
History of IgA Nephropathy Mouse Models



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. IgAN Epidemiology
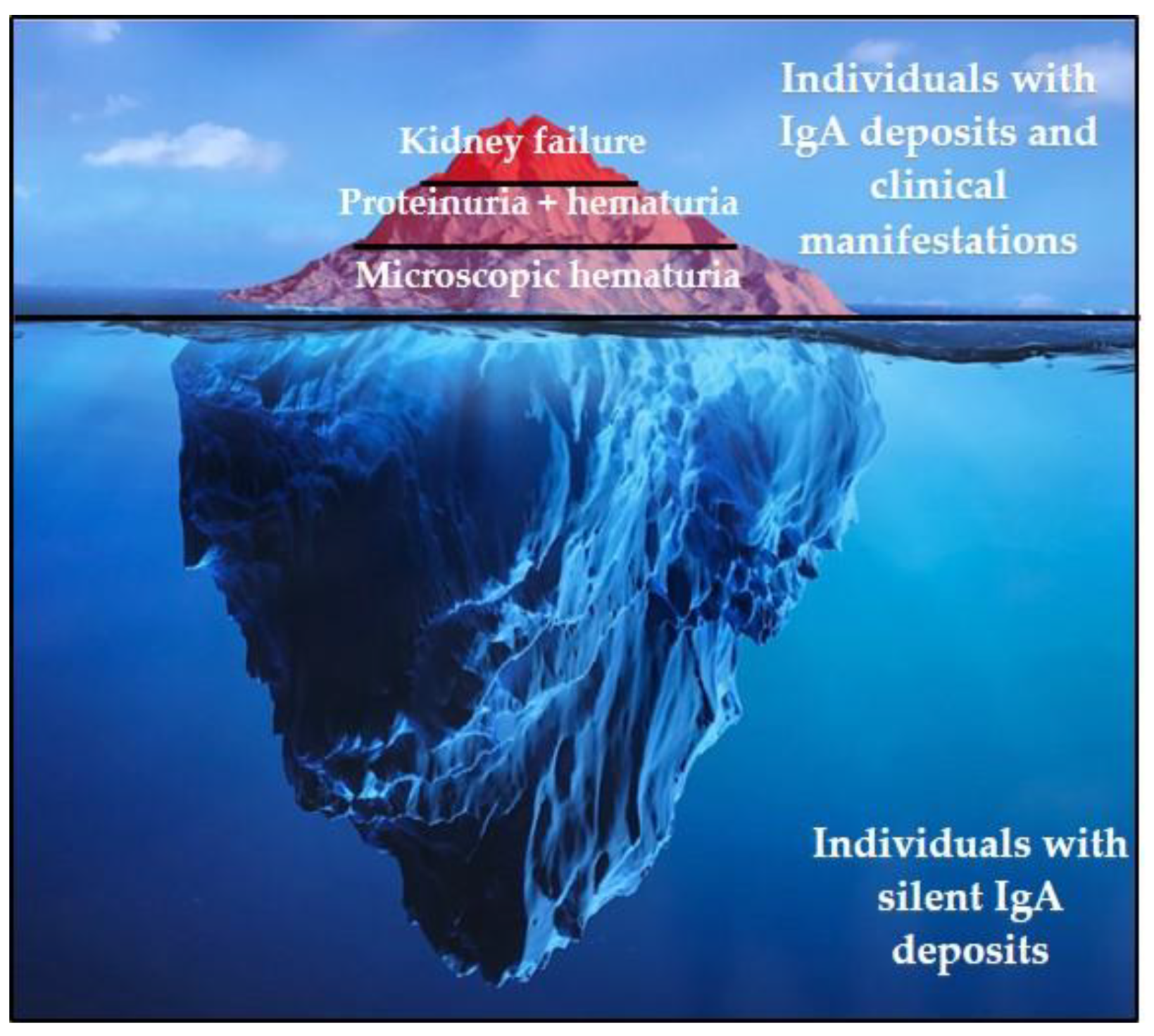
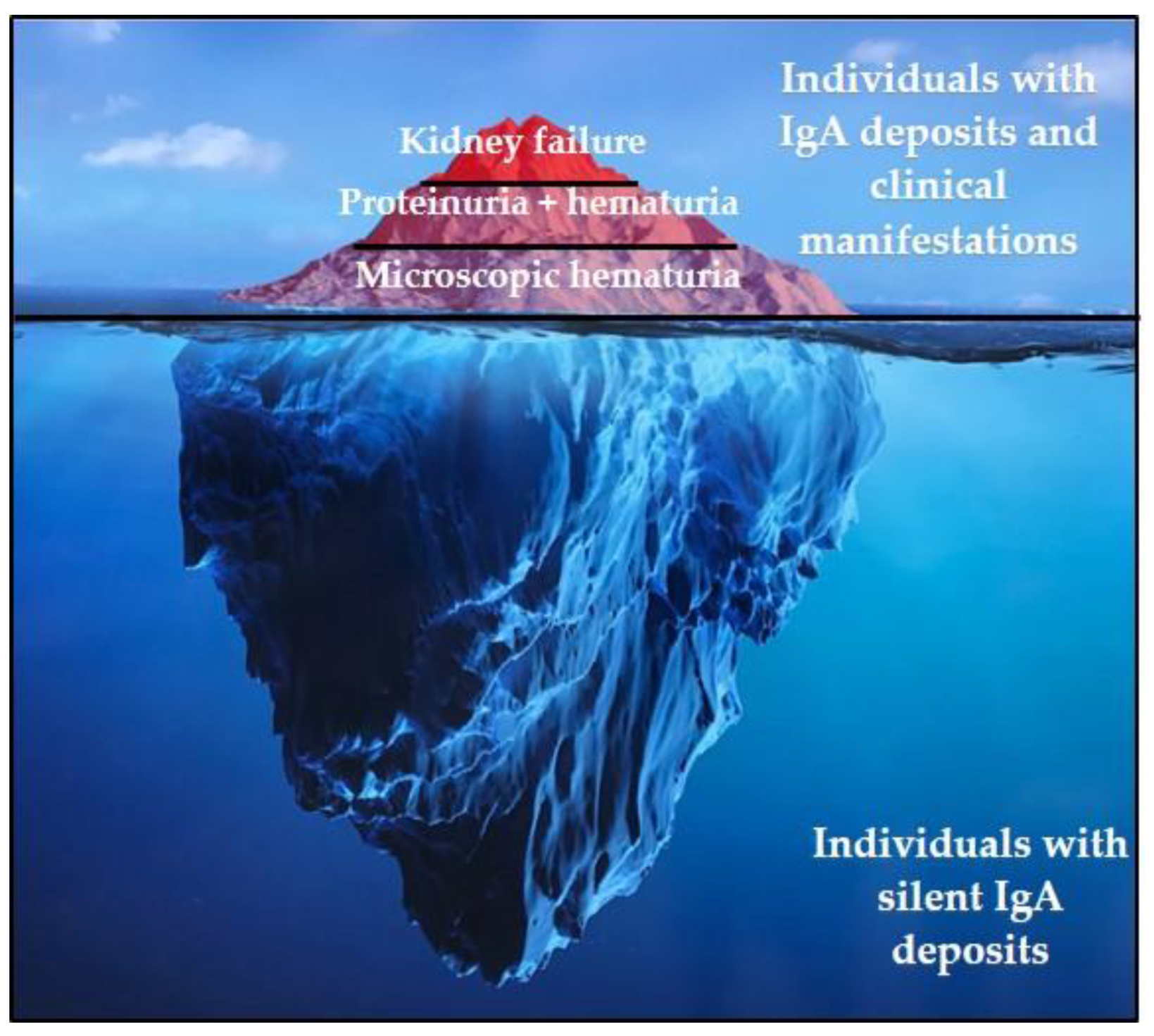
2. IgAN: Clinical and Histopathological Presentation
3. Enigmatic Functions of IgA
4. IgAN: A Multifactorial Disease
5. Genetic Factors
6. Aberrant IgA Glycosylation
7. Environmental Factors
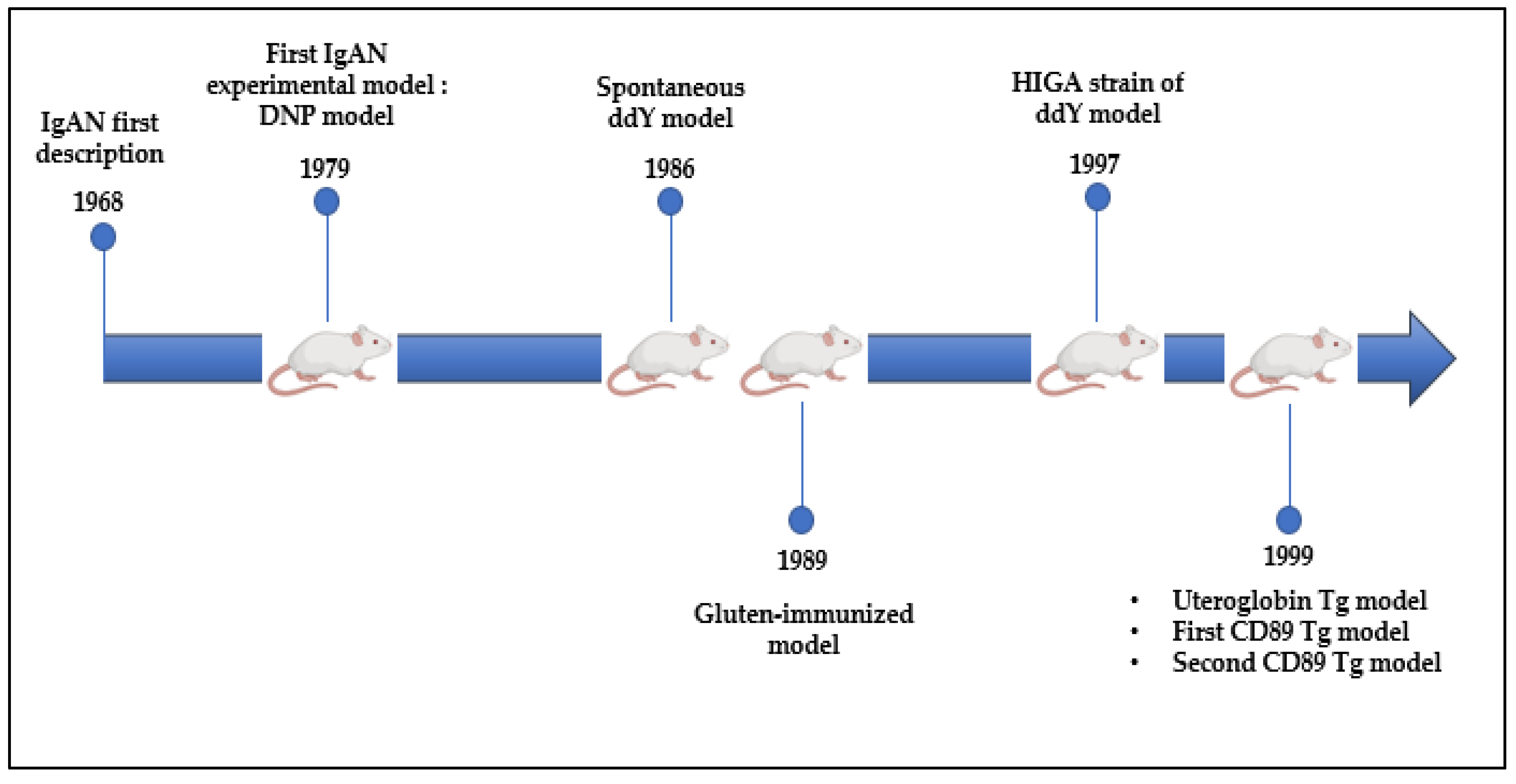
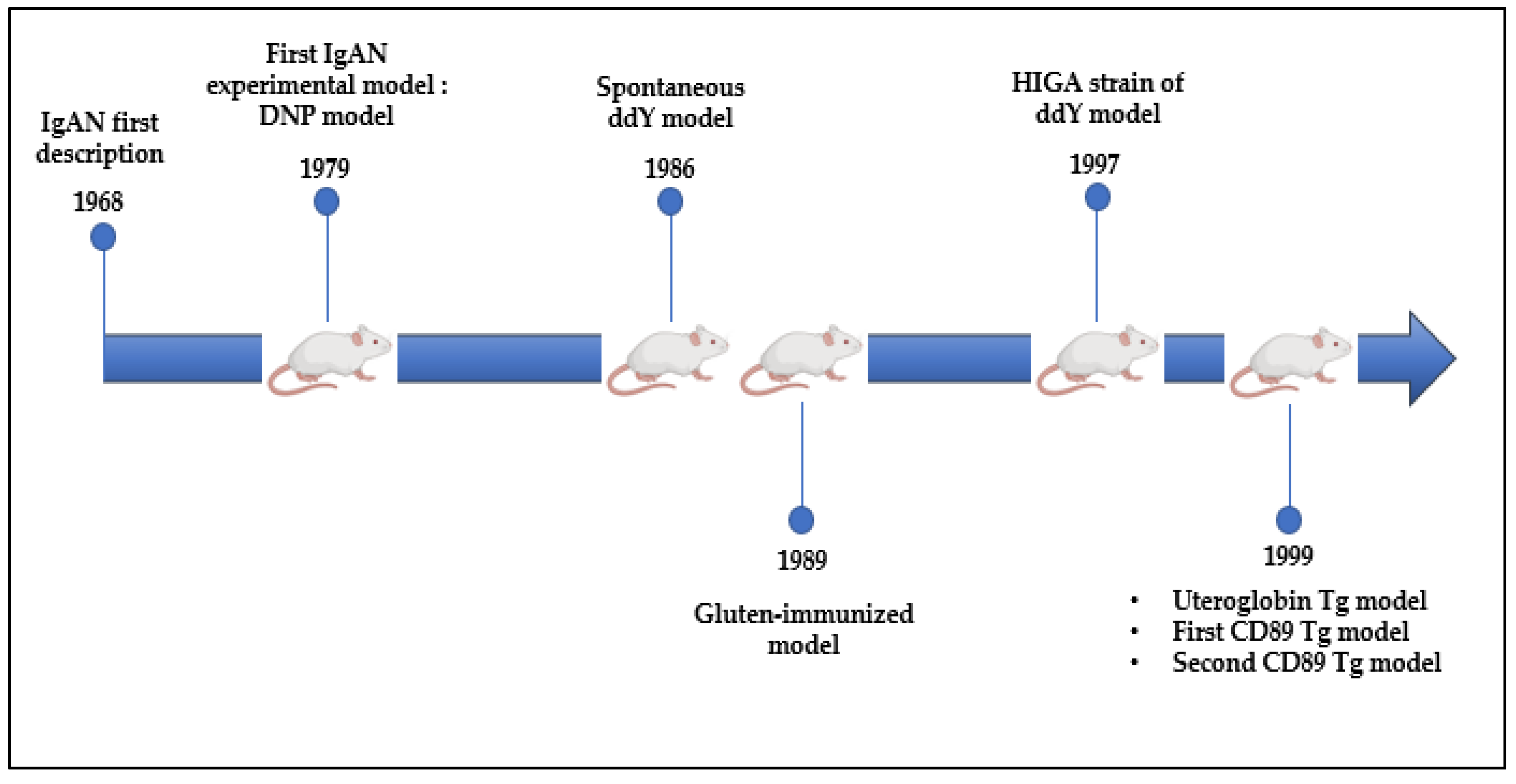
8. What Do We Learn from Experimental Models?
8.1. DNP and ddY Models
8.2. Uteroglobin Tg Model
8.3. CD89 Tg Models
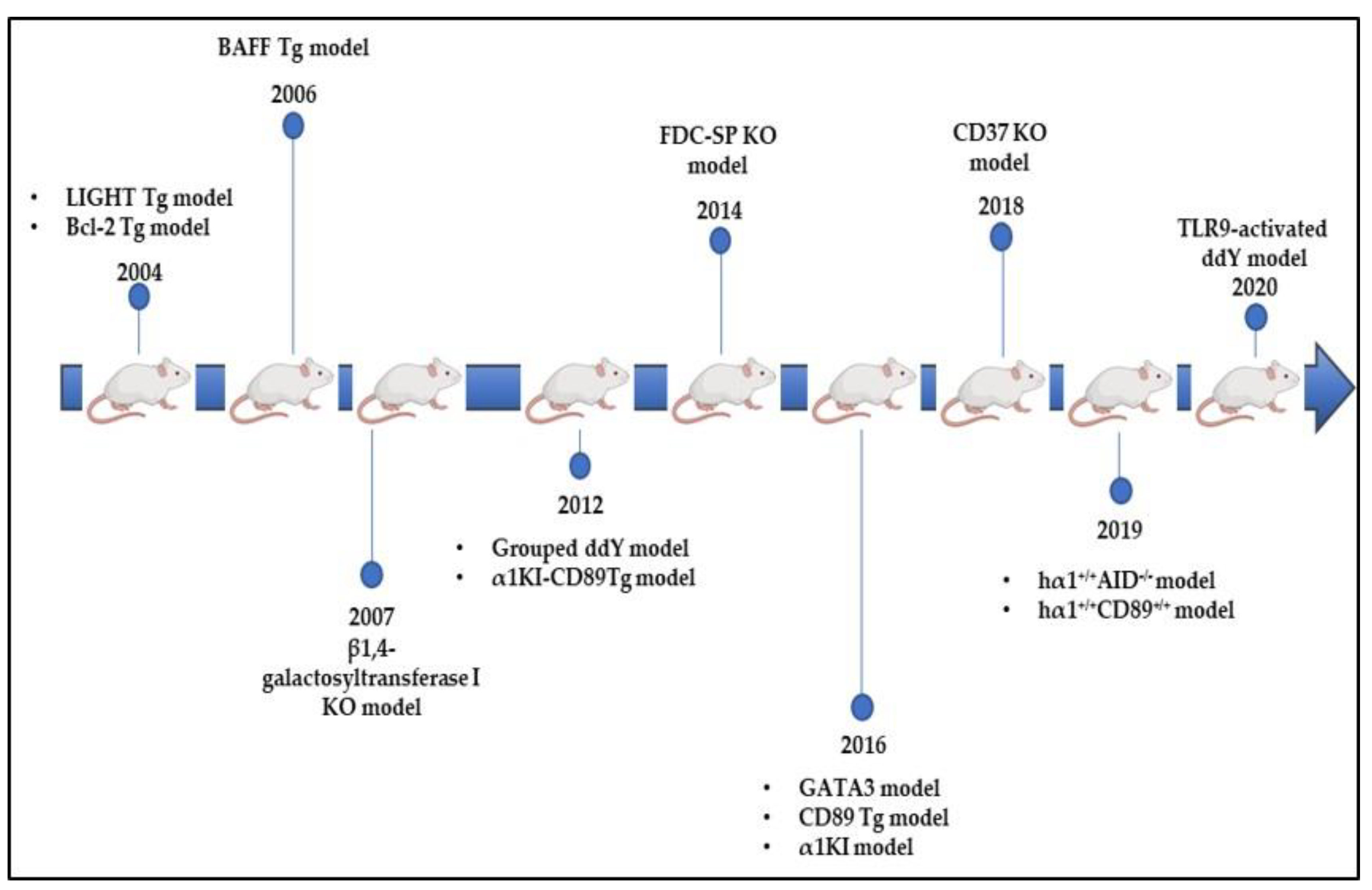
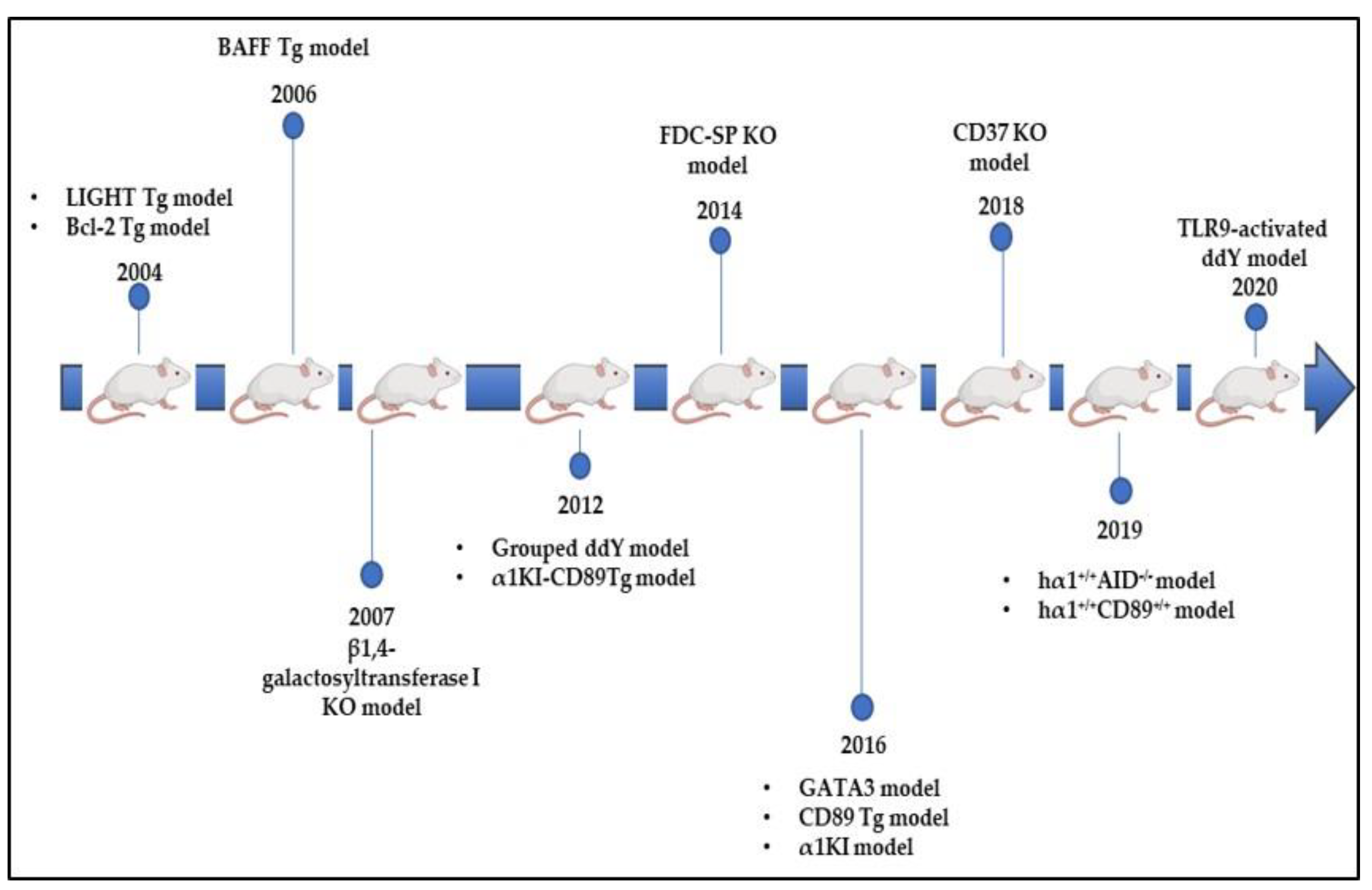
8.4. Bcl-2 Tg Model
8.5. LIGHT Tg Model
8.6. BAFF and CD37 Tg Models
8.7. β4GalT-I KO Model
8.8. FDC-SP KO Model
8.9. hα1+/+AID−/− Model
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Suzuki, K.; Honda, K.; Tanabe, K.; Toma, H.; Nihei, H.; Yamagushi, Y. Incidence of Latent Mesangial IgA Deposition in Renal Allograft Donors in Japan. Kidney Int. 2003, 63, 2286–2294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, C.K.; Barratt, J. Is IgA Nephropathy a Single Disease. In Pathogenesis and Treatment in IgA Nephropathy; Springer: Tokyo, Japan, 2016; pp. 3–17. ISBN 978-4-431-55587-2. [Google Scholar]
- Mestecky, J.; Raska, M.; Julian, B.A.; Gharavi, A.G.; Renfrow, M.B.; Moldoveanu, Z.; Novak, L.; Matousovic, K.; Novak, J. IgA Nephropathy: Molecular Mechanisms of the Disease. Annu. Rev. Pathol. Mech. Dis. 2013, 8, 217–240. [Google Scholar] [CrossRef] [Green Version]
- Wyatt, R.J.; Julian, B.A. IgA Nephropathy. N. Engl. J. Med. 2013, 368, 2402–2414. [Google Scholar] [CrossRef] [Green Version]
- Noel, L.-H. Atlas de Pathologie Rénale; FLAMMARION MEDECINE-SCIENCE Edition; LAVOISIER MSP: Paris, France, 2008. [Google Scholar]
- Roberts, I.S.; Cook, H.T.; Troyanov, S.; Alpers, C.E.; Amore, A.; Barratt, J.; Berthoux, F.; Bonsib, S.; Bruijn, J.A.; Cattran, D.C. The Oxford Classification of IgA Nephropathy: Pathology Definitions, Correlations, and Reproducibility. Kidney Int. 2009, 76, 546–556. [Google Scholar] [CrossRef] [Green Version]
- Lai, K.-N.; Chui, S.-H.; Lai, F.M.-M.; Lam, C.W. Predominant Synthesis of IgA with Lambda Light Chain in IgA Nephropathy. Kidney Int. 1988, 33, 584–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunker, J.J.; Drees, C.; Watson, A.R.; Plunkett, C.H.; Nagler, C.R.; Schneewind, O.; Eren, A.M.; Bendelac, A. B Cell Superantigens in the Human Intestinal Microbiota. Sci. Transl. Med. 2019, 11, 507. [Google Scholar] [CrossRef]
- Pascal, V.; Hiblot, M.; Wehbi, B.; Aldigier, J.-C.; Cogné, M. Homéostasie de la réponse IgA et microbiote. Med. Sci. 2021, 37, 35–40. [Google Scholar] [CrossRef]
- Bunker, J.J.; Bendelac, A. IgA Responses to Microbiota. Immunity 2018, 49, 211–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knoppova, B.; Reily, C.; Maillard, N.; Rizk, D.V.; Moldoveanu, Z.; Mestecky, J.; Raska, M.; Renfrow, M.B.; Julian, B.A.; Novak, J. The Origin and Activities of IgA1-Containing Immune Complexes in IgA Nephropathy. Front. Immunol. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Floege, J.; Feehally, J. The Mucosa–Kidney Axis in IgA Nephropathy. Nat. Rev. Nephrol. 2016, 12, 147–156. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, H. Insights into the Role of Mucosal Immunity in IgA Nephropathy. Clin. J. Am. Soc. Nephrol. 2018, CJN.04370418. [Google Scholar] [CrossRef]
- Liu, L.; Wang, L.; Jiang, Y.; Yao, L.; Dong, L.; Li, Z.; Li, X. Tonsillectomy for IgA Nephropathy: A Meta-Analysis. Am. J. Kidney Dis. 2015, 65, 80–87. [Google Scholar] [CrossRef]
- Kiryluk, K.; Novak, J.; Gharavi, A.G. The Genetics and Immunobiology of IgA Nephropathy. J. Clin. Investig. 2014, 124, 2325–2332. [Google Scholar] [CrossRef] [Green Version]
- Kiryluk, K.; Novak, J.; Gharavi, A.G. Pathogenesis of Immunoglobulin A Nephropathy: Recent Insight from Genetic Studies. Annu. Rev. Med. 2013, 64, 339–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Wang, L.; Shi, D.-C.; Foo, J.-N.; Zhong, Z.; Khor, C.-C.; Lanzani, C.; Citterio, L.; Salvi, E.; Yin, P.-R.; et al. Genome-Wide Meta-Analysis Identifies Three Novel Susceptibility Loci and Reveals Ethnic Heterogeneity of Genetic Susceptibility for IgA Nephropathy. J. Am. Soc. Nephrol. 2020, 31, 2949–2963. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-N.; Zhou, X.-J.; Chen, P.; Yu, G.-Z.; Zhang, X.; Hou, P.; Liu, L.-J.; Shi, S.-F.; Lv, J.-C.; Zhang, H. Interaction between G ALNT12 and C1GALT1 Associates with Galactose-Deficient IgA1 and IgA Nephropathy. J. Am. Soc. Nephrol. 2021, 32, 545–552. [Google Scholar] [CrossRef]
- Suzuki, H.; Suzuki, Y.; Yamanaka, T.; Hirose, S.; Nishimura, H.; Toei, J.; Horikoshi, S.; Tomino, Y. Genome-Wide Scan in a Novel IgA Nephropathy Model Identifies a Susceptibility Locus on Murine Chromosome 10, in a Region Syntenic to Human IGAN1 on Chromosome 6q22–23. J. Am. Soc. Nephrol. 2005, 16, 1289–1299. [Google Scholar] [CrossRef] [Green Version]
- Rifai, A.; Fadden, K.; Morrison, S.L.; Chintalacharuvu, K.R. The N-Glycans Determine the Differential Blood Clearance and Hepatic Uptake of Human Immunoglobulin (Ig)A1 and Iga2 Isotypes. J. Exp. Med. 2000, 191, 2171–2182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andre, P.M.; Le Pogamp, P.; Chevet, D. Impairment of Jacalin Binding to Serum IgA in IgA Nephropathy. J. Clin. Lab. Anal. 1990, 4, 115–119. [Google Scholar] [CrossRef]
- Moldoveanu, Z.; Wyatt, R.J.; Lee, J.Y.; Tomana, M. Patients with IgA Nephropathy Have Increased Serum Galactose-Deficient IgA1 Levels. Kidney Int. 2007, 71, 1148–1154. [Google Scholar] [CrossRef] [Green Version]
- Shimozato, S.; Hiki, Y.; Odani, H.; Takahashi, K.; Yamamoto, K.; Sugiyama, S. Serum Under-Galactosylated IgA1 Is Increased in Japanese Patients with IgA Nephropathy. Nephrol. Dial. Transplant. 2008, 23, 1931–1939. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Moldoveanu, Z.; Hall, S.; Brown, R.; Vu, H.L.; Novak, L.; Julian, B.A.; Tomana, M.; Wyatt, R.J.; Edberg, J.C.; et al. IgA1-Secreting Cell Lines from Patients with IgA Nephropathy Produce Aberrantly Glycosylated IgA1. J. Clin. Investig. 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagawa, H.; Suzuki, H.; Suzuki, Y.; Kiryluk, K.; Gharavi, A.G.; Matsuoka, K.; Makita, Y.; Julian, B.A.; Novak, J.; Tomino, Y. A Panel of Serum Biomarkers Differentiates IgA Nephropathy from Other Renal Diseases. PLoS ONE 2014, 9, e98081. [Google Scholar] [CrossRef] [Green Version]
- Gharavi, A.G.; Moldoveanu, Z.; Wyatt, R.J.; Barker, C.V.; Woodford, S.Y.; Lifton, R.P.; Mestecky, J.; Novak, J.; Julian, B.A. Aberrant IgA1 Glycosylation Is Inherited in Familial and Sporadic IgA Nephropathy. J. Am. Soc. Nephrol. 2008, 19, 1008–1014. [Google Scholar] [CrossRef] [PubMed]
- Lehoux, S.; Mi, R.; Aryal, R.P.; Wang, Y.; Schjoldager, K.T.-B.G.; Clausen, H.; van Die, I.; Han, Y.; Chapman, A.B.; Cummings, R.D.; et al. Identification of Distinct Glycoforms of IgA1 in Plasma from Patients with Immunoglobulin A (IgA) Nephropathy and Healthy Individuals. Mol. Cell. Proteom. 2014, 13, 3097–3113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiki, Y.; Takahashi, K.; Shimozato, S.; Odani, H.; Yamamoto, K.; Tomita, M.; Hasegawa, M.; Murakami, K.; Nabeshima, K.; Nakai, S.; et al. Protective Role of Anti-Synthetic Hinge Peptide Antibody for Glomerular Deposition of Hypoglycosylated IgA1. Clin. Exp. Nephrol. 2008, 12, 20–27. [Google Scholar] [CrossRef]
- Schmitt, R.; Carlsson, F.; Mörgelin, M.; Tati, R.; Lindahl, G.; Karpman, D. Tissue Deposits of IgA-Binding Streptococcal M Proteins in IgA Nephropathy and Henoch-Schönlein Purpura. Am. J. Pathol. 2010, 176, 608–618. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Z.; Tan, J.; Tan, L.; Tang, Y.; Qiu, Z.; Pei, G.; Qin, W. Modifications of Gut Microbiota Are Associated with the Severity of IgA Nephropathy in the Chinese Population. Int. Immunopharmacol. 2020, 89, 107085. [Google Scholar] [CrossRef]
- Duchez, S.; Amin, R.; Cogne, N.; Delpy, L.; Sirac, C.; Pascal, V.; Corthesy, B.; Cogne, M. Premature Replacement of with Immunoglobulin Chains Impairs Lymphopoiesis and Mucosal Homing but Promotes Plasma Cell Maturation. Proc. Natl. Acad. Sci. USA 2010, 107, 3064–3069. [Google Scholar] [CrossRef] [Green Version]
- Oruc, Z.; Oblet, C.; Boumediene, A.; Druilhe, A.; Pascal, V.; Le Rumeur, E.; Cuvillier, A.; El Hamel, C.; Lecardeur, S.; Leanderson, T.; et al. IgA Structure Variations Associate with Immune Stimulations and IgA Mesangial Deposition. J. Am. Soc. Nephrol. 2016. [Google Scholar] [CrossRef] [Green Version]
- Chemouny, J.M.; Gleeson, P.J.; Abbad, L.; Lauriero, G.; Boedec, E.; Le Roux, K.; Monot, C.; Bredel, M.; Bex-Coudrat, J.; Sannier, A.; et al. Modulation of the Microbiota by Oral Antibiotics Treats Immunoglobulin A Nephropathy in Humanized Mice. Nephrol. Dial. Transplant. 2018. [Google Scholar] [CrossRef]
- Papista, C.; Lechner, S.; Mkaddem, S.B. Gluten Exacerbates IgA Nephropathy in Humanizedmice through Gliadin–CD89 Interaction. Kidney Int. 2015, 88, 276–285. [Google Scholar] [CrossRef]
- Rifai, A. Complement Activation in Experimental IgA Nephropathy: An Antigen-Mediated Process. Kidney Int. 1987, 32, 838–844. [Google Scholar] [CrossRef] [Green Version]
- Coppo, R.; Roccatello, D.; Amore, A.; Quattrocchio, G.; Molino, A.; Gianoglio, B.; Amoroso, A.; Bajardi, P.; Piccoli, G. Effects of a Gluten-Free Diet in Primary IgA Nephropathy. Clin. Nephrol. 1990, 33, 72–86. [Google Scholar] [PubMed]
- Yagame, M.; Tomino, Y.; Eguchi, K.; Miura, M. Levels of Circulating IgA Immune Complexes after Gluten-Rich Diet in Patients with IgA Nephropathy. Nephron 1988, 49, 104–106. [Google Scholar] [CrossRef] [PubMed]
- Coppo, R.; Mazzucco, G.; Martina, G.; Roccatello, D. Gluten-Induced Experimental IgA Glomerulopathy. Lab. Investig. 1989, 60, 499–506. [Google Scholar]
- Petska, J.; Moorman, M.; Warner, R. Dysregulation of IgA Production and IgA Nephropathy Induced by the Trichothecene Vomitoxin. Food Chem. Toxicol. 1989, 27, 361–368. [Google Scholar]
- Chintalacharuvu, S.R.; Nagy, N.U.; Sigmund, N.; Nedrud, J.G.; Amm, M.L.; Emancipator, S.N. T Cell Cytokines Determine the Severity of Experimental IgA Nephropathy by Regulating IgA Glycosylation. Clin. Exp. Immunol. 2001, 126, 326–333. [Google Scholar] [CrossRef]
- Imai, H.; Nakamoto, Y.; Asakura, K. Spontaneous Glomerular IgA Deposition in DdY Mice: An Animal Model of IgA Nephritis. Kidney Int. 1985, 27, 756–761. [Google Scholar] [CrossRef] [Green Version]
- Muso, E.; Yoshida, H.; Takeuchi, E.; Yashiro, M.; Matsushima, H.; Oyama, A.; Suyama, K.; Kawamura, T.; Kamata, T.; Miyawaki, S.; et al. Enhanced Production of Glomerular Extracellular Matrix in a New Mouse Strain of High Serum IgA DdY Mice. Kidney Int. 1996, 50, 1946–1957. [Google Scholar] [CrossRef]
- Okazaki, K.; Suzuki, Y.; Otsuji, M.; Suzuki, H.; Kihara, M.; Kajiyama, T.; Hashimoto, A.; Nishimura, H.; Brown, R.; Hall, S.; et al. Development of a Model of Early-Onset IgA Nephropathy. J. Am. Soc. Nephrol. 2012, 23, 1364–1374. [Google Scholar] [CrossRef]
- Nishie, T.; Miyaishi, O.; Azuma, H.; Kameyama, A.; Naruse, C.; Hashimoto, N.; Yokoyama, H.; Narimatsu, H.; Wada, T.; Asano, M. Development of Immunoglobulin A Nephropathy- Like Disease in β-1,4-Galactosyltransferase-I-Deficient Mice. Am. J. Pathol. 2007, 170, 447–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makita, Y.; Suzuki, H.; Kano, T.; Takahata, A.; Julian, B.A.; Novak, J.; Suzuki, Y. TLR9 Activation Induces Aberrant IgA Glycosylation via APRIL-and IL-6–Mediated Pathways in IgA Nephropathy. Kidney Int. 2020, 97, 340–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, F.; Kundu, G.C.; Zhang, Z.; Ward, J.; DeMayo, F.; Mukherjee, A.B. Uteroglobin Is Essential in Preventing Immunoglobulin A Nephropathy in Mice. Nat. Med. 1999, 5, 1018. [Google Scholar] [CrossRef] [PubMed]
- Launay, P.; Grossetete, B.; Arcos-Fajardo, M.; Gaudin, E.; Torres, S.P.; Beaudoin, L.; Patey-Mariaud de Serre, N. Fca Receptor (CD89) Mediates the Development of Immunoglobulin A (IgA) Nephropathy (Berger’s Disease): Evidence for Pathogenic Soluble Receptor–IgA Complexes in Patients and CD89 Transgenic Mice. J. Exp. Med. 2000, 191, 1999–2010. [Google Scholar] [CrossRef] [Green Version]
- Berthelot, L.; Papista, C.; Maciel, T.T.; Biarnes-Pelicot, M.; Tissandie, E.; Wang, P.H.M.; Tamouza, H.; Jamin, A.; Bex-Coudrat, J.; Gestin, A.; et al. Transglutaminase Is Essential for IgA Nephropathy Development Acting through IgA Receptors. J. Exp. Med. 2012, 209, 793–806. [Google Scholar] [CrossRef]
- Xu, L.; Li, B.; Huang, M.; Xie, K.; Li, D.; Li, Y.; Gu, H.; Fang, J. Critical Role of Kupffer Cell CD89 Expression in Experimental IgA Nephropathy. PLoS ONE 2016, 11, e0159426. [Google Scholar] [CrossRef] [Green Version]
- Van Egmond, M.; van Vuuren, A.H.; Morton, H.C.; van Spriel, A.B.; Shen, L.; Hofhuis, F.M.A.; Saito, T.; Mayadas, T.N.; Verbeek, J.S.; van de Winkel, J.G. Human Immunoglobulin A Receptor (FcaRI, CD89) Function in Transgenic Mice Requires Both FcR g Chain and CR3 (CD11b/CD18). Blood 1999, 93, 4387–4394. [Google Scholar] [CrossRef]
- Wehbi, B.; Oblet, C.; Boyer, F.; Huard, A.; Druilhe, A.; Paraf, F.; Cogné, E.; Moreau, J.; El Makhour, Y.; Badran, B.; et al. Mesangial Deposition Can Strongly Involve Innate-Like IgA Molecules Lacking Affinity Maturation. J. Am. Soc. Nephrol. 2019, 30, 1238–1249. [Google Scholar] [CrossRef]
- Marquina, R.; Díez, M.A.; López-Hoyos, M.; Buelta, L.; Kuroki, A.; Kikuchi, S.; Villegas, J.; Pihlgren, M.; Siegrist, C.-A.; Arias, M.; et al. Inhibition of B Cell Death Causes the Development of an IgA Nephropathy in (New Zealand White × C57BL/6)F 1-Bcl-2 Transgenic Mice. J. Immunol. 2004, 172, 7177–7185. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Anders, R.A.; Wu, Q.; Peng, D.; Cho, J.H.; Sun, Y.; Karaliukas, R.; Kang, H.-S.; Turner, J.R.; Fu, Y.-X. Dysregulated LIGHT Expression on T Cells Mediates Intestinal Inflammation and Contributes to IgA Nephropathy. J. Clin. Investig. 2004, 113, 826–835. [Google Scholar] [CrossRef]
- McCarthy, D.D.; Chiu, S.; Gao, Y.; Summers-deLuca, L.E.; Gommerman, J.L. BAFF Induces a Hyper-IgA Syndrome in the Intestinal Lamina Propria Concomitant with IgA Deposition in the Kidney Independent of LIGHT. Cell. Immunol. 2006, 241, 85–94. [Google Scholar] [CrossRef]
- McCarthy, D.D.; Kujawa, J.; Wilson, C.; Papandile, A.; Poreci, U.; Porfilio, E.A.; Ward, L.; Lawson, M.A.E.; Macpherson, A.J.; McCoy, K.D.; et al. Mice Overexpressing BAFF Develop a Commensal Flora–Dependent, IgA-Associated Nephropathy. J. Clin. Investig. 2011, 121, 3991–4002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rops, A.L.; Figdor, C.G.; van der Schaaf, A.; Tamboer, W.P.; Bakker, M.A.; Berden, J.H.; Dijkman, H.B.P.M.; Steenbergen, E.J.; van der Vlag, J.; van Spriel, A.B. The Tetraspanin CD37 Protects Against Glomerular IgA Deposition and Renal Pathology. Am. J. Pathol. 2010, 176, 2188–2197. [Google Scholar] [CrossRef] [PubMed]
- Rops, A.L.; Jansen, E.; van der Schaaf, A.; Pieterse, E.; Rother, N.; Hofstra, J.; Dijkman, H.B.; van de Logt, A.-E.; Wetzels, J.; van der Vlag, J. Interleukin-6 Is Essential for Glomerular Immunoglobulin A Deposition and the Development of Renal Pathology in Cd37-Deficient Mice. Kidney Int. 2018, 93, 1356–1366. [Google Scholar] [CrossRef]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal Developmental Pathways for the Generation of Pathogenic Effector TH17 and Regulatory T Cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef]
- Linterman, M.A.; Vinuesa, C.G. Signals That Influence T Follicular Helper Cell Differentiation and Function. Semin. Immunopathol. 2010, 32, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Molyneux, K.; Wimbury, D.; Pawluczyk, I.; Muto, M.; Bhachu, J.; Mertens, P.R.; Feehally, J.; Barratt, J. Β1,4-Galactosyltransferase 1 Is a Novel Receptor for IgA in Human Mesangial Cells. Kidney Int. 2017, 92, 1458–1468. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Liu, P.; Gao, L.; Zhang, X.; Lan, P.; Bijol, V.; Lv, J.; Zhang, H.; Jin, J. Renal Deposition and Clearance of Recombinant Poly- IgA Complexes in a Model of IgA Nephropathy. J. Pathol. 2021. [Google Scholar] [CrossRef]
- Hou, S.; Landego, I.; Jayachandran, N.; Miller, A.; Gibson, I.W.; Ambrose, C.; Marshall, A.J. Follicular Dendritic Cell Secreted Protein FDC-SP Controls IgA Production. Mucosal Immunol. 2014, 7, 948–957. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wehbi, B.; Pascal, V.; Zawil, L.; Cogné, M.; Aldigier, J.-C. History of IgA Nephropathy Mouse Models. J. Clin. Med. 2021, 10, 3142. https://doi.org/10.3390/jcm10143142
Wehbi B, Pascal V, Zawil L, Cogné M, Aldigier J-C. History of IgA Nephropathy Mouse Models. Journal of Clinical Medicine. 2021; 10(14):3142. https://doi.org/10.3390/jcm10143142
Chicago/Turabian StyleWehbi, Batoul, Virginie Pascal, Lina Zawil, Michel Cogné, and Jean-Claude Aldigier. 2021. "History of IgA Nephropathy Mouse Models" Journal of Clinical Medicine 10, no. 14: 3142. https://doi.org/10.3390/jcm10143142
APA StyleWehbi, B., Pascal, V., Zawil, L., Cogné, M., & Aldigier, J.-C. (2021). History of IgA Nephropathy Mouse Models. Journal of Clinical Medicine, 10(14), 3142. https://doi.org/10.3390/jcm10143142