
Acute Movement Disorders in Childhood


Abstract
1. Introduction
2. Methods
3. Approach to Acute Movement Disorders in Childhood
4. Immune-Mediated Movement Disorders
4.1. Sydenham Chorea
4.2. Systemic Lupus Erythematosus (SLE) and Antiphospholipid Antibody Syndrome (APS)
4.3. Anti-N-methyl-d-aspartate Receptor (NMDAR) and Other Autoimmune Encephalitis (AE)
4.4. Acute Disseminated Encephalopathy (ADEM)
4.5. Opsoclonus–Myoclonus Syndrome (OMS) and Paraneoplastic MDs
5. Infectious and Para-Infectious Disorders
5.1. Viral Infections
5.2. Bacterial Infections
5.3. Protozoal, Fungal and Helmintic Infections
5.4. Infectious and Post-Infectious Bilateral Striatal Necrosis (BSN)
5.5. Acute Necrotizing Encephalopathy (ANEC)
6. Vascular Diseases
7. Drug-Induced and Toxic Movement Disorders
7.1. Acute Dystonic Rreactions
7.2. Neuroleptic Malignant Syndrome
7.3. Serotonin Syndrome
7.4. Other Iatrogenic Movement Disorders
7.5. Systemic Intoxications
8. Inborn Errors of Metabolism and Genetic Disorders
8.1. Organic Acidurias
8.2. Mitochondrial Disorders
8.3. Other Genetic Disorders
9. Metabolic and Endocrine Disorders
10. Paroxysmal Sympathetic Hyperactivity
11. Tics Disorders and Tourette Syndrome
12. Functional Movement Disorders
13. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gandhi, S.E.; Newman, E.J.; Marshall, V.L. Emergency presentations of movement disorders. Pract. Neurol. 2020, 20. [Google Scholar] [CrossRef] [PubMed]
- Kirkham, F.J.; Haywood, P.; Kashyape, P.; Borbone, J.; Lording, A.; Pryde, K.; Cox, M.; Keslake, J.; Smith, M.; Cuthbertson, L.; et al. Movement disorder emergencies in childhood. Eur. J. Paediatr. Neurol. 2011, 15, 390–404. [Google Scholar] [CrossRef] [PubMed]
- Dale, R.C.; Singh, H.; Troedson, C.; Pillai, S.; Gaikiwari, S.; Kozlowska, K. A prospective study of acute movement disorders in children. Dev. Med. Child Neurol. 2010, 52, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Goraya, J.S. Acute movement disorders in children: Experience from a developing country. J. Child Neurol. 2015, 30, 406–411. [Google Scholar] [CrossRef]
- Raucci, U.; Parisi, P.; Vanacore, N.; Garone, G.; Bondone, C.; Palmieri, A.; Calistri, L.; Suppiej, A.; Falsaperla, R.; Capuano, A.; et al. Acute hyperkinetic movement disorders in italian paediatric emergency departments. Arch. Dis. Child. 2018, 103, 790–794. [Google Scholar] [CrossRef] [PubMed]
- Garone, G.; Capuano, A.; Travaglini, L.; Graziola, F.; Stregapede, F.; Zanni, G.; Vigevano, F.; Bertini, E.; Nicita, F. Clinical and genetic overview of paroxysmal movement disorders and episodic ataxias. Int. J. Mol. Sci. 2020, 21, 3603. [Google Scholar] [CrossRef]
- McGovern, E.M.; Roze, E.; Counihan, T.J. The expanding spectrum of paroxysmal movement disorders: Update from clinical features to therapeutics. Curr. Opin. Neurol. 2018, 31, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.M.; Lin, J.-P.; Lynch, T.; King, M.D. Status dystonicus: A practice guide. Dev. Med. Child Neurol. 2014, 56, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Lumsden, D.E.; King, M.D.; Allen, N.M. Status dystonicus in childhood. Curr. Opin. Pediatr. 2017, 29, 674–682. [Google Scholar] [CrossRef]
- Brandsma, R.; van Egmond, M.E.; Tijssen, M.A.J. Diagnostic approach to paediatric movement disorders: A clinical practice guide. Dev. Med. Child Neurol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Hussain, E.; Nordli, D. EEG patterns in acute pediatric encephalopathies. J. Clin. Neurophysiol. 2013, 30, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F. Movement disorders in childhood. Parkinsonism Relat. Disord. 2014, 20 (Suppl. S1), S13–S16. [Google Scholar] [CrossRef]
- Eshel, G.; Lahat, E.; Azizi, E.; Gross, B.; Aladjem, M. Chorea as a manifestation of rheumatic fever—A 30-year survey (1960–1990). Eur. J. Pediatr. 1993, 152, 645–646. [Google Scholar] [CrossRef] [PubMed]
- Zomorrodi, A.; Wald, E.R. Sydenham’s chorea in Western Pennsylvania. Pediatrics 2006, 117, e675–e679. [Google Scholar] [CrossRef]
- Cardoso, F. Autoimmune choreas. J. Neurol. Neurosurg. Psychiatry 2017, 88, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Baizabal-Carvallo, J.F.; Cardoso, F. Chorea in children: Etiology, diagnostic approach and management. J. Neural Transm. Vienna Austria 1996 2020, 127, 1323–1342. [Google Scholar] [CrossRef]
- Ben-Pazi, H.; Jaworowski, S.; Shalev, R.S. Cognitive and psychiatric phenotypes of movement disorders in children: A systematic review. Dev. Med. Child Neurol. 2011, 53, 1077–1084. [Google Scholar] [CrossRef] [PubMed]
- Punukollu, M.; Mushet, N.; Linney, M.; Hennessy, C.; Morton, M. Neuropsychiatric manifestations of sydenham’s chorea: A systematic review. Dev. Med. Child Neurol. 2016, 58, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Gewitz, M.H.; Baltimore, R.S.; Tani, L.Y.; Sable, C.A.; Shulman, S.T.; Carapetis, J.; Remenyi, B.; Taubert, K.A.; Bolger, A.F.; Beerman, L.; et al. Revision of the Jones Criteria for the diagnosis of acute rheumatic fever in the era of doppler echocardiography: A scientific statement from the american heart association. Circulation 2015, 131, 1806–1818. [Google Scholar] [CrossRef]
- Dean, S.L.; Singer, H.S. Treatment of sydenham’s chorea: A review of the current evidence. Tremor Hyperkinetic Mov. N. Y. N 2017, 7, 456. [Google Scholar] [CrossRef]
- Cardoso, F.; Vargas, A.P.; Oliveira, L.D.; Guerra, A.A.; Amaral, S.V. Persistent sydenham’s chorea. Mov. Disord. Off. J. Mov. Disord. Soc. 1999, 14, 805–807. [Google Scholar] [CrossRef]
- Korn-Lubetzki, I.; Brand, A.; Steiner, I. Recurrence of sydenham chorea: Implications for pathogenesis. Arch. Neurol. 2004, 61, 1261–1264. [Google Scholar] [CrossRef] [PubMed]
- Gurkas, E.; Karalok, Z.S.; Taskin, B.D.; Aydogmus, U.; Guven, A.; Degerliyurt, A.; Bektas, O.; Yilmaz, C. Predictors of recurrence in sydenham’s chorea: Clinical observation from a single center. Brain Dev. 2016, 38, 827–834. [Google Scholar] [CrossRef]
- Demiroren, K.; Yavuz, H.; Cam, L.; Oran, B.; Karaaslan, S.; Demiroren, S. Sydenham’s chorea: A clinical follow-up of 65 patients. J. Child Neurol. 2007, 22, 550–554. [Google Scholar] [CrossRef]
- Tumas, V.; Caldas, C.T.; Santos, A.C.; Nobre, A.; Fernandes, R.M.F. Sydenham’s chorea: Clinical observations from a Brazilian movement disorder clinic. Parkinsonism Relat. Disord. 2007, 13, 276–283. [Google Scholar] [CrossRef]
- Berrios, X.; Quesney, F.; Morales, A.; Blazquez, J.; Bisno, A.L. Are all recurrences of “pure” sydenham chorea true recurrences of acute rheumatic fever? J. Pediatr. 1985, 107, 867–872. [Google Scholar] [CrossRef]
- Peluso, S.; Antenora, A.; De Rosa, A.; Roca, A.; Maddaluno, G.; Brescia Morra, V.; De Michele, G. Antiphospholipid-related chorea. Front. Neurol. 2012, 3, 150. [Google Scholar] [CrossRef] [PubMed]
- Soybilgic, A.; Avcin, T. Pediatric APS: State of the art. Curr. Rheumatol. Rep. 2020, 22, 9. [Google Scholar] [CrossRef] [PubMed]
- Cervera, R.; Asherson, R.A.; Font, J.; Tikly, M.; Pallarés, L.; Chamorro, A.; Ingelmo, M. Chorea in the antiphospholipid syndrome. Clinical, radiologic, and immunologic characteristics of 50 patients from our clinics and the recent literature. Medicine 1997, 76, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Kiechl-Kohlendorfer, U.; Ellemunter, H.; Kiechl, S. Chorea as the presenting clinical feature of primary antiphospholipid syndrome in childhood. Neuropediatrics 1999, 30, 96–98. [Google Scholar] [CrossRef] [PubMed]
- Angelini, L.; Zibordi, F.; Zorzi, G.; Nardocci, N.; Caporali, R.; Ravelli, A.; Martini, A. Neurological disorders, other than stroke, associated with antiphospholipid antibodies in childhood. Neuropediatrics 1996, 27, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Martino, D.; Chew, N.-K.; Mir, P.; Edwards, M.J.; Quinn, N.P.; Bhatia, K.P. Atypical movement disorders in antiphospholipid syndrome. Mov. Disord. 2006, 21, 944–949. [Google Scholar] [CrossRef]
- Dale, R.C.; Yin, K.; Ding, A.; Merheb, V.; Varadkhar, S.; McKay, D.; Singh-Grewal, D.; Brilot, F. Antibody binding to neuronal surface in movement disorders associated with lupus and antiphospholipid antibodies. Dev. Med. Child Neurol. 2011, 53, 522–528. [Google Scholar] [CrossRef] [PubMed]
- Dale, R.C. Immune-mediated extrapyramidal movement disorders, including sydenham chorea. Handb. Clin. Neurol. 2013, 112, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Brogna, C.; Mariotti, P.; Manna, R. Conventional and intravenous immunoglobulin therapy in paediatric antiphospholipid antibodies-related chorea. Lupus 2014, 23, 1449–1451. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, K.; Mori, M.; Yoshida, A. Mycophenolate mofetil therapy for two cases of antiphospholipid antibody-associated chorea. Mod. Rheumatol. 2018, 28, 709–711. [Google Scholar] [CrossRef] [PubMed]
- Baglan, E.; Ozdel, S.; Uysal Yazıcı, M.; Azapağası, E.; Çelik, H.; Yüksel, D.; Uçan, B.; Karakaya, D.; Bulbul, M. A novel therapeutic approach using the zipper method to treat chorea in a pediatric-onset systemic lupus erythematosus patient. Lupus 2021. [Google Scholar] [CrossRef]
- Balint, B.; Vincent, A.; Meinck, H.-M.; Irani, S.R.; Bhatia, K.P. Movement disorders with neuronal antibodies: Syndromic approach, genetic parallels and pathophysiology. Brain J. Neurol. 2018, 141, 13–36. [Google Scholar] [CrossRef]
- Dalmau, J.; Graus, F. Antibody-Mediated Encephalitis. N. Engl. J. Med. 2018, 378, 840–851. [Google Scholar] [CrossRef]
- Goenka, A.; Jain, V.; Nariai, H.; Spiro, A.; Steinschneider, M. Extended clinical spectrum of anti-n-methyl-d-aspartate receptor encephalitis in children: A case series. Pediatr. Neurol. 2017, 72, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, S.S.; Sinclair, K.; Pillai, S.; Merheb, V.; Aumann, T.D.; Gill, D.; Dale, R.C.; Brilot, F. Herpes simplex encephalitis relapse with chorea is associated with autoantibodies to n-methyl-d-aspartate receptor or dopamine-2 receptor. Mov. Disord. Off. J. Mov. Disord. Soc. 2014, 29, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Nosadini, M.; Mohammad, S.S.; Corazza, F.; Ruga, E.M.; Kothur, K.; Perilongo, G.; Frigo, A.C.; Toldo, I.; Dale, R.C.; Sartori, S. Herpes simplex virus-induced anti-n-methyl-d-aspartate receptor encephalitis: A systematic literature review with analysis of 43 cases. Dev. Med. Child Neurol. 2017, 59, 796–805. [Google Scholar] [CrossRef]
- Goldberg, E.M.; Titulaer, M.; de Blank, P.M.; Sievert, A.; Ryan, N. Anti-n-methyl-d-aspartate receptor-mediated encephalitis in infants and toddlers: Case report and review of the literature. Pediatr. Neurol. 2014, 50, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Cantalupo, G.; Fasano, A. Pediatric freezing of gait caused by Anti-NMDAR encephalitis. Mov. Disord. Off. J. Mov. Disord. Soc. 2019, 34, 756–757. [Google Scholar] [CrossRef]
- Baizabal-Carvallo, J.F.; Stocco, A.; Muscal, E.; Jankovic, J. The spectrum of movement disorders in children with anti-NMDA receptor encephalitis. Mov. Disord. 2013, 28, 543–547. [Google Scholar] [CrossRef]
- Mohammad, S.S.; Fung, V.S.C.; Grattan-Smith, P.; Gill, D.; Pillai, S.; Ramanathan, S.; Brilot, F.; Dale, R.C. Movement disorders in children with anti-NMDAR encephalitis and other autoimmune encephalopathies. Mov. Disord. 2014, 29, 1539–1542. [Google Scholar] [CrossRef]
- Granata, T.; Matricardi, S.; Ragona, F.; Freri, E.; Zibordi, F.; Andreetta, F.; Binelli, S.; Nardocci, N. Pediatric NMDAR encephalitis: A single center observation study with a closer look at movement disorders. Eur. J. Paediatr. Neurol. 2018, 22, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Varley, J.A.; Webb, A.J.S.; Balint, B.; Fung, V.S.C.; Sethi, K.D.; Tijssen, M.A.J.; Lynch, T.; Mohammad, S.S.; Britton, F.; Evans, M.; et al. The movement disorder associated with NMDAR antibody-encephalitis is complex and characteristic: An expert video-rating study. J. Neurol. Neurosurg. Psychiatry 2019, 90, 724–726. [Google Scholar] [CrossRef]
- Shimazaki, H.; Morita, M.; Nakano, I.; Dalmau, J. Inverse ocular bobbing in a patient with encephalitis associated with antibodies to the N-Methyl-D-aspartate receptor. Arch. Neurol. 2008, 65, 1251. [Google Scholar] [CrossRef]
- Mohammad, S.S.; Ramanathan, S.; Brilot, F.; Dale, R.C. Autoantibody-associated movement disorders. Neuropediatrics 2013, 44, 336–345. [Google Scholar] [CrossRef]
- Dale, R.C.; Merheb, V.; Pillai, S.; Wang, D.; Cantrill, L.; Murphy, T.K.; Ben-Pazi, H.; Varadkar, S.; Aumann, T.D.; Horne, M.K.; et al. Antibodies to surface dopamine-2 receptor in autoimmune movement and psychiatric disorders. Brain J. Neurol. 2012, 135, 3453–3468. [Google Scholar] [CrossRef] [PubMed]
- Clardy, S.L.; Lennon, V.A.; Dalmau, J.; Pittock, S.J.; Jones, H.R.; Renaud, D.L.; Harper, C.M.; Matsumoto, J.Y.; McKeon, A. Childhood onset of stiff-man syndrome. JAMA Neurol. 2013, 70, 1531–1536. [Google Scholar] [CrossRef]
- Damásio, J.; Leite, M.I.; Coutinho, E.; Waters, P.; Woodhall, M.; Santos, M.A.; Carrilho, I.; Vincent, A. Progressive encephalomyelitis with rigidity and myoclonus: The first pediatric case with glycine receptor antibodies. JAMA Neurol. 2013, 70, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Kruer, M.C.; Hoeftberger, R.; Lim, K.Y.; Coryell, J.C.; Svoboda, M.D.; Woltjer, R.L.; Dalmau, J. Aggressive course in encephalitis with opsoclonus, ataxia, chorea, and seizures: The first pediatric case of γ-aminobutyric acid type b receptor autoimmunity. JAMA Neurol. 2014, 71, 620–623. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.-Y.; Gelfand, J.M.; Geschwind, M.D. Anti-gamma-aminobutyric acid receptor type a encephalitis: A review. Curr. Opin. Neurol. 2020, 33, 372–380. [Google Scholar] [CrossRef]
- Tobin, W.O.; Lennon, V.A.; Komorowski, L.; Probst, C.; Clardy, S.L.; Aksamit, A.J.; Appendino, J.P.; Lucchinetti, C.F.; Matsumoto, J.Y.; Pittock, S.J.; et al. DPPX potassium channel antibody: Frequency, clinical accompaniments, and outcomes in 20 patients. Neurology 2014, 83, 1797–1803. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, S.S.; Dale, R.C. Principles and approaches to the treatment of immune-mediated movement disorders. Eur. J. Paediatr. Neurol. 2018, 22, 292–300. [Google Scholar] [CrossRef]
- Mehanna, R.; Jankovic, J. Movement disorders in multiple sclerosis and other demyelinating diseases. J. Neurol. Sci. 2013, 328, 1–8. [Google Scholar] [CrossRef]
- Tenembaum, S.; Chamoles, N.; Fejerman, N. Acute disseminated encephalomyelitis: A long-term follow-up study of 84 pediatric patients. Neurology 2002, 59, 1224–1231. [Google Scholar] [CrossRef]
- Kabakus, N.; Taskin, E.; Aydin, M. Segmental myoclonus as the presenting symptom of an acute disseminated encephalomyelitis: A case report. Eur. J. Paediatr. Neurol. 2006, 10, 45–48. [Google Scholar] [CrossRef]
- Hacohen, Y.; Nishimoto, Y.; Fukami, Y.; Lang, B.; Waters, P.; Lim, M.J.; Yuki, N.; Vincent, A. Paediatric brainstem encephalitis associated with glial and neuronal autoantibodies. Dev. Med. Child Neurol. 2016, 58, 836–841. [Google Scholar] [CrossRef]
- Sa, M.; Thornton, R.; Chong, W.K.; Kaliakatsos, M.; Hacohen, Y. Paediatric MOG antibody-associated ADEM with complex movement disorder: A case report. Mult. Scler. Houndmills Basingstoke Engl. 2019, 25, 125–128. [Google Scholar] [CrossRef] [PubMed]
- Tate, E.D.; Allison, T.J.; Pranzatelli, M.R.; Verhulst, S.J. Neuroepidemiologic trends in 105 US cases of pediatric opsoclonus-myoclonus syndrome. J. Pediatr. Oncol. Nurs. Off. J. Assoc. Pediatr. Oncol. Nurses 2005, 22, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Pang, K.K.; de Sousa, C.; Lang, B.; Pike, M.G. A prospective study of the presentation and management of dancing eye syndrome/opsoclonus-myoclonus syndrome in the United Kingdom. Eur. J. Paediatr. Neurol. EJPN Off. J. Eur. Paediatr. Neurol. Soc. 2010, 14, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, P.; Heim, J.; Cornejo, P.; Kane, L.; Santiago, J.; Kruer, M.C. Opsoclonus-myoclonus-ataxia syndrome in children. J. Neurol. 2021. [Google Scholar] [CrossRef]
- Rudnick, E.; Khakoo, Y.; Antunes, N.L.; Seeger, R.C.; Brodeur, G.M.; Shimada, H.; Gerbing, R.B.; Stram, D.O.; Matthay, K.K. Opsoclonus-myoclonus-ataxia syndrome in neuroblastoma: Clinical outcome and antineuronal antibodies-a report from the children’s cancer group study. Med. Pediatr. Oncol. 2001, 36, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Aljabban, I.; Fanburg-Smith, J.C.; Grant, C.; Moore, M. Pediatric whole body MRI detects causative ovarian teratoma in opsoclonus myoclonus syndrome. Radiol. Case Rep. 2020, 15, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Pranzatelli, M.R.; Tate, E.D.; McGee, N.R. Demographic, clinical, and immunologic features of 389 children with opsoclonus-myoclonus syndrome: A cross-sectional study. Front. Neurol. 2017, 8. [Google Scholar] [CrossRef]
- McCarthy, V.P.; Zimmerman, A.W.; Miller, C.A. Central nervous system manifestations of parainfluenza virus type 3 infections in childhood. Pediatr. Neurol. 1990, 6, 197–201. [Google Scholar] [CrossRef]
- Guedes, B.F.; Vieira Filho, M.A.A.; Listik, C.; Carra, R.B.; Pereira, C.B.; da Silva, E.R.; Gomes, H.R.; Vidal, J.E. HIV-associated opsoclonus-myoclonus-ataxia syndrome: Early infection, immune reconstitution syndrome or secondary to other diseases? Case report and literature review. J. Neurovirol. 2018, 24, 123–127. [Google Scholar] [CrossRef]
- Ichiba, N.; Miyake, Y.; Sato, K.; Oda, M.; Kimoto, H. Mumps-induced opsoclonus-myoclonus and ataxia. Pediatr. Neurol. 1988, 4, 224–227. [Google Scholar] [CrossRef]
- Huber, B.M.; Strozzi, S.; Steinlin, M.; Aebi, C.; Fluri, S. Mycoplasma pneumoniae associated opsoclonus-myoclonus syndrome in three cases. Eur. J. Pediatr. 2010, 169, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Syrbe, S.; Merkenschlager, A.; Bernhard, M.K.; Grosche, J.; Liebert, U.G.; Hirsch, W.; Härtig, W. Opsoclonus-myoclonus syndrome after adenovirus infection. SpringerPlus 2015, 4, 636. [Google Scholar] [CrossRef]
- Ertekin, V.; Tan, H. Opsoclonus-myoclonus syndrome attributable to hepatitis C infection. Pediatr. Neurol. 2010, 42, 441–442. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Sinha, M.; Kumar, R.; Shukla, R.; Ahuja, R.C. Opsoclonus-myoclonus syndrome caused by varicella-zoster virus. Ann. Indian Acad. Neurol. 2010, 13, 211–212. [Google Scholar] [CrossRef] [PubMed]
- Gurkas, E.; Gucuyener, K.; Yılmaz, U.; Havalı, C.; Demir, E. Opsoclonus-myoclonus syndrome following rotavirus gastroenteritis. Pediatr. Int. 2014, 56, e86–e87. [Google Scholar] [CrossRef]
- Vukelic, D.; Bozinovic, D.; Morovic, M.; Tesovic, G.; Ruzic Sabljic, E.; Barisic, N.; Knezovic, I. Opsoclonus-myoclonus syndrome in a child with neuroborreliosis. J. Infect. 2000, 40, 189–191. [Google Scholar] [CrossRef]
- Leen, W.G.; Weemaes, C.M.; Verbeek, M.M.; Willemsen, M.A.; Rotteveel, J.J. Rituximab and intravenous immunoglobulins for relapsing postinfectious opsoclonus-myoclonus syndrome. Pediatr. Neurol. 2008, 39, 213–217. [Google Scholar] [CrossRef]
- Kuban, K.C.; Ephros, M.A.; Freeman, R.L.; Laffell, L.B.; Bresnan, M.J. Syndrome of opsoclonus-myoclonus caused by coxsackie B3 infection. Ann. Neurol. 1983, 13, 69–71. [Google Scholar] [CrossRef]
- Pranzatelli, M.R.; Tate, E.D.; McGee, N.R. Multifactorial analysis of opsoclonus-myoclonus syndrome etiology (“Tumor” vs. “No Tumor”) in a cohort of 356 US children. Pediatr. Blood Cancer 2018, 65, e27097. [Google Scholar] [CrossRef]
- Armangué, T.; Sabater, L.; Torres-Vega, E.; Martínez-Hernández, E.; Ariño, H.; Petit-Pedrol, M.; Planagumà, J.; Bataller, L.; Dalmau, J.; Graus, F. Clinical and Immunological features of opsoclonus-myoclonus syndrome in the era of neuronal cell surface antibodies. JAMA Neurol. 2016, 73, 417–424. [Google Scholar] [CrossRef]
- Antunes, N.L.; Khakoo, Y.; Matthay, K.K.; Seeger, R.C.; Stram, D.O.; Gerstner, E.; Abrey, L.E.; Dalmau, J. Antineuronal antibodies in patients with neuroblastoma and paraneoplastic opsoclonus-myoclonus. J. Pediatr. Hematol. Oncol. 2000, 22, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Connolly, A.M.; Pestronk, A.; Mehta, S.; Pranzatelli, M.R.; Noetzel, M.J. Serum autoantibodies in childhood opsoclonus-myoclonus syndrome: An analysis of antigenic targets in neural tissues. J. Pediatr. 1997, 130, 878–884. [Google Scholar] [CrossRef]
- Kirsten, A.; Beck, S.; Fühlhuber, V.; Kaps, M.; Kreutz, T.; Korfei, M.; Schmitt, S.; Preissner, K.T.; Blaes, F. New autoantibodies in pediatric opsoclonus myoclonus syndrome. Ann. N. Y. Acad. Sci. 2007, 1110, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Blaes, F.; Fühlhuber, V.; Preissner, K.T. Identification of autoantigens in pediatric opsoclonus-myoclonus syndrome. Expert Rev. Clin. Immunol. 2007, 3, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Blaes, F.; Fühlhuber, V.; Korfei, M.; Tschernatsch, M.; Behnisch, W.; Rostasy, K.; Hero, B.; Kaps, M.; Preissner, K.T. Surface-binding autoantibodies to cerebellar neurons in opsoclonus syndrome. Ann. Neurol. 2005, 58, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Korfei, M.; Fühlhuber, V.; Schmidt-Wöll, T.; Kaps, M.; Preissner, K.T.; Blaes, F. Functional characterisation of autoantibodies from patients with pediatric opsoclonus-myoclonus-syndrome. J. Neuroimmunol. 2005, 170, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Berridge, G.; Menassa, D.A.; Moloney, T.; Waters, P.J.; Welding, I.; Thomsen, S.; Zuberi, S.; Fischer, R.; Aricescu, A.R.; Pike, M.; et al. Glutamate receptor Δ2 serum antibodies in pediatric opsoclonus myoclonus ataxia syndrome. Neurology 2018, 91, e714–e723. [Google Scholar] [CrossRef]
- Panzer, J.A.; Anand, R.; Dalmau, J.; Lynch, D.R. Antibodies to dendritic neuronal surface antigens in opsoclonus myoclonus ataxia syndrome. J. Neuroimmunol. 2015, 286, 86–92. [Google Scholar] [CrossRef]
- Petit-Pedrol, M.; Guasp, M.; Armangue, T.; Lavarino, C.; Morales La Madrid, A.; Saiz, A.; Graus, F.; Dalmau, J. Absence of GluD2 antibodies in patients with opsoclonus-myoclonus syndrome. Neurology 2021, 96, e1082–e1087. [Google Scholar] [CrossRef] [PubMed]
- Kurian, M.; Lalive, P.H.; Dalmau, J.O.; Horvath, J. Opsoclonus-myoclonus syndrome in anti-N-Methyl-D-aspartate receptor encephalitis. Arch. Neurol. 2010, 67, 118–121. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Petit-Pedrol, M.; Armangue, T.; Peng, X.; Bataller, L.; Cellucci, T.; Davis, R.; McCracken, L.; Martinez-Hernandez, E.; Mason, W.P.; Kruer, M.C.; et al. Encephalitis with refractory seizures, status epilepticus, and antibodies to the GABAA receptor: A case series, characterisation of the antigen, and analysis of the effects of antibodies. Lancet Neurol. 2014, 13, 276–286. [Google Scholar] [CrossRef]
- Markakis, I.; Alexiou, E.; Xifaras, M.; Gekas, G.; Rombos, A. Opsoclonus-myoclonus-ataxia syndrome with autoantibodies to glutamic acid decarboxylase. Clin. Neurol. Neurosurg. 2008, 110, 619–621. [Google Scholar] [CrossRef]
- Bohrer, S.; De La Villeon, G.; Carneiro, M.; Fernandez, C.; Garbi, D.; Mace, L.; Milh, M.; Guillaumont, S.; Echenne, B.; Honnorat, J.; et al. Acute-onset chorea, dystonia, and cardiac fibroelastoma in a child: A paraneoplastic association? Mov. Disord. 2013, 28, 250–252. [Google Scholar] [CrossRef] [PubMed]
- Hoashi, T.; Florentine, M.S.; Gordon, D.; Ohye, R.G. Cardiac papillary fibroelastoma presenting as chorea in childhood. Pediatr. Cardiol. 2009, 30, 995–997. [Google Scholar] [CrossRef]
- Poewe, W.; Djamshidian-Tehrani, A. Movement disorders in systemic diseases. Neurol. Clin. 2015, 33, 269–297. [Google Scholar] [CrossRef] [PubMed]
- Misra, U.K.; Kalita, J. Spectrum of movement disorders in encephalitis. J. Neurol. 2010, 257, 2052–2058. [Google Scholar] [CrossRef] [PubMed]
- Dimova, P.S.; Bojinova, V.; Georgiev, D.; Milanov, I. Acute reversible parkinsonism in epstein-barr virus-related encephalitis lethargica-like illness. Mov. Disord. 2006, 21, 564–566. [Google Scholar] [CrossRef]
- Hsieh, J.-C.; Lue, K.-H.; Lee, Y.-L. Parkinson-like syndrome as the major presenting symptom of Epstein-Barr virus encephalitis. Arch. Dis. Child. 2002, 87, 358. [Google Scholar] [CrossRef] [PubMed]
- Ryan, M.M.; Procopis, P.G.; Ouvrier, R.A. Influenza a encephalitis with movement disorder. Pediatr. Neurol. 1999, 21, 669–673. [Google Scholar] [CrossRef]
- Pulickal, A.S.; Ramachandran, S.; Rizek, P.; Narula, P.; Schubert, R. Chorea and developmental regression associated with human herpes virus-6 encephalitis. Pediatr. Neurol. 2013, 48, 249–251. [Google Scholar] [CrossRef] [PubMed]
- Leavell, R.; Ray, C.G.; Ferry, P.C.; Minnich, L.L. Unusual acute neurologic presentations with Epstein-Barr virus infection. Arch. Neurol. 1986, 43, 186–188. [Google Scholar] [CrossRef]
- Dale, R.C.; Church, A.J.; Heyman, I. Striatal encephalitis after varicella zoster infection complicated by tourettism. Mov. Disord. 2003, 18, 1554–1556. [Google Scholar] [CrossRef]
- Kalita, J.; Misra, U.K.; Pandey, S.; Dhole, T.N. A comparison of clinical and radiological findings in adults and children with Japanese Encephalitis. Arch. Neurol. 2003, 60, 1760–1764. [Google Scholar] [CrossRef] [PubMed]
- Kalita, J.; Misra, U.K. Markedly severe dystonia in Japanese Encephalitis. Mov. Disord. 2000, 15, 1168–1172. [Google Scholar] [CrossRef]
- Kalita, J.; Misra, U.K.; Pradhan, P.K. Oromandibular dystonia in encephalitis. J. Neurol. Sci. 2011, 304, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Titulaer, M.J.; Leypoldt, F.; Dalmau, J. Antibodies to N-Methyl-D-aspartate and other synaptic receptors in choreoathetosis and relapsing symptoms post-herpes virus encephalitis. Mov. Disord. 2014, 29, 3–6. [Google Scholar] [CrossRef]
- Mintz, M.; Tardieu, M.; Hoyt, L.; McSherry, G.; Mendelson, J.; Oleske, J. Levodopa therapy improves motor function in HIV-infected children with extrapyramidal syndromes. Neurology 1996, 47, 1583–1585. [Google Scholar] [CrossRef]
- Solomons, R.; Slogrove, A.; Schoeman, J.; Marais, B.; van Zyl, G.; Maritz, J.; van Toorn, R. Acute extrapyramidal dysfunction in two HIV-infected children. J. Trop. Pediatr. 2011, 57, 227–231. [Google Scholar] [CrossRef][Green Version]
- VanDongen-Trimmer, H.; Sannagowdara, K.; Balakrishnan, B.; Farias-Moeller, R. A case of HIV seroconversion presenting similarly to Anti-N-Methyl-D-aspartate receptor encephalitis. Neurocrit. Care 2019, 31, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Alarcón, F.; Dueñas, G.; Cevallos, N.; Lees, A.J. Movement disorders in 30 patients with tuberculous meningitis. Mov. Disord. 2000, 15, 561–569. [Google Scholar] [CrossRef]
- Babikian, V.L.; Heydemann, P.T.; Swisher, C.N. Extrapyramidal movements in a patient with tuberculous meningitis. An early clue to diagnosis. Clin. Pediatr. 1985, 24, 113–115. [Google Scholar] [CrossRef] [PubMed]
- Garone, G.; Graziola, F.; Nicita, F.; Frascarelli, F.; Randi, F.; Zazza, M.; Cantonetti, L.; Cossu, S.; Marras, C.E.; Capuano, A. Prestatus and status dystonicus in children and adolescents. Dev. Med. Child Neurol. 2020, 62, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Burstein, L.; Breningstall, G.N. Movement disorders in bacterial meningitis. J. Pediatr. 1986, 109, 260–264. [Google Scholar] [CrossRef]
- Riedel, M.; Straube, A.; Schwarz, M.J.; Wilske, B.; Müller, N. Lyme disease presenting as Tourette’s syndrome. Lancet 1998, 351, 418–419. [Google Scholar] [CrossRef]
- Micheli, R.; Perini, A.; Duse, M. Hemidystonia secondary to acquired toxoplasmosis in a non-immunodeficient patient. Eur. J. Pediatr. 1994, 153, 731–733. [Google Scholar] [CrossRef]
- Patel, R.; Jha, S.; Yadav, R.K. Pleomorphism of the clinical manifestations of neurocysticercosis. Trans. R. Soc. Trop. Med. Hyg. 2006, 100, 134–141. [Google Scholar] [CrossRef]
- Verma, R.; Agarwal, A.; Kar, A.M. Hemichorea resulting from single enhancing computed tomography lesion. J. Assoc. Physicians India 2006, 54, 735–737. [Google Scholar] [PubMed]
- Karnik, P.S.; Tullu, M.S.; David, J.J.E.; Ghildiyal, R.G. Neurocysticercosis-induced hemiballismus in a child. J. Child Neurol. 2011, 26, 904–906. [Google Scholar] [CrossRef]
- Tonduti, D.; Chiapparini, L.; Moroni, I.; Ardissone, A.; Zorzi, G.; Zibordi, F.; Raspante, S.; Panteghini, C.; Garavaglia, B.; Nardocci, N. Neurological disorders associated with striatal lesions: Classification and diagnostic approach. Curr. Neurol. Neurosci. Rep. 2016, 16, 54. [Google Scholar] [CrossRef] [PubMed]
- Goutières, F.; Aicardi, J. Acute neurological dysfunction associated with destructive lesions of the basal ganglia in children. Ann. Neurol. 1982, 12, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Larsen, P.D.; Crisp, D. Acute bilateral striatal necrosis associated with mycoplasma pneumoniae infection. Pediatr. Infect. Dis. J. 1996, 15, 1124–1126. [Google Scholar] [CrossRef]
- Van Buiren, M.; Uhl, M. Images in clinical medicine. Bilateral striatal necrosis associated with mycoplasma pneumoniae infection. N. Engl. J. Med. 2003, 348, 720. [Google Scholar] [CrossRef] [PubMed]
- Murakami, A.; Morimoto, M.; Adachi, S.; Ishimaru, Y.; Sugimoto, T. Infantile bilateral striatal necrosis associated with human Herpes Virus-6 (HHV-6) infection. Brain Dev. 2005, 27, 527–530. [Google Scholar] [CrossRef]
- Cambonie, G.; Houdon, L.; Rivier, F.; Bongrand, A.F.; Echenne, B. Infantile bilateral striatal necrosis following measles. Brain Dev. 2000, 22, 221–223. [Google Scholar] [CrossRef]
- Brandel, J.P.; Vidailhet, M.; Noseda, G.; Harpey, J.P.; Agid, Y. Mycoplasma pneumoniae postinfectious encephalomyelitis with bilateral striatal necrosis. Mov. Disord. 1996, 11, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Dale, R.C.; Church, A.J.; Benton, S.; Surtees, R.A.; Lees, A.; Thompson, E.J.; Giovannoni, G.; Neville, B.G. Post-streptococcal autoimmune dystonia with isolated bilateral striatal necrosis. Dev. Med. Child Neurol. 2002, 44, 485–489. [Google Scholar] [CrossRef]
- Karagulle Kendi, A.T.; Krenzel, C.; Ott, F.W.; Brace, J.R.; Norberg, S.K.; Kieffer, S.A. Poststreptococcal dystonia with bilateral striatal enlargement: MR imaging and spectroscopic findings. AJNR Am. J. Neuroradiol. 2008, 29, 1276–1278. [Google Scholar] [CrossRef]
- Voudris, K.A.; Skardoutsou, A.; Hasiotou, M.; Theodoropoulos, B.; Vagiakou, E.A. Long-term findings on brain magnetic resonance imaging in acute encephalopathy with bilateral striatal necrosis associated with measles. J. Child Neurol. 2002, 17, 776–777. [Google Scholar] [CrossRef]
- Wu, X.; Wu, W.; Pan, W.; Wu, L.; Liu, K.; Zhang, H.-L. Acute necrotizing encephalopathy: An underrecognized clinicoradiologic disorder. Mediat. Inflamm. 2015, 2015, 792578. [Google Scholar] [CrossRef]
- Mizuguchi, M.; Abe, J.; Mikkaichi, K.; Noma, S.; Yoshida, K.; Yamanaka, T.; Kamoshita, S. Acute necrotising encephalopathy of childhood: A new syndrome presenting with multifocal, symmetric brain lesions. J. Neurol. Neurosurg. Psychiatry 1995, 58, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Weng, W.-C.; Peng, S.S.-F.; Lee, W.-T. Acute necrotizing encephalopathy of childhood with spinal cord involvement: A case report. J. Child Neurol. 2010, 25, 1539–1541. [Google Scholar] [CrossRef] [PubMed]
- Mizuguchi, M.; Ichiyama, T.; Imataka, G.; Okumura, A.; Goto, T.; Sakuma, H.; Takanashi, J.-I.; Murayama, K.; Yamagata, T.; Yamanouchi, H.; et al. Guidelines for the diagnosis and treatment of acute encephalopathy in childhood. Brain Dev. 2021, 43, 2–31. [Google Scholar] [CrossRef]
- Kothare, S.V.; Pollack, P.; Kulberg, A.G.; Ravin, P.D. Gabapentin treatment in a child with delayed-onset hemichorea/hemiballismus. Pediatr. Neurol. 2000, 22, 68–71. [Google Scholar] [CrossRef]
- Baik, J.S.; Lee, M.S. Movement disorders associated with moyamoya disease: A report of 4 new cases and a review of literatures. Mov. Disord. 2010, 25, 1482–1486. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.; Bell-Stephens, T.; Steinberg, G.K. Patients with moyamoya disease presenting with movement disorder. J. Neurosurg. Pediatr. 2010, 6, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, S.; Houkin, K. Moyamoya disease: Current concepts and future perspectives. Lancet Neurol. 2008, 7, 1056–1066. [Google Scholar] [CrossRef]
- Medlock, M.D.; Cruse, R.S.; Winek, S.J.; Geiss, D.M.; Horndasch, R.L.; Schultz, D.L.; Aldag, J.C. A 10-year experience with postpump chorea. Ann. Neurol. 1993, 34, 820–826. [Google Scholar] [CrossRef] [PubMed]
- Przekop, A.; McClure, C.; Ashwal, S. Postoperative encephalopathy with choreoathetosis. Handb. Clin. Neurol. 2011, 100, 295–305. [Google Scholar] [CrossRef]
- Rodnitzky, R.L. Drug-induced movement disorders in children. Semin. Pediatr. Neurol. 2003, 10, 80–87. [Google Scholar] [CrossRef]
- Derinoz, O.; Caglar, A.A. Drug-induced movement disorders in children at paediatric emergency department: “Dystonia”. Emerg. Med. J. EMJ 2013, 30, 130–133. [Google Scholar] [CrossRef]
- Russell, S.A.; Hennes, H.M.; Herson, K.J.; Stremski, E.S. Upper airway compromise in acute chlorpromazine ingestion. Am. J. Emerg. Med. 1996, 14, 467–468. [Google Scholar] [CrossRef]
- Moosavi, S.M.; Ahmadi, M.; Monajemi, M.B. Acute dystonia due to citalopram treatment: A case series. Glob. J. Health Sci. 2014, 6, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Muthusami, S.; Basu, S.; Kumar, A.; Dash, A. Acute dyskinesia and extrapyramidal disorder in a child after ingestion of escitalopram. J. Child Adolesc. Psychopharmacol. 2009, 19, 317–318. [Google Scholar] [CrossRef]
- Najjar, F.; Price, L.H. Citalopram and dystonia. J. Am. Acad. Child Adolesc. Psychiatry 2004, 43, 8–9. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.; Salek, S. Stimulant-atypical antipsychotic interaction and acute dystonia. J. Am. Acad. Child Adolesc. Psychiatry 2005, 44, 510–512. [Google Scholar] [CrossRef]
- Levine, J.B.; Deneys, M.L.; Benjamin, S. Dystonia with combined antipsychotic and stimulant treatment. J. Am. Acad. Child Adolesc. Psychiatry 2007, 46, 665–666. [Google Scholar] [CrossRef] [PubMed]
- Hollis, C.P.; Thompson, A. Acute dyskinesia on starting methylphenidate after risperidone withdrawal. Pediatr. Neurol. 2007, 37, 287–288. [Google Scholar] [CrossRef]
- Tekin, U.; Soyata, A.Z.; Oflaz, S. Acute focal dystonic reaction after acute methylphenidate treatment in an adolescent patient. J. Clin. Psychopharmacol. 2015, 35, 209–211. [Google Scholar] [CrossRef]
- Waugh, J.L. Acute dyskinetic reaction in a healthy toddler following methylphenidate ingestion. Pediatr. Neurol. 2013, 49, 58–60. [Google Scholar] [CrossRef]
- Yilmaz, A.E.; Donmez, A.; Orun, E.; Tas, T.; Isik, B.; Sonmez, F.M. Methylphenidate-induced acute orofacial and extremity dyskinesia. J. Child Neurol. 2013, 28, 781–783. [Google Scholar] [CrossRef]
- Keresztény, Á.; Ferenczi-Dallos, G.; Velő, S.; Gádoros, J.; Balázs, J. Dyskinesia in treatment-naive and stimulant-treated children with ADHD. J. Atten. Disord. 2020, 24, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Balázs, J.; Dallos, G.; Keresztény, A.; Czobor, P.; Gádoros, J. Methylphenidate treatment and dyskinesia in children with attention-deficit/hyperactivity disorder. J. Child Adolesc. Psychopharmacol. 2011, 21, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Basil, B.; Mathews, M.; Budur, K. Methylphenidate-induced neuroleptic malignant syndrome: A case report. Prim. Care Companion J. Clin. Psychiatry 2006, 8, 47. [Google Scholar] [CrossRef] [PubMed]
- Squires, L.A.; Neumeyer, A.M.; Bloomberg, J.; Krishnamoorthy, K.S. Hyperpyrexia in an adolescent on desipramine treatment. Clin. Pediatr. 1992, 31, 635–636. [Google Scholar] [CrossRef]
- Ehara, H.; Maegaki, Y.; Takeshita, K. Neuroleptic malignant syndrome and methylphenidate. Pediatr. Neurol. 1998, 19, 299–301. [Google Scholar] [CrossRef]
- Croarkin, P.E.; Emslie, G.J.; Mayes, T.L. Neuroleptic malignant syndrome associated with atypical antipsychotics in pediatric patients: A review of published cases. J. Clin. Psychiatry 2008, 69, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.R.; Munoz, D.M.; Alpert, M.; Perlmutter, I.R.; Diaz, J. Neuroleptic malignant syndrome in children and adolescents. J. Am. Acad. Child Adolesc. Psychiatry 1999, 38, 187–194. [Google Scholar] [CrossRef]
- Yaman, A.; Kendirli, T.; Ödek, Ç.; Yıldız, C.; Beğde, F.; Erkol, H.; İnce, E. Neuroleptic malignant syndrome associated with metoclopramide in a child. Turk. J. Pediatr. 2014, 56, 535–537. [Google Scholar]
- Connor, D.F.; Fletcher, K.E.; Wood, J.S. Neuroleptic-related dyskinesias in children and adolescents. J. Clin. Psychiatry 2001, 62, 967–974. [Google Scholar] [CrossRef]
- Friedman, J.H. Movement disorders induced by psychiatric drugs that do not block dopamine receptors. Parkinsonism Relat. Disord. 2020, 79, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Factor, S.A.; Burkhard, P.R.; Caroff, S.; Friedman, J.H.; Marras, C.; Tinazzi, M.; Comella, C.L. Recent developments in drug-induced movement disorders: A mixed picture. Lancet Neurol. 2019, 18, 880–890. [Google Scholar] [CrossRef]
- Ford, J.B.; Albertson, T.E.; Owen, K.P.; Sutter, M.E.; McKinney, W.B. Acute, sustained chorea in children after supratherapeutic dosing of amphetamine-derived medications. Pediatr. Neurol. 2012, 47, 216–218. [Google Scholar] [CrossRef]
- Sukhudyan, B.G.; Dimova, P.S.; Capuano, A.; Vigevano, F. Dyskinesia as a new adverse effect of hormonal treatment in west syndrome. Epileptic Disord. Int. Epilepsy J. Videotape 2014, 16, 5–12. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Arita, J.H.; Vale, T.C.; Pedroso, J.L.; Faria, E.C.; Arita, F.N.; Masruha, M.R.; Barsottini, O.G.P. ACTH-induced dyskinesia in a child with west syndrome (Infantile Spasms). Parkinsonism Relat. Disord. 2016, 24, 145–146. [Google Scholar] [CrossRef] [PubMed]
- Dill, P.; Datta, A.N.; Weber, P.; Schneider, J. Are vigabatrin induced T2 hyperintensities in cranial MRI associated with acute encephalopathy and extrapyramidal symptoms? Eur. J. Paediatr. Neurol. 2013, 17, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Schonstedt, V.; Stecher, X.; Venegas, V.; Silva, C. Vigabatrin-induced MRI changes associated with extrapyramidal symptoms in a child with infantile spasms. Neuroradiol. J. 2015, 28, 515–518. [Google Scholar] [CrossRef]
- Zaatreh, M.; Tennison, M.; D’Cruz, O.; Beach, R.L. Anticonvulsants-induced chorea: A role for pharmacodynamic drug interaction? Seizure 2001, 10, 596–599. [Google Scholar] [CrossRef]
- Montenegro, M.A.; Scotoni, A.E.; Cendes, F. Dyskinesia induced by phenytoin. Arq. Neuropsiquiatr. 1999, 57, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, N.; Ravat, S.H.; Shah, P.U. Phenytoin induced dyskinesia. Indian Pediatr. 1998, 35, 274–276. [Google Scholar] [PubMed]
- Pranzatelli, M.R.; Albin, R.L.; Cohen, B.H. Acute dyskinesias in young asthmatics treated with theophylline. Pediatr. Neurol. 1991, 7, 216–219. [Google Scholar] [CrossRef]
- Ferreyros, J.; Alfonso, I. Dyskinesia secondary to aminophylline. Pediatr. Neurol. 1992, 8, 157. [Google Scholar] [CrossRef]
- Epstein, D.; Difazio, M. Orofacial automatisms induced by acute withdrawal from high-dose midazolam mimicking nonconvulsive status epilepticus in a child. Mov. Disord. 2007, 22, 712–715. [Google Scholar] [CrossRef]
- Bergman, I.; Steeves, M.; Burckart, G.; Thompson, A. Reversible neurologic abnormalities associated with prolonged intravenous midazolam and fentanyl administration. J. Pediatr. 1991, 119, 644–649. [Google Scholar] [CrossRef]
- Nomoto, M.; Thompson, P.D.; Sheehy, M.P.; Quinn, N.P.; Marsden, C.D. Anticholinergic-induced chorea in the treatment of focal dystonia. Mov. Disord. 1987, 2, 53–56. [Google Scholar] [CrossRef]
- Antelmi, E.; Erro, R.; Pisani, A.; Mencacci, N.; Bhatia, K.P. Persistent chorea in DYT6, due to anticholinergic therapy. Parkinsonism Relat. Disord. 2015, 21, 1282–1283. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.J.; Vaughan, C.L.; Atayee, R.; Hirst, J.M.; O’Donnell, K.; Edmonds, K.P. Hydromorphone-induced chorea as an atypical presentation of opioid neurotoxicity: A case report and review of the literature. Palliat. Med. 2018, 32, 1529–1532. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, R. Opioid-induced neurotoxicity. Can. Fam. Phys. Med. Fam. Can. 2007, 53, 426–427. [Google Scholar]
- Wasserman, S.; Yahr, M.D. Choreic movements induced by the use of methadone. Arch. Neurol. 1980, 37, 727–728. [Google Scholar] [CrossRef]
- Grebosky, J. Case report: A movement disorder related to use of oxycodone. Am. Fam. Phys. 2005, 71, 1. [Google Scholar]
- Van der Plas, A.A.; van Rijn, M.A.; van Hilten, J.J. Baclofen-Induced chorea in complex regional pain syndrome-related dystonia. Mov. Disord. 2010, 25, 959–960. [Google Scholar] [CrossRef] [PubMed]
- Lahlou, A.; Benlamkaddem, S.; Berdai, M.A.; Harandou, M. Accidental modopar© poisoning in a two-year-old child: A case report. J. Crit. Care Med. 2019, 5, 157–160. [Google Scholar] [CrossRef]
- Eggink, H.; Kuiper, A.; Delnooz, C.C.S.; Sival, D.A.; de Koning, T.J.; Tijssen, M.A.J. Patience is the key: Contraceptive induced chorea in a girl with down syndrome. Eur. J. Paediatr. Neurol. 2016, 20, 671–673. [Google Scholar] [CrossRef] [PubMed]
- Sharmila, V.; Babu, T.A. Oral contraceptive pills induced hemichorea in an adolescent female with polycystic ovarian disease. Indian J. Pharmacol. 2015, 47, 232–233. [Google Scholar] [CrossRef] [PubMed]
- McClain, B.C.; Probst, L.A.; Pinter, E.; Hartmannsgruber, M. Intravenous clonidine use in a neonate experiencing opioid-induced myoclonus. Anesthesiology 2001, 95, 549–550. [Google Scholar] [CrossRef]
- Gilbert, D.L. Drug-induced movement disorders in children. Ann. N. Y. Acad. Sci. 2008, 1142, 72–84. [Google Scholar] [CrossRef]
- Rissardo, J.P.; Caprara, A.L.F. Carbamazepine-, oxcarbazepine-, eslicarbazepine-associated movement disorder: A literature review. Clin. Neuropharmacol. 2020, 43, 66–80. [Google Scholar] [CrossRef]
- Isitemiz, I.; Uzman, S.; Toptaş, M.; Vahapoglu, A.; Gül, Y.G.; Inal, F.Y.; Akkoc, I. Prevention of etomidate-induced myoclonus: Which is superior: Fentanyl, midazolam, or a combination? A retrospective comparative study. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2014, 20, 262–267. [Google Scholar] [CrossRef]
- Robertson, P.L.; Garofalo, E.A.; Silverstein, F.S.; Komarynski, M.A. Carbamazepine-induced tics. Epilepsia 1993, 34, 965–968. [Google Scholar] [CrossRef]
- Pranzatelli, M.R.; Mott, S.H.; Pavlakis, S.G.; Conry, J.A.; Tate, E.D. Clinical spectrum of secondary parkinsonism in childhood: A reversible disorder. Pediatr. Neurol. 1994, 10, 131–140. [Google Scholar] [CrossRef]
- Mott, S.H.; Packer, R.J.; Vezina, L.G.; Kapur, S.; Dinndorf, P.A.; Conry, J.A.; Pranzatelli, M.R.; Quinones, R.R. Encephalopathy with parkinsonian features in children following bone marrow transplantations and high-dose amphotericin B. Ann. Neurol. 1995, 37, 810–814. [Google Scholar] [CrossRef]
- Chutorian, A.M.; Bojko, A.; Heier, L.; Frucht, S.; Nygaard, T.; Edelberg, D. Toxic pediatric Parkinsonism: Report of a child with metabolic studies and response to treatment. J. Child Neurol. 2003, 18, 812–815. [Google Scholar] [CrossRef]
- Manley, T.J.; Chusid, M.J.; Rand, S.D.; Wells, D.; Margolis, D.A. Reversible Parkinsonism in a child after bone marrow transplantation and lipid-based amphotericin B therapy. Pediatr. Infect. Dis. J. 1998, 17, 433–434. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.S.; Cheon, H.Y. Delayed movement disorders after carbon monoxide poisoning. Eur. Neurol. 1999, 42, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Cury, R.G.; Marin, J.H.; Contreras Lopez, W.O. Lead poisoning: Myoclonus following welding exposure. Acta Med. Port. 2017, 30, 889. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, X.P.; Yang, R.M. Movement disorders possibly induced by traditional chinese herbs. Eur. Neurol. 2003, 50, 153–159. [Google Scholar] [CrossRef]
- Ortigoza-Escobar, J.D. A proposed diagnostic algorithm for inborn errors of metabolism presenting with movements disorders. Front. Neurol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Galosi, S.; Nardecchia, F.; Leuzzi, V. Treatable inherited movement disorders in children: Spotlight on clinical and biochemical features. Mov. Disord. Clin. Pract. 2020, 7, 154–166. [Google Scholar] [CrossRef]
- Ebrahimi-Fakhari, D.; Van Karnebeek, C.; Münchau, A. Movement disorders in treatable inborn errors of metabolism. Mov. Disord. 2019, 34, 598–613. [Google Scholar] [CrossRef] [PubMed]
- Fecek, C.; Samanta, D. Subacute Necrotizing Encephalomyelopathy. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Baertling, F.; Rodenburg, R.J.; Schaper, J.; Smeitink, J.A.; Koopman, W.J.H.; Mayatepek, E.; Morava, E.; Distelmaier, F. A guide to diagnosis and treatment of leigh syndrome. J. Neurol. Neurosurg. Psychiatry 2014, 85, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Schubert Baldo, M.; Vilarinho, L. Molecular basis of leigh syndrome: A current look. Orphanet J. Rare Dis. 2020, 15. [Google Scholar] [CrossRef]
- Carecchio, M.; Zorzi, G.; Ragona, F.; Zibordi, F.; Nardocci, N. ATP1A3-related disorders: An update. Eur. J. Paediatr. Neurol. EJPN Off. J. Eur. Paediatr. Neurol. Soc. 2018, 22, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Sabouraud, P.; Riquet, A.; Spitz, M.-A.; Deiva, K.; Nevsimalova, S.; Mignot, C.; Lesca, G.; Bednarek, N.; Doummar, D.; Pietrement, C.; et al. Relapsing encephalopathy with cerebellar ataxia are caused by variants involving p.Arg756 in ATP1A3. Eur. J. Paediatr. Neurol. 2019, 23, 448–455. [Google Scholar] [CrossRef]
- Lorincz, M.T. Neurologic Wilson’s Disease. Ann. N. Y. Acad. Sci. 2010, 1184, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Li, L.-Y.; Zhu, X.-Q.; Tao, W.-W.; Yang, W.-M.; Chen, H.-Z.; Wang, Y. Acute onset neurological symptoms in wilson disease after traumatic, surgical or emotional events: A cross-sectional study. Medicine 2019, 98, e15917. [Google Scholar] [CrossRef]
- Di Lazzaro, G.; Graziola, F.; Sancesario, A.; Insalaco, A.; Moneta, G.M.; Castelli, E.; Bertini, E.; Travaglini, L.; Stregapede, F.; Capuano, A.; et al. Movement disorders in ADAR1 disease: Insights from a comprehensive cohort. Parkinsonism Relat. Disord. 2020, 79, 100–104. [Google Scholar] [CrossRef]
- Saini, A.G.; Sankhyan, N.; Singhi, P. Chorea in late-infantile neuronal ceroid lipofuscinosis: An atypical presentation. Pediatr. Neurol. 2016, 60, 75–78. [Google Scholar] [CrossRef]
- Alves, C.; Sampaio, S.; Barbosa, V.; Machado, M. Acute chorea and type 1 diabetes mellitus: Clinical and neuroimaging findings. Pediatr. Diabetes 2012, 13, e30–e34. [Google Scholar] [CrossRef]
- Aquino, J.H.W.; Spitz, M.; Pereira, J.S. Hemichorea-hemiballismus as the first sign of type 1b diabetes during adolescence and its recurrence in the setting of infection. J. Child Neurol. 2015, 30, 1362–1365. [Google Scholar] [CrossRef]
- Parke, J. Acute encephalopathies. In Oski’s Pediatrics. Principles and Practice; McMillan, J.A., Feigin, R.D., DeAngelis, C., Jones, M.D., Eds.; Lippincott, Williams & Wilkins: Philadelphia, PA, USA, 2006; p. 2258. [Google Scholar]
- Baluarte, J.H. Neurological complications of renal disease. Semin. Pediatr. Neurol. 2017, 24, 25–32. [Google Scholar] [CrossRef]
- Seeherunvong, T.; Diamantopoulos, S.; Berkovitz, G.D. A nine year old girl with thyrotoxicosis, ataxia, and chorea. Brain Dev. 2007, 29, 660–661. [Google Scholar] [CrossRef]
- Blackman, J.A.; Patrick, P.D.; Buck, M.L.; Rust, R.S. Paroxysmal autonomic instability with dystonia after brain injury. Arch. Neurol. 2004, 61, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Baguley, I.J.; Perkes, I.E.; Fernandez-Ortega, J.-F.; Rabinstein, A.A.; Dolce, G.; Hendricks, H.T.; Consensus Working Group. Paroxysmal sympathetic hyperactivity after acquired brain injury: Consensus on conceptual definition, nomenclature, and diagnostic criteria. J. Neurotrauma 2014, 31, 1515–1520. [Google Scholar] [CrossRef]
- Buerger, K.J.; Salazar, R. Alternating hemidystonia following traumatic brain injury as an unusual presentation of paroxysmal autonomic instability with dystonia syndrome. BMJ Case Rep. 2014, 2014. [Google Scholar] [CrossRef]
- Safadieh, L.; Sharara-Chami, R.; Dabbagh, O. Paroxysmal autonomic instability with dystonia after pneumococcal meningoencephalitis. Case Rep. Med. 2012, 2012, 965932. [Google Scholar] [CrossRef]
- Singh, D.K.; Singh, N. Paroxysmal autonomic instability with dystonia in a child: Rare manifestation of an interpeduncular tuberculoma. Pediatr. Neurosurg. 2011, 47, 275–278. [Google Scholar] [CrossRef] [PubMed]
- Ramdhani, N.A.; Sikma, M.A.; Witkamp, T.D.; Slooter, A.J.; de Lange, D.W. Paroxysmal autonomic instability with dystonia in a patient with tuberculous meningitis: A case report. J. Med. Case Rep. 2010, 4, 304. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, H.; Nishimura, A.; Okano, S.; Miyanomae, Y.; Takeuchi, Y.; Sawada, T. Neuroleptic malignant syndrome-like state in a patient with down syndrome and basal ganglia calcification. Brain Dev. 1992, 14, 400–403. [Google Scholar] [CrossRef]
- Acampa, M.; Guideri, F.; Hayek, J.; Blardi, P.; De Lalla, A.; Zappella, M.; Auteri, A. Sympathetic overactivity and plasma leptin levels in rett syndrome. Neurosci. Lett. 2008, 432, 69–72. [Google Scholar] [CrossRef]
- Goddeau, R.P.; Silverman, S.B.; Sims, J.R. Dexmedetomidine for the treatment of paroxysmal autonomic instability with dystonia. Neurocrit. Care 2007, 7, 217–220. [Google Scholar] [CrossRef]
- Stern, J.S. Tourette’s Syndrome and its borderland. Pract. Neurol. 2018, 18, 262–270. [Google Scholar] [CrossRef]
- Leckman, J.F. Tourette’s Syndrome. Lancet Lond. Engl. 2002, 360, 1577–1586. [Google Scholar] [CrossRef]
- Mittal, S.O.; Klassen, B.T.; Hassan, A.; Bower, J.H.; Coon, E.A. Acute worsening of tics on varenicline. Clin. Neuropharmacol. 2017, 40, 231–232. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, N.; Herold, R.; Janszky, J.; Komoly, S.; Nagy, F. Tics status: A movement disorder emergency: Observations. J. Neurol. 2011, 258, 143–145. [Google Scholar] [CrossRef] [PubMed]
- Cossu, G.; Colosimo, C. Hyperkinetic movement disorder emergencies. Curr. Neurol. Neurosci. Rep. 2017, 17, 6. [Google Scholar] [CrossRef]
- Szejko, N.; Jakubczyk, A.; Dunalska, A.; Janik, P. Dystonic tics in patients with Gilles de la Tourette syndrome. Neurol. Neurochir. Pol. 2019, 53, 335–340. [Google Scholar] [CrossRef]
- Cheung, M.-Y.C.; Shahed, J.; Jankovic, J. Malignant Tourette Syndrome. Mov. Disord. 2007, 22, 1743–1750. [Google Scholar] [CrossRef]
- Wilbur, C.; Bitnun, A.; Kronenberg, S.; Laxer, R.M.; Levy, D.M.; Logan, W.J.; Shouldice, M.; Yeh, E.A. PANDAS/PANS in childhood: Controversies and evidence. Paediatr. Child Health 2019, 24, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Swedo, S.E.; Leonard, H.L.; Garvey, M.; Mittleman, B.; Allen, A.J.; Perlmutter, S.; Lougee, L.; Dow, S.; Zamkoff, J.; Dubbert, B.K. Pediatric autoimmune neuropsychiatric disorders associated with streptococcal infections: Clinical description of the first 50 cases. Am. J. Psychiatry 1998, 155, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Martino, D.; Schrag, A.; Anastasiou, Z.; Apter, A.; Benaroya-Milstein, N.; Buttiglione, M.; Cardona, F.; Creti, R.; Efstratiou, A.; Hedderly, T.; et al. Association of group a streptococcus exposure and exacerbations of chronic tic disorders: A multinational prospective cohort study. Neurology 2021. [Google Scholar] [CrossRef]
- Chouksey, A.; Pandey, S. Functional movement disorders in children. Front. Neurol. 2020, 11, 570151. [Google Scholar] [CrossRef] [PubMed]
- Heyman, I.; Liang, H.; Hedderly, T. COVID-19 related increase in childhood tics and tic-like attacks. Arch. Dis. Child. 2021, 106, 420–421. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
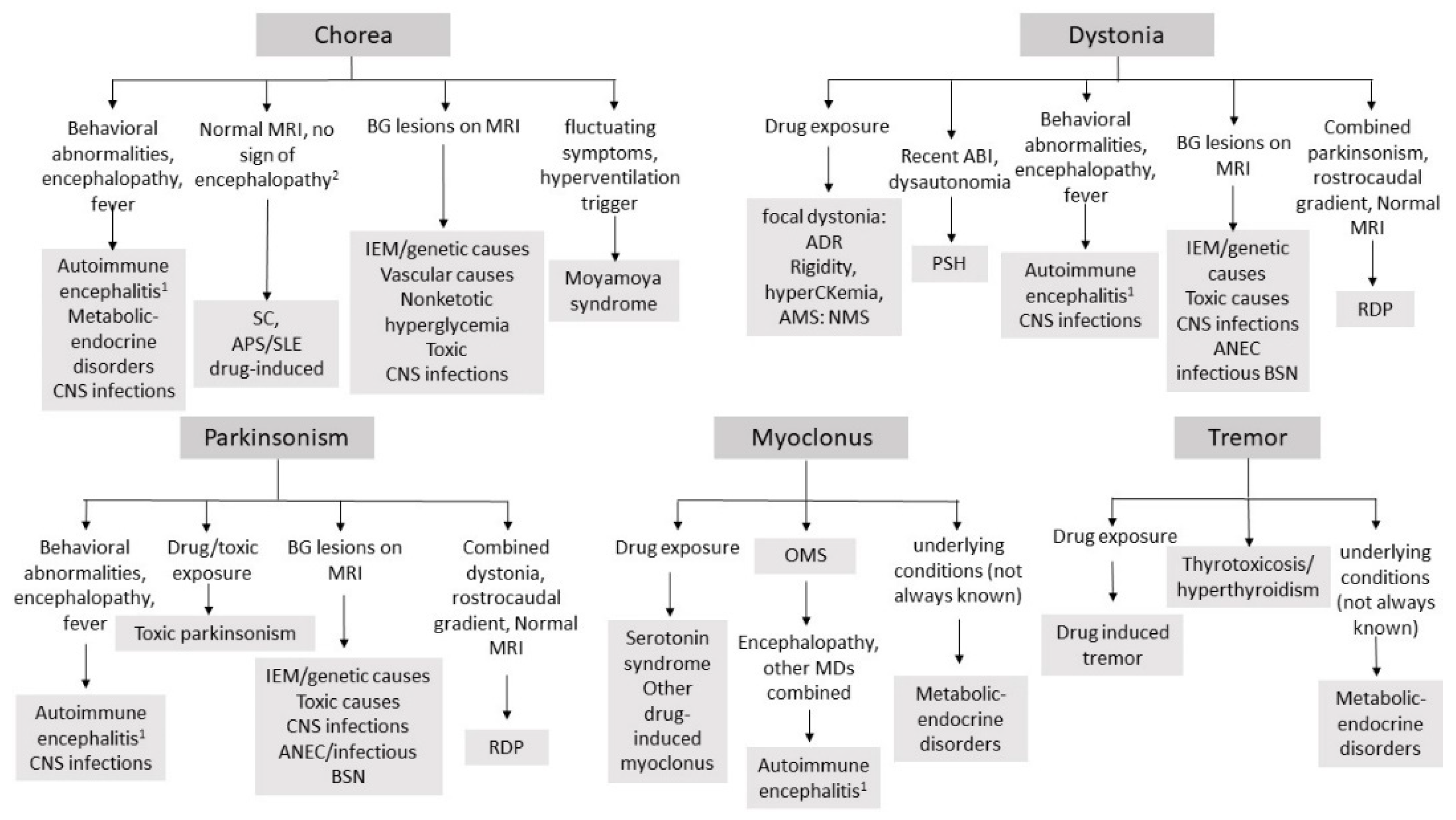
| Chorea |
| First-line tests throat swab culture, ASO and antiDNAse titer, cardiac US, EKG, CRP, ESR Routine blood test Neuroimaging |
| Second-line tests: aPL (anti GP2 IgG and IgM, anti cardiolipin IgG and IgM, LAC assay), to be always performed if chorea does not fulfil Jones’ criteria for rheumatic fever, ANA, ENA, complement fractions (C3–C4), consider joint US if signs of arthritis TSH/fT4 If signs of encephalitis i/encephalopathy: EEG, CSF sampling for cell count, proteins, CSF culture, PCR for neurotropic viruses, consider OCB, specific aAb testing on CSF and serum (NMDAR, D2R) Consider studies for IEM 1 if suggestive findings on MRI, young age, encephalopathy induced by catabolic states or high protein intake Consider MRA to exclude vasculopathy (especially if fluctuating symptoms or in presence of vascular lesions) |
| Dystonia |
| First-line tests Exclude drug/toxic exposure (toxic screening if needed) Routine blood test Neuroimaging |
| Second-line tests: If signs of encephalitis/encephalopathy: EEG, CSF sampling for cell count, proteins, CSF culture, PCR for neurotropic viruses, consider OCB, specific aAb testing on CSF and serum (NMDAR, D2R) Consider screening for IEM 1 (including ceruloplasmin/copper studies), especially if suggestive MRI findings. If severe dystonia: CPK, myoglobin, creatinine, urea. |
| Myoclonus |
| First-line tests Tumor screening (HVA/VMA urine levels, pelvis/abdomen/chest CT or MRI, MIBG scan). Exclude metabolic or endocrine disorders (uremia/hepatic encephalopathy). Exclude drug/toxic exposure (toxic screening if needed) Routine blood test Neuroimaging |
| Second-line tests: If rigidity, ataxia, or signs of encephalopathy: EEG, CSF sampling for OCB, cell count, proteins and specific aAb testing on CSF and serum (anti-DPPX, GlyR, GAD, amphysin). Consider work-up for IEM 1, according with MRI findings, especially if young age |
| Parkinsonism |
| First-line tests Routine blood test Neuroimaging Exclude drug/toxic exposure (toxic screening if needed) |
| Second-line tests: If signs of encephalitis/encephalopathy/behavioral abnormalities i: EEG, CSF sampling for OCB, cell count, proteins, CSF culture, PCR for neurotropic viruses, specific aAb testing on CSF and serum (NMDAR, D2R). consider work-up for IEM 1 (including ceruloplasmin/copper studies), especially if suggestive MRI findings |
| Tremor |
| First-line tests Exclude toxic causes (drug/toxins) and metabolic/endocrine disorders: electrolyte imbalances, TSH/fT4 testing; blood glucose levels; toxic screening (if needed) Consider Neuroimaging |
| Second-line tests: If signs of encephalopathy/encephalopathy: EEG, CSF sampling for OCB, cell count, proteins, CSF culture, PCR for neurotropic viruses, specific aAb testing on CSF and serum (NMDAR, D2R). Consider Screening for IEM 1 (including ceruloplasmin/copper studies), according with MRI finding |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garone, G.; Graziola, F.; Grasso, M.; Capuano, A. Acute Movement Disorders in Childhood. J. Clin. Med. 2021, 10, 2671. https://doi.org/10.3390/jcm10122671
Garone G, Graziola F, Grasso M, Capuano A. Acute Movement Disorders in Childhood. Journal of Clinical Medicine. 2021; 10(12):2671. https://doi.org/10.3390/jcm10122671
Chicago/Turabian StyleGarone, Giacomo, Federica Graziola, Melissa Grasso, and Alessandro Capuano. 2021. "Acute Movement Disorders in Childhood" Journal of Clinical Medicine 10, no. 12: 2671. https://doi.org/10.3390/jcm10122671
APA StyleGarone, G., Graziola, F., Grasso, M., & Capuano, A. (2021). Acute Movement Disorders in Childhood. Journal of Clinical Medicine, 10(12), 2671. https://doi.org/10.3390/jcm10122671