ONE 2016, 11, e0166700. [Google Scholar] [CrossRef]
- Suzuki, Y.; Matsuzaki, K.; Suzuki, H.; Okazaki, K.; Yanagawa, H.; Ieiri, N.; Sato, M.; Sato, T.; Taguma, Y.; Matsuoka, J.; et al. Serum levels of galactose-deficient immunoglobulin (Ig) A1 and related immune complex are associated with disease activity of IgA nephropathy. Clin. Exp. Immunol. 2014, 18, 770–777. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Hou, P.; Lv, J.; Moldoveanu, Z.; Li, Y.; Kiryluk, K.; Gharavi, A.G.; Novak, J.; Zhang, H. The level of galactose-deficient IgA1 in the sera of patients with IgA nephropathy is associated with disease progression. Kidney Int. 2012, 82, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Maixnerova, D.; Ling, C.; Hall, S.; Reily, C.; Brown, R.; Neprasova, M.; Suchanek, M.; Honsova, E.; Zima, T.; Novak, J.; et al. Galactose-deficient IgA1 and the corresponding IgG autoantibodies predict IgA nephropathy progression. PLoS ONE 2019, 14, e0212254. [Google Scholar] [CrossRef]
- Lafayette, R.A.; Canetta, P.A.; Rovin, B.H.; Appel, G.B.; Novak, J.; Nath, K.A.; Sethi, S.; Tumlin, J.A.; Mehta, K.; Hogan, M.; et al. A Randomized, controlled trial of Rituximab in IgA nephropathy with proteinuria and renal dysfunction. J. Am. Soc. Nephrol. 2017, 28, 1306–1313. [Google Scholar] [CrossRef]
- Wada, Y.; Matsumoto, K.; Suzuki, T.; Saito, T.; Kanazawa, N.; Tachibana, S.; Iseri, K.; Sugiyama, M.; Iyoda, M.; Shibata, T. Clinical significance of serum and mesangial galactose-deficient IgA1 in patients with IgA nephropathy. PLoS ONE 2018, 13, e0206865. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Yasutake, J.; Makita, Y.; Tanbo, Y.; Yamasaki, K.; Sofue, T.; Kano, T.; Suzuki, Y. IgA nephropathy and IgA vasculitis with nephritis have a shared feature involving galactose-deficient IgA1-oriented pathogenesis. Kidney Int. 2018, 93, 700–705. [Google Scholar] [CrossRef]
- Temurhan, S.; Akgul, S.U.; Caliskan, Y.; Artan, A.S.; Kekik, C.; Yazici, H.; Demir, E.; Caliskan, B.; Turkmen, A.; Oguz, F.S.; et al. A Novel biomarker for post-transplant recurrent IgA nephropathy. Transplant. Proc. 2017, 49, 541–545. [Google Scholar] [CrossRef]
- Yamasaki, K.; Suzuki, H.; Yasutake, J.; Yamazaki, Y.; Suzuki, Y. Galactose-Deficient IgA1-Specific Antibody Recognizes GalNAc-Modified Unique Epitope on Hinge Region of IgA1. Monoclon. Antib. Immunodiagn. Immunother. 2018, 37, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Lv, J.; Zhang, X.; Chen, P.; Zhao, M.; Zhang, H. Secondary IgA Nephropathy shares the same immune features with primary IgA nephropathy. Kidney Int. Rep. 2020, 5, 165–172. [Google Scholar] [CrossRef]
- Zhao, L.; Peng, L.; Yang, D.; Chen, S.; Lan, Z.; Zhu, X.; Yuan, S.; Chen, G.; Liu, Y.; Liu, H. Immunostaining of galactose-deficient IgA1 by KM55 is not specific for immunoglobulin A nephropathy. Clin. Immunol. 2020, 217, 108483. [Google Scholar] [CrossRef]
- Cassol, C.A.; Bott, C.; Nadasdy, G.M.; Alberton, V.; Malvar, A.; Nagaraja, H.N.; Nadasdy, T.; Rovin, B.H.; Satoskar, A.A. Immunostaining for galactose-deficient immunoglobulin A is not specific for primary immunoglobulin A nephropathy. Nephrol. Dial. Transplant. 2020, 35, 2123–2129. [Google Scholar] [CrossRef]
- Hiki, Y.; Tanaka, A.; Kokubo, T.; Iwase, H.; Nishikido, J.; Hotta, K.; Kobayashi, Y. Analyses of IgA1 hinge glycopeptides in IgA nephropathy by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. J. Am. Soc. Nephrol. 1998, 9, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Iijima, H.; Tajiri, M.; Shinzaki, S.; Shiraishi, E.; Hiyama, S.; Mukai, A.; Nakajima, S.; Iwatani, H.; Nishida, T.; et al. Deficiency of N-acetylgalactosamine in O-linked oligosaccharides of IgA is a novel biologic marker for Crohn’s disease. Inflamm. Bowel Dis. 2012, 18, 1723–1734. [Google Scholar] [CrossRef]
- Nakazawa, S.; Imamura, R.; Kawamura, M.; Kato, T.; Abe, T.; Namba, T.; Iwatani, H.; Yamanaka, K.; Uemura, M.; Kishikawa, H.; et al. Difference in IgA1 O-glycosylation between IgA deposition donors and IgA nephropathy recipients. Biochem. Biophys. Res. Commun. 2019, 508, 1106–1112. [Google Scholar] [CrossRef] [PubMed]
- Odani, H.; Hiki, Y.; Takahashi, M.; Nishimoto, A.; Yasuda, Y.; Iwase, H.; Shinzato, T.; Maeda, K. Direct evidence for decreased sialylation and galactosylation of human serum IgA1 Fc O-glycosylated hinge peptides in IgA nephropathy by mass spectrometry. Biochem. Biophys. Res. Commun. 2000, 271, 268–274. [Google Scholar] [CrossRef]
- Buck, K.S.; Smith, A.C.; Molyneux, K.; El-Barbary, H.; Feehally, J.; Barratt, J. B-cell O-galactosyltransferase activity, and expression of O-glycosylation genes in bone marrow in IgA nephropathy. Kidney Int. 2008, 73, 1128–1136. [Google Scholar] [CrossRef][Green Version]
- Xing, Y.; Li, L.; Zhang, Y.; Wang, F.; He, D.; Liu, Y.; Jia, J.; Yan, T.; Lin, S. C1GALT1 expression is associated with galactosylation of IgA1 in peripheral B lymphocyte in immunoglobulin a nephropathy. BMC Nephrol. 2020, 21, 18. [Google Scholar] [CrossRef]
- Novak, J.; Raskova Kafkova, L.; Suzuki, H.; Tomana, M.; Matousovic, K.; Brown, R.; Hall, S.; Sanders, J.T.; Eison, T.M.; Moldoveanu, Z.; et al. IgA1 immune complexes from pediatric patients with IgA nephropathy activate cultured human mesangial cells. Nephrol. Dial. Transplant. 2011, 26, 3451–3457. [Google Scholar] [CrossRef] [PubMed]
- Iwatani, H.; Inoue, T.; Wada, Y.; Nagasawa, Y.; Yamamoto, R.; Iijima, H.; Takehara, T.; Imai, E.; Rakugi, H.; Isaka, Y. Quantitative change of IgA hinge O-glycan composition is a novel marker of therapeutic responses of IgA nephropathy. Biochem. Biophys. Res. Commun. 2012, 428, 339–342. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics of serum IgA1 in IgAN | ||
| High reactivity with lectin from Helix aspersa (HAA) | Moldoveanu,2007 [32], Shimozato, 2008 [33] | |
| High reactivity with monoclonal antibodies | Yasutake, 2015 [152], Hiki, 2015 [154] | |
| IgA1 carbohydrates in IgAN assessed by mass spectrometry | ||
| Decrease of galactose (Gal) | Hiki, 1998 [167], Inoue, 2012 [168], Nakazawa, 2019 [169] | |
| Decrease of N-acetylgalactosamine (GalNAc) | Hiki,1998 [167], Odani, 2000 [170], Inoue, 2012 [168], Nakazawa, 2019 [169] | |
| Decrease of sialic acid | Odani, 2000 [170] | |
| Sites of glycan attachment | ||
| Occur in specific sites | Renfrow, 2005 [50], Iwasaki, 2003 [51], Takahashi, 2010 [52], Takahashi, 2012 [53] | |
| Disease in specific sites | Needs to be investigated | |
| Expression of O-glycosyltransferases in B cells | ||
| No decrease in the ratio of C1GALT1: GALNT2 or C1GALT1: C1GALT1C1 | Buck, 2008 [171] | |
| Expression of C1GALT1 and ST6GALNAC2 altered in IgA1-producing cell lines from IgAN patients | Suzuki, 2008 [76] | |
| Decreased C1GALT1 expression in IgAN CD19+ B cells | Xing, 2020 [172] | |
| O-Glycosylation characteristics of polymeric IgA1 | ||
| Polymeric IgA1 shows higher reactivity with HAA than in monomeric IgA1 | Oortwijin, 2006 [149], Suzuki, 2008 [76], Novak, 2011 [173] | |
| Polymeric IgA1 interacted with CD71 and its interaction is enhanced by sialidase and β-galactosidase | Moura, 2004 [139] | |
| Epitopes recognized by IgG/A autoantibodies | ||
| Needs to be investigated | ||
| Clinical factors related to glycosylation of IgA1 | ||
| Genetic influences | Gd-IgA1 levels are highly inherited and affected by variants of glycosyltransferase | Tam,2009 [82], Kiryluk, 2011 [83], Gharavi, 2008 [79], Lin, 2009 [84], Hastings, 2010 [85], Gale, 2017 [95], Kiryluk, 2017 [96] |
| Susceptibility to IgAN is affected by variants of glycosyltransferase | Li 2007 [86,89], Pirulli, 2009 [87], Zhu, 2009, [88] | |
| Race differences | Gd-IgA1 levels elevated in Caucasian patients | Gale,2017 [95] |
| Gd-IgA1-increasing allele is common in Europeans | Kiryluk, 2017 [96] | |
| MicroRNA regulating C1GALT1 and GALNT2 overexpressed in Caucasian patients | Serino, 2016 [100] | |
| Age differences | Needs to be investigated | |
| Disease activity | No association of Gd-IgA1 levels measured by HAA-based ELISA with disease activity | Moldoveanu, 2007 [32], Shimozato, 2008 [33] |
| Positive association of Gd-IgA1 level measured by HAA-based ELISA with disease activity | Suzuki, 2014 [156], Sun, 2016 [155], Zhao, 2012 [157], Maixnerova, 2019 [158] | |
| Gd-IgA1 levels measured by KM55 associated with disease progression or recurrence | Wada, 2018 [160], Temurhan, 2017 [162] | |
| Longitudinal changes | Longitudinal changes of Gd-IgA1 serum levels by HAA ELISA before and after therapy need additional studies | Shimozato, 2008 [33], Suzuki, 2014 [156], Lafayette, 2017 [159] |
| O-Glycoform analyzed by MS changes between, before and after therapy | Iwatani, 2012 [174] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohyama, Y.; Renfrow, M.B.; Novak, J.; Takahashi, K. Aberrantly Glycosylated IgA1 in IgA Nephropathy: What We Know and What We Don’t Know. J. Clin. Med. 2021, 10, 3467. https://doi.org/10.3390/jcm10163467
Ohyama Y, Renfrow MB, Novak J, Takahashi K. Aberrantly Glycosylated IgA1 in IgA Nephropathy: What We Know and What We Don’t Know. Journal of Clinical Medicine. 2021; 10(16):3467. https://doi.org/10.3390/jcm10163467
Chicago/Turabian StyleOhyama, Yukako, Matthew B. Renfrow, Jan Novak, and Kazuo Takahashi. 2021. "Aberrantly Glycosylated IgA1 in IgA Nephropathy: What We Know and What We Don’t Know" Journal of Clinical Medicine 10, no. 16: 3467. https://doi.org/10.3390/jcm10163467
APA StyleOhyama, Y., Renfrow, M. B., Novak, J., & Takahashi, K. (2021). Aberrantly Glycosylated IgA1 in IgA Nephropathy: What We Know and What We Don’t Know. Journal of Clinical Medicine, 10(16), 3467. https://doi.org/10.3390/jcm10163467