
Myasthenia Gravis: Epidemiology, Pathophysiology and Clinical Manifestations


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Epidemiology
3. Subtypes of MG and Their Clinical Manifestations
3.1. MG Due to Antibodies against AChR (AChR-MG)
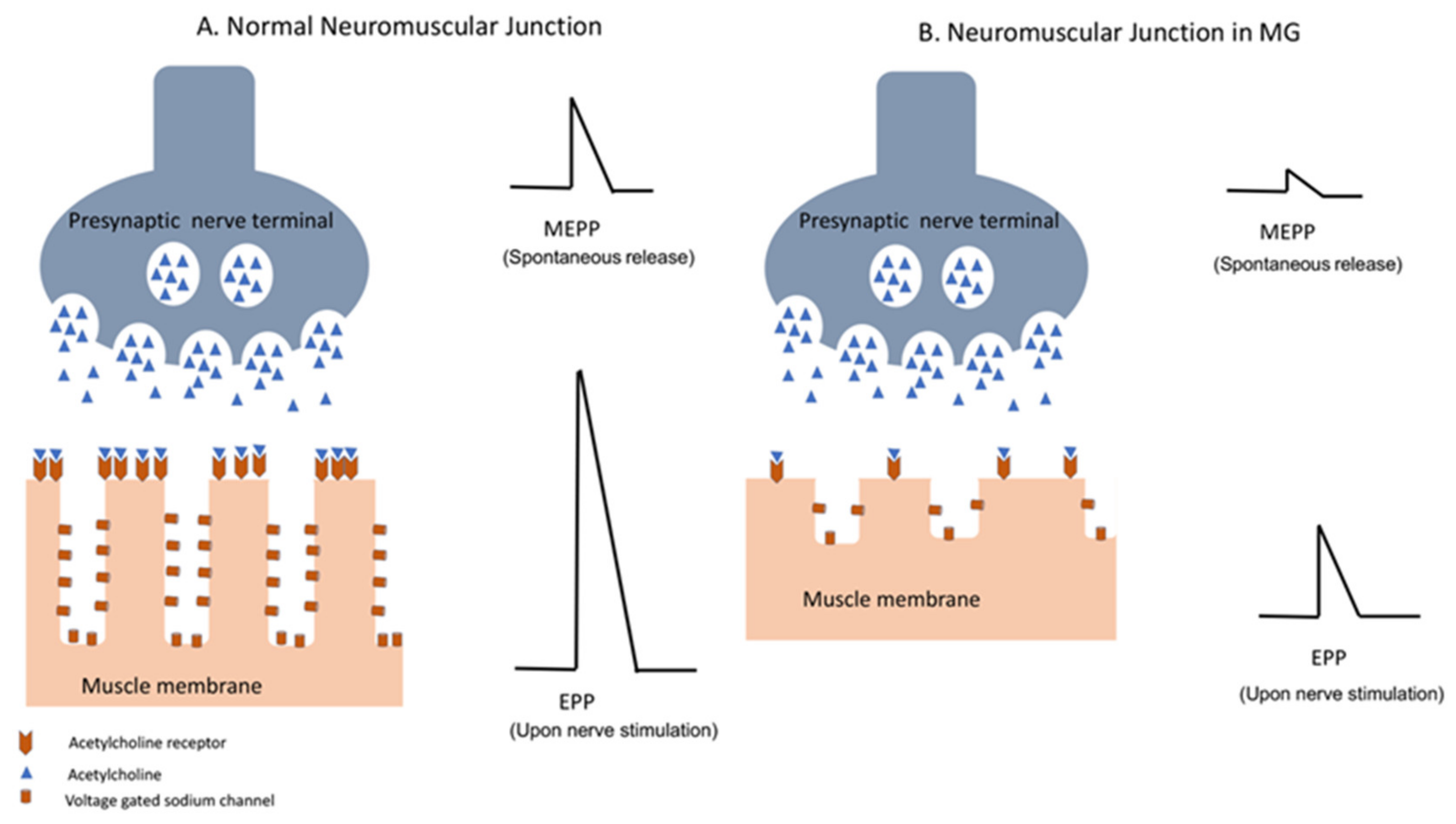
3.1.1. Effector Mechanisms
3.1.2. Clinical Manifestations
3.1.3. AChR MG Subtypes
Ocular MG
Generalized AChR Ab Positive MG (AChR-MG): Early vs. Late Onset
Thymoma-Associated MG
3.2. MuSK Antibody-Associated MG (MuSK-MG)
3.2.1. Effector Mechanisms
3.2.2. Clinical Manifestations
3.3. Double-Seronegative Generalized MG
3.4. Lrp4 Antibody-Associated MG (Lrp4-MG)
3.4.1. Effector Mechanisms
3.4.2. Clinical Manifestations
4. Pediatric MG
5. Pathophysiology
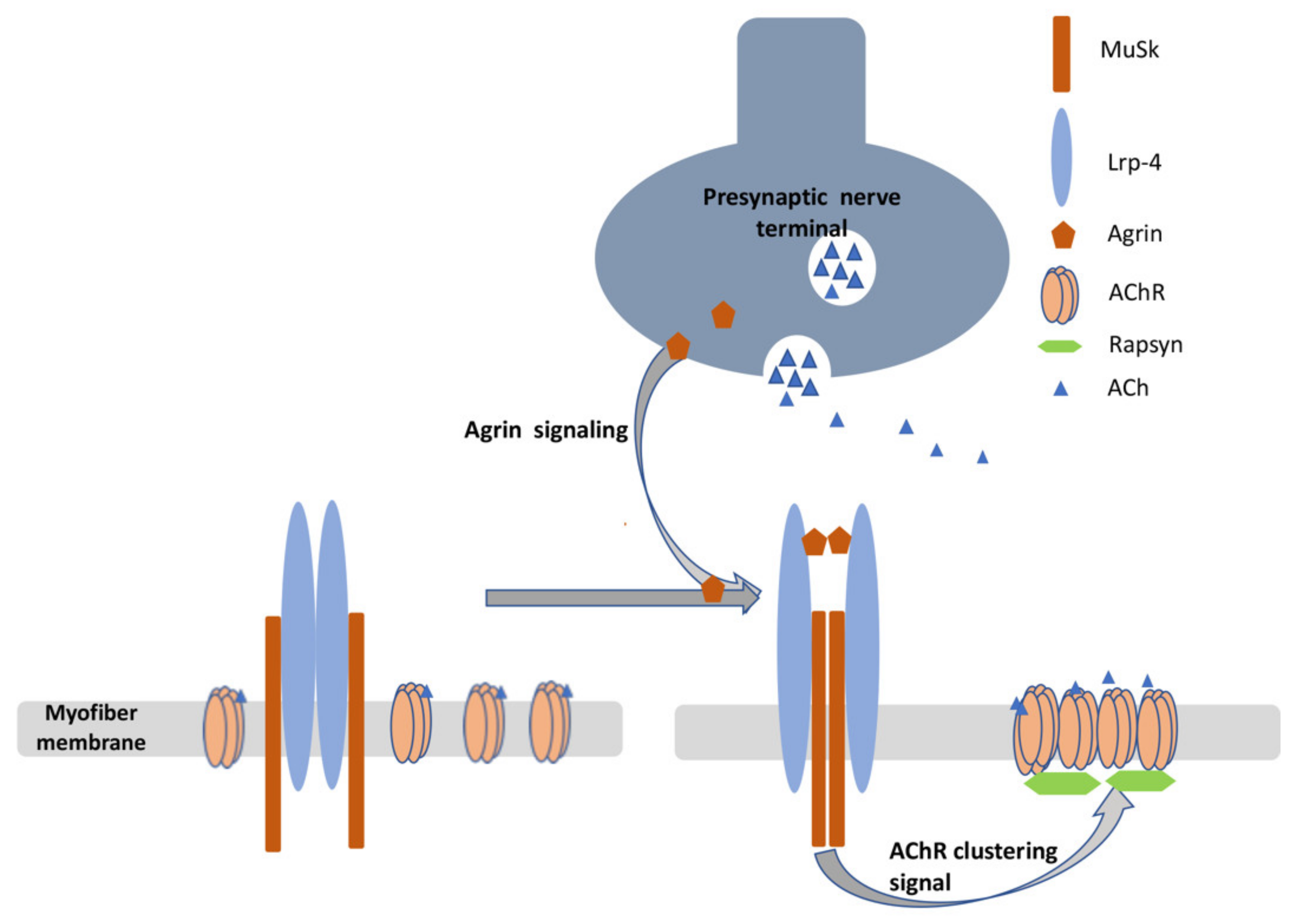
5.1. Physiology and Organization of the Neuromuscular Junction
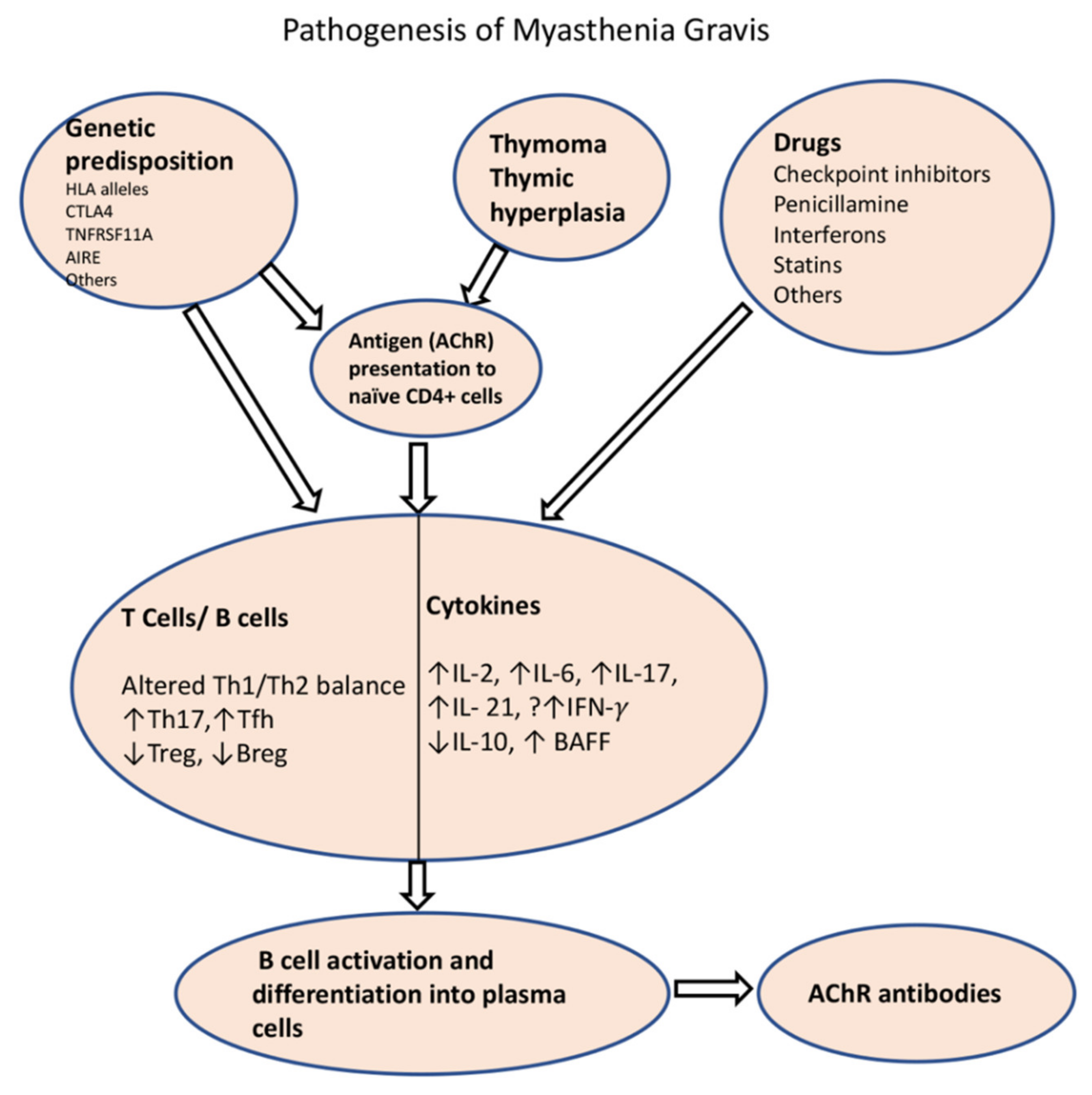
5.2. Immune Dysregulation in MG
Defective B Cell Tolerance
5.3. Role of Thymus in MG
5.3.1. Role of T Cells and Cytokines in the Development of MG
5.3.2. Regulatory T Cells (Tregs), Regulatory B Cells (Bregs), and B Cell-Activating Factor (BAFF) Signaling in MG
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McGrogan, A.; Sneddon, S.; de Vries, C.S. The Incidence of Myasthenia Gravis: A Systematic Literature Review. Neuroepidemiology 2010, 34, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Patrick, J.; Lindstrom, J. Autoimmune Response to Acetylcholine Receptor. Science 1973, 180, 871–872. [Google Scholar] [CrossRef] [PubMed]
- Fambrough, D.M.; Drachman, D.B.; Satyamurti, S. Neuromuscular Junction in Myasthenia Gravis: Decreased Acetylcholine Receptors. Science 1973, 182, 293–295. [Google Scholar] [CrossRef] [PubMed]
- McConville, J.; Farrugia, M.E.; Beeson, D.; Kishore, U.; Metcalfe, R.; Newsom-Davis, J.; Vincent, A. Detection and Characterization of MuSK Antibodies in Seronegative Myasthenia Gravis. Ann. Neurol. 2004, 55, 580–584. [Google Scholar] [CrossRef]
- Hoch, W.; McConville, J.; Helms, S.; Newsom-Davis, J.; Melms, A.; Vincent, A. Auto-Antibodies to the Receptor Tyrosine Kinase MuSK in Patients with Myasthenia Gravis without Acetylcholine Receptor Antibodies. Nat. Med. 2001, 7, 365–368. [Google Scholar] [CrossRef]
- Higuchi, O.; Hamuro, J.; Motomura, M.; Yamanashi, Y. Autoantibodies to Low-Density Lipoprotein Receptor-Related Protein 4 in Myasthenia Gravis. Ann. Neurol. 2011, 69, 418–422. [Google Scholar] [CrossRef]
- Pevzner, A.; Schoser, B.; Peters, K.; Cosma, N.-C.; Karakatsani, A.; Schalke, B.; Melms, A.; Kröger, S. Anti-LRP4 Autoantibodies in AChR- and MuSK-Antibody-Negative Myasthenia Gravis. J. Neurol. 2012, 259, 427–435. [Google Scholar] [CrossRef]
- Gasperi, C.; Melms, A.; Schoser, B.; Zhang, Y.; Meltoranta, J.; Risson, V.; Schaeffer, L.; Schalke, B.; Kröger, S. Anti-Agrin Autoantibodies in Myasthenia Gravis. Neurology 2014, 82, 1976–1983. [Google Scholar] [CrossRef]
- Zhang, B.; Shen, C.; Bealmear, B.; Ragheb, S.; Xiong, W.-C.; Lewis, R.A.; Lisak, R.P.; Mei, L. Autoantibodies to Agrin in Myasthenia Gravis Patients. PLoS ONE 2014, 9, e91816. [Google Scholar] [CrossRef]
- Szczudlik, P.; Szyluk, B.; Lipowska, M.; Ryniewicz, B.; Kubiszewska, J.; Dutkiewicz, M.; Gilhus, N.E.; Kostera-Pruszczyk, A. Antititin Antibody in Early- and Late-Onset Myasthenia Gravis. Acta Neurol. Scand. 2014, 130, 229–233. [Google Scholar] [CrossRef]
- Kufukihara, K.; Watanabe, Y.; Inagaki, T.; Takamatsu, K.; Nakane, S.; Nakahara, J.; Ando, Y.; Suzuki, S. Cytometric Cell-Based Assays for Anti-Striational Antibodies in Myasthenia Gravis with Myositis and/or Myocarditis. Sci. Rep. 2019, 9, 5284. [Google Scholar] [CrossRef]
- Grob, D.; Brunner, N.; Namba, T.; Pagala, M. Lifetime Course of Myasthenia Gravis. Muscle Nerve 2008, 37, 141–149. [Google Scholar] [CrossRef]
- Phillips, L.H. The Epidemiology of Myasthenia Gravis. Ann. N. Y. Acad. Sci. 2003, 998, 407–412. [Google Scholar] [CrossRef]
- Somnier, F.E.; Engel, P.J.H. The Occurrence of Anti-Titin Antibodies and Thymomas: A Population Survey of MG 1970–1999. Neurology 2002, 59, 92–98. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, B.K.; Cockerell, O.C.; Sander, J.W.; Shorvon, S.D. The Incidence and Lifetime Prevalence of Neurological Disorders in a Prospective Community-Based Study in the UK. Brain 2000, 123 (Pt 4), 665–676. [Google Scholar] [CrossRef]
- Fang, W.; Li, Y.; Mo, R.; Wang, J.; Qiu, L.; Ou, C.; Lin, Z.; Huang, Z.; Feng, H.; He, X.; et al. Hospital and Healthcare Insurance System Record–Based Epidemiological Study of Myasthenia Gravis in Southern and Northern China. Neurol. Sci. 2020, 41, 1211–1223. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Lee, H.S.; Shin, H.Y.; Choi, Y.C.; Kim, S.M. The Epidemiology of Myasthenia Gravis in Korea. Yonsei Med. J. 2016, 57, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Lee, J.Y.; Lim, N.G.; Hong, Y.H. Incidence and Prevalence of Myasthenia Gravis in Korea: A Population-Based Study Using the National Health Insurance Claims Database. J. Clin. Neurol. 2016, 12, 340–344. [Google Scholar] [CrossRef]
- Bettini, M.; Chaves, M.; Cristiano, E.; Pagotto, V.; Perez, L.; Giunta, D.; Rugiero, M. Incidence of Autoimmune Myasthenia Gravis in a Health Maintenance Organization in Buenos Aires, Argentina. Neuroepidemiology 2017, 48, 119–123. [Google Scholar] [CrossRef]
- Carr, A.S.; Cardwell, C.R.; McCarron, P.O.; McConville, J. A Systematic Review of Population Based Epidemiological Studies in Myasthenia Gravis. Bmc Neurol. 2010, 10, 46. [Google Scholar] [CrossRef]
- Ciafaloni, E. Myasthenia Gravis and Congenital Myasthenic Syndromes. Contin. Lifelong Learn. Neurol. 2019, 25, 1767–1784. [Google Scholar] [CrossRef]
- Oh, S.J.; Morgan, M.B.; Lu, L.; Hatanaka, Y.; Hemmi, S.; Young, A.; Claussen, G.C. Racial Differences in Myasthenia Gravis in Alabama. Muscle Nerve 2009, 39, 328–332. [Google Scholar] [CrossRef]
- Phillips, L.H.; Torner, J.C.; Anderson, M.S.; Cox, G.M. The Epidemiology of Myasthenia Gravis in Central and Western Virginia. Neurology 1992, 42, 1888–1893. [Google Scholar] [CrossRef] [PubMed]
- Alshekhlee, A.; Miles, J.D.; Katirji, B.; Preston, D.C.; Kaminski, H.J. Incidence and Mortality Rates of Myasthenia Gravis and Myasthenic Crisis in US Hospitals. Neurology 2009, 72, 1548–1554. [Google Scholar] [CrossRef]
- Heckmann, J.M.; Owen, E.P.; Little, F. Myasthenia Gravis in South Africans: Racial Differences in Clinical Manifestations. Neuromuscul. Disord. 2007, 17, 929–934. [Google Scholar] [CrossRef] [PubMed]
- Peragallo, J.H.; Bitrian, E.; Kupersmith, M.J.; Zimprich, F.; Whittaker, T.J.; Lee, M.S.; Bruce, B.B. Relationship between Age, Gender, and Race in Patients Presenting with Myasthenia Gravis with Only Ocular Manifestations. J. Neuroophthalmol. 2016, 36, 29–32. [Google Scholar] [CrossRef]
- Boldingh, M.I.; Maniaol, A.; Brunborg, C.; Dekker, L.; Lipka, A.; Niks, E.H.; Verschuuren, J.; Tallaksen, C. Prevalence and Clinical Aspects of Immigrants with Myasthenia Gravis in Northern Europe. Muscle Nerve 2017, 55, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Abukhalil, F.; Mehta, B.; Saito, E.; Mehta, S.; McMurtray, A. Gender and Ethnicity Based Differences in Clinical and Laboratory Features of Myasthenia Gravis. Autoimmune Dis. 2015, 2015, 197893. [Google Scholar] [CrossRef]
- Deymeer, F.; Gungor-Tuncer, O.; Yilmaz, V.; Parman, Y.; Serdaroglu, P.; Ozdemir, C.; Vincent, A.; Saruhan-Direskeneli, G. Clinical Comparison of Anti-MuSK- vs Anti-AChR-Positive and Seronegative Myasthenia Gravis. Neurology 2007, 68, 609–611. [Google Scholar] [CrossRef]
- Albuquerque, E.X.; Pereira, E.F.R.; Alkondon, M.; Rogers, S.W. Mammalian Nicotinic Acetylcholine Receptors: From Structure to Function. Physiol. Rev. 2009, 89, 73–120. [Google Scholar] [CrossRef]
- Tzartos, S.J.; Barkas, T.; Cung, M.T.; Mamalaki, A.; Marraud, M.; Orlewski, P.; Papanastasiou, D.; Sakarellos, C.; Sakarellos-Daitsiotis, M.; Tsantili, P.; et al. Anatomy of the Antigenic Structure of a Large Membrane Autoantigen, the Muscle-Type Nicotinic Acetylcholine Receptor. Immunol. Rev. 1998, 163, 89–120. [Google Scholar] [CrossRef]
- Kordas, G.; Lagoumintzis, G.; Sideris, S.; Poulas, K.; Tzartos, S.J. Direct Proof of the In Vivo Pathogenic Role of the AChR Autoantibodies from Myasthenia Gravis Patients. PLoS ONE 2014, 9, e108327. [Google Scholar] [CrossRef]
- Lennon, V.A.; McCormick, D.J.; Lambert, E.H.; Griesmann, G.E.; Atassi, M.Z. Region of Peptide 125-147 of Acetylcholine Receptor Alpha Subunit Is Exposed at Neuromuscular Junction and Induces Experimental Autoimmune Myasthenia Gravis, T-Cell Immunity, and Modulating Autoantibodies. Proc. Natl. Acad. Sci. USA 1985, 82, 8805–8809. [Google Scholar] [CrossRef]
- Fostieri, E.; Beeson, D.; Tzartos, S.J. The Conformation of the Main Immunogenic Region on the α-Subunit of Muscle Acetylcholine Receptor Is Affected by Neighboring Receptor Subunits. Febs Lett. 2000, 481, 127–130. [Google Scholar] [CrossRef]
- Morgan, B.P.; Chamberlain-Banoub, J.; Neal, J.W.; Song, W.; Mizuno, M.; Harris, C.L. The Membrane Attack Pathway of Complement Drives Pathology in Passively Induced Experimental Autoimmune Myasthenia Gravis in Mice. Clin. Exp. Immunol. 2006, 146, 294–302. [Google Scholar] [CrossRef]
- Rødgaard, A.; Nielsen, F.C.; Djurup, R.; Somnier, F.; Gammeltoft, S. Acetylcholine Receptor Antibody in Myasthenia Gravis: Predominance of IgG Subclasses 1 and 3. Clin. Exp. Immunol. 1987, 67, 82–88. [Google Scholar]
- Drachman, D.B.; Angus, C.W.; Adams, R.N.; Michelson, J.D.; Hoffman, G.J. Myasthenic Antibodies Cross-Link Acetylcholine Receptors to Accelerate Degradation. N. Engl. J. Med. 1978, 298, 1116–1122. [Google Scholar] [CrossRef]
- Hara, H.; Hayashi, K.; Ohta, K.; Itoh, N.; Nishitani, H.; Ohta, M. Detection and Characterization of Blocking-Type Anti-Acetylcholine Receptor Antibodies in Sera from Patients with Myasthenia Gravis. Clin. Chem. 1993, 39, 2053–2057. [Google Scholar] [CrossRef]
- Wintzen, A.R.; Plomp, J.J.; Molenaar, P.C.; van Dijk, J.G.; van Kempen, G.T.; Vos, R.M.; Wokke, J.H.; Vincent, A. Acquired Slow-Channel Syndrome: A Form of Myasthenia Gravis with Prolonged Open Time of the Acetylcholine Receptor Channel. Ann. Neurol. 1998, 44, 657–664. [Google Scholar] [CrossRef]
- Howard, F.M.; Lennon, V.A.; Finley, J.; Matsumoto, J.; Elveback, L.R. Clinical Correlations of Antibodies That Bind, Block, or Modulate Human Acetylcholine Receptors in Myasthenia Gravis. Ann. N. Y. Acad. Sci. 1987, 505, 526–538. [Google Scholar] [CrossRef]
- Gomez, C.M.; Richman, D.P. Anti-Acetylcholine Receptor Antibodies Directed against the Alpha-Bungarotoxin Binding Site Induce a Unique Form of Experimental Myasthenia. Proc. Natl. Acad. Sci. USA 1983, 80, 4089–4093. [Google Scholar] [CrossRef]
- Oosterhuis, H.J. The Natural Course of Myasthenia Gravis: A Long Term Follow up Study. J. Neurol. Neurosurg. Psychiatry 1989, 52, 1121–1127. [Google Scholar] [CrossRef]
- Hong, Y.-H.; Kwon, S.-B.; Kim, B.-J.; Kim, B.J.; Kim, S.H.; Kim, J.K.; Park, K.-S.; Park, K.-J.; Sung, J.-J.; Sohn, E.H.; et al. Prognosis of Ocular Myasthenia in Korea: A Retrospective Multicenter Analysis of 202 Patients. J. Neurol. Sci. 2008, 273, 10–14. [Google Scholar] [CrossRef]
- Hendricks, T.M.; Bhatti, M.T.; Hodge, D.O.; Chen, J.J. Incidence, Epidemiology, and Transformation of Ocular Myasthenia Gravis: A Population-Based Study. Am. J. Ophthalmol. 2019, 205, 99–105. [Google Scholar] [CrossRef]
- Gilhus, N.E. Myasthenia Gravis. N. Engl. J. Med. 2016, 375, 2570–2581. [Google Scholar] [CrossRef]
- Sih, M.; Soliven, B.; Mathenia, N.; Jacobsen, J.; Rezania, K. Head-Drop: A Frequent Feature of Late-Onset Myasthenia Gravis: Head-Drop in Myasthenia Gravis. Muscle Nerve 2017, 56, 441–444. [Google Scholar] [CrossRef]
- Grob, D. Course and Management of Myasthenia Gravis. J. Am. Med. Assoc. 1953, 153, 529–532. [Google Scholar] [CrossRef]
- Kaminski, H.J.; Maas, E.; Spiegel, P.; Ruff, R.L. Why Are Eye Muscles Frequently Involved in Myasthenia Gravis? Neurology 1990, 40, 1663–1669. [Google Scholar] [CrossRef]
- MacLennan, C.; Beeson, D.; Buijs, A.-M.; Vincent, A.; Newsom-Davis, J. Acetylcholine Receptor Expression in Human Extraocular Muscles and Their Susceptibility to Myasthenia Gravis. Ann. Neurol. 1997, 41, 423–431. [Google Scholar] [CrossRef]
- Narayanaswami, P.; Sanders, D.B.; Wolfe, G.; Benatar, M.; Cea, G.; Evoli, A.; Gilhus, N.E.; Illa, I.; Kuntz, N.L.; Massey, J.; et al. International Consensus Guidance for Management of Myasthenia Gravis: 2020 Update. Neurology 2021, 96, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Gilhus, N.E.; Verschuuren, J.J. Myasthenia Gravis: Subgroup Classification and Therapeutic Strategies. Lancet Neurol. 2015, 14, 1023–1036. [Google Scholar] [CrossRef]
- Vandiedonck, C.; Beaurain, G.; Giraud, M.; Hue-Beauvais, C.; Eymard, B.; Tranchant, C.; Gajdos, P.; Dausset, J.; Garchon, H.-J. Pleiotropic Effects of the 8.1 HLA Haplotype in Patients with Autoimmune Myasthenia Gravis and Thymus Hyperplasia. Proc. Natl. Acad. Sci. USA 2004, 101, 15464–15469. [Google Scholar] [CrossRef] [PubMed]
- Spagni, G.; Todi, L.; Monte, G.; Valentini, M.; Di Sante, G.; Damato, V.; Marino, M.; Evoli, A.; Lantieri, F.; Provenzano, C. Human Leukocyte Antigen Class II Associations in Late-onset Myasthenia Gravis. Ann. Clin. Transl. Neurol. 2021, 8, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Saruhan-Direskeneli, G.; Hughes, T.; Yilmaz, V.; Durmus, H.; Adler, A.; Alahgholi-Hajibehzad, M.; Aysal, F.; Yentür, S.P.; Akalin, M.A.; Dogan, O.; et al. Genetic Heterogeneity within the HLA Region in Three Distinct Clinical Subgroups of Myasthenia Gravis. Clin. Immunol. 2016, 166–167, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Maniaol, A.H.; Elsais, A.; Lorentzen, Å.R.; Owe, J.F.; Viken, M.K.; Sæther, H.; Flåm, S.T.; Bråthen, G.; Kampman, M.T.; Midgard, R.; et al. Late Onset Myasthenia Gravis Is Associated with HLA DRB1*15:01 in the Norwegian Population. PLoS ONE 2012, 7, e36603. [Google Scholar] [CrossRef]
- Renton, A.E.; Pliner, H.A.; Provenzano, C.; Evoli, A.; Ricciardi, R.; Nalls, M.A.; Marangi, G.; Abramzon, Y.; Arepalli, S.; Chong, S.; et al. A Genome-Wide Association Study of Myasthenia Gravis. Jama Neurol. 2015, 72, 396–404. [Google Scholar] [CrossRef]
- Gilhus, N.E.; Skeie, G.O.; Romi, F.; Lazaridis, K.; Zisimopoulou, P.; Tzartos, S. Myasthenia Gravis—Autoantibody Characteristics and Their Implications for Therapy. Nat Rev Neurol. 2016, 12, 259–268. [Google Scholar] [CrossRef]
- Bernard, C.; Frih, H.; Pasquet, F.; Kerever, S.; Jamilloux, Y.; Tronc, F.; Guibert, B.; Isaac, S.; Devouassoux, M.; Chalabreysse, L.; et al. Thymoma Associated with Autoimmune Diseases: 85 Cases and Literature Review. Autoimmun. Rev. 2016, 15, 82–92. [Google Scholar] [CrossRef]
- Muñiz-Castrillo, S.; Vogrig, A.; Honnorat, J. Associations between HLA and Autoimmune Neurological Diseases with Autoantibodies. Auto Immun. Highlights 2020, 11, 2. [Google Scholar] [CrossRef]
- Kim, N.; Stiegler, A.L.; Cameron, T.O.; Hallock, P.T.; Gomez, A.M.; Huang, J.H.; Hubbard, S.R.; Dustin, M.L.; Burden, S.J. Lrp4 Is a Receptor for Agrin and Forms a Complex with MuSK. Cell 2008, 135, 334–342. [Google Scholar] [CrossRef]
- Guptill, J.T.; Sanders, D.B.; Evoli, A. Anti-Musk Antibody Myasthenia Gravis: Clinical Findings and Response to Treatment in Two Large Cohorts. Muscle Nerve 2011, 44, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Cole, R.N.; Reddel, S.W.; Gervásio, O.L.; Phillips, W.D. Anti-MuSK Patient Antibodies Disrupt the Mouse Neuromuscular Junction. Ann. Neurol. 2008, 63, 782–789. [Google Scholar] [CrossRef]
- Niks, E.H.; van Leeuwen, Y.; Leite, M.I.; Dekker, F.W.; Wintzen, A.R.; Wirtz, P.W.; Vincent, A.; van Tol, M.J.D.; Jol-van der Zijde, C.M.; Verschuuren, J.J.G.M. Clinical Fluctuations in MuSK Myasthenia Gravis Are Related to Antigen-Specific IgG4 Instead of IgG1. J. Neuroimmunol. 2008, 195, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Poulas, K.; Koutsouraki, E.; Kordas, G.; Kokla, A.; Tzartos, S.J. Anti-MuSK- and Anti-AChR-Positive Myasthenia Gravis Induced by d-Penicillamine. J. Neuroimmunol. 2012, 250, 94–98. [Google Scholar] [CrossRef]
- Plomp, J.J.; Huijbers, M.G.; van der Maarel, S.M.; Verschuuren, J.J. Pathogenic IgG4 Subclass Autoantibodies in MuSK Myasthenia Gravis: MuSK Myasthenia Gravis IgG4. Ann. N. Y. Acad. Sci. 2012, 1275, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Inoue, A.; Okada, M.; Murata, Y.; Kakuta, S.; Jigami, T.; Kubo, S.; Shiraishi, H.; Eguchi, K.; Motomura, M.; et al. The Muscle Protein Dok-7 Is Essential for Neuromuscular Synaptogenesis. Science 2006, 312, 1802–1805. [Google Scholar] [CrossRef]
- Huijbers, M.G.; Zhang, W.; Klooster, R.; Niks, E.H.; Friese, M.B.; Straasheijm, K.R.; Thijssen, P.E.; Vrolijk, H.; Plomp, J.J.; Vogels, P.; et al. MuSK IgG4 Autoantibodies Cause Myasthenia Gravis by Inhibiting Binding between MuSK and Lrp4. Proc. Natl. Acad. Sci. USA 2013, 110, 20783–20788. [Google Scholar] [CrossRef] [PubMed]
- Koneczny, I.; Cossins, J.; Waters, P.; Beeson, D.; Vincent, A. MuSK Myasthenia Gravis IgG4 Disrupts the Interaction of LRP4 with MuSK but Both IgG4 and IgG1-3 Can Disperse Preformed Agrin-Independent AChR Clusters. PLoS ONE 2013, 8, e80695. [Google Scholar] [CrossRef]
- Sanders, D.B.; El-Salem, K.; Massey, J.M.; McConville, J.; Vincent, A. Clinical Aspects of MuSK Antibody Positive Seronegative MG. Neurology 2003, 60, 1978–1980. [Google Scholar] [CrossRef]
- Pasnoor, M.; Wolfe, G.I.; Nations, S.; Trivedi, J.; Barohn, R.J.; Herbelin, L.; McVey, A.; Dimachkie, M.; Kissel, J.; Walsh, R.; et al. Clinical Findings in MuSK-Antibody Positive Myasthenia Gravis: A U.S. Experience. Muscle Nerve 2010, 41, 370–374. [Google Scholar] [CrossRef]
- Bartoccioni, E.; Scuderi, F.; Augugliaro, A.; Chiatamone Ranieri, S.; Sauchelli, D.; Alboino, P.; Marino, M.; Evoli, A. HLA Class II Allele Analysis in MuSK-Positive Myasthenia Gravis Suggests a Role for DQ5. Neurology 2009, 72, 195–197. [Google Scholar] [CrossRef]
- Alahgholi-Hajibehzad, M.; Yilmaz, V.; Gülsen-Parman, Y.; Aysal, F.; Oflazer, P.; Deymeer, F.; Saruhan-Direskeneli, G. Association of HLA-DRB1∗14, -DRB1∗16 and -DQB1∗05 with MuSK-Myasthenia Gravis in Patients from Turkey. Hum. Immunol. 2013, 74, 1633–1635. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, Y.; Chen, J.; Huang, X.; Wang, H.; Li, Y.; Liu, W.; Feng, H. AChRAb and MuSKAb Double-Seropositive Myasthenia Gravis: A Distinct Subtype? Neurol. Sci. 2021, 42, 863–869. [Google Scholar] [CrossRef]
- Cortés-Vicente, E.; Gallardo, E.; Martínez, M.Á.; Díaz-Manera, J.; Querol, L.; Rojas-García, R.; Illa, I. Clinical Characteristics of Patients With Double-Seronegative Myasthenia Gravis and Antibodies to Cortactin. Jama Neurol. 2016, 73, 1099. [Google Scholar] [CrossRef] [PubMed]
- Rivner, M.H.; Quarles, B.M.; Pan, J.; Yu, Z.; Howard, J.F.; Corse, A.; Dimachkie, M.M.; Jackson, C.; Vu, T.; Small, G.; et al. Clinical Features of LRP4 /Agrin-antibody–Positive Myasthenia Gravis: A Multicenter Study. Muscle Nerve 2020, 62, 333–343. [Google Scholar] [CrossRef]
- Zhang, B.; Tzartos, J.S.; Belimezi, M.; Ragheb, S.; Bealmear, B.; Lewis, R.A.; Xiong, W.-C.; Lisak, R.P.; Tzartos, S.J.; Mei, L. Autoantibodies to Lipoprotein-Related Protein 4 in Patients with Double-Seronegative Myasthenia Gravis. Arch. Neurol. 2012, 69, 445–451. [Google Scholar] [CrossRef]
- Rivner, M.H.; Liu, S.; Quarles, B.; Fleenor, B.; Shen, C.; Pan, J.; Mei, L. Agrin and Low-Density Lipoprotein-Related Receptor Protein 4 Antibodies in Amyotrophic Lateral Sclerosis Patients. Muscle Nerve 2017, 55, 430–432. [Google Scholar] [CrossRef]
- Tzartos, J.S.; Zisimopoulou, P.; Rentzos, M.; Karandreas, N.; Zouvelou, V.; Evangelakou, P.; Tsonis, A.; Thomaidis, T.; Lauria, G.; Andreetta, F.; et al. LRP4 Antibodies in Serum and CSF from Amyotrophic Lateral Sclerosis Patients. Ann. Clin. Transl. Neurol. 2014, 1, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.-H.; Tseng, H.-F. Nationwide Population-Based Epidemiological Study of Myasthenia Gravis in Taiwan. Neuroepidemiology 2010, 35, 66–71. [Google Scholar] [CrossRef]
- Chiu, H.C.; Vincent, A.; Newsom-Davis, J.; Hsieh, K.H.; Hung, T. Myasthenia Gravis: Population Differences in Disease Expression and Acetylcholine Receptor Antibody Titers between Chinese and Caucasians. Neurology 1987, 37, 1854–1857. [Google Scholar] [CrossRef]
- Chiang, L.M.; Darras, B.T.; Kang, P.B. Juvenile Myasthenia Gravis. Muscle Nerve 2009, 39, 423–431. [Google Scholar] [CrossRef]
- Castro, D.; Derisavifard, S.; Anderson, M.; Greene, M.; Iannaccone, S. Juvenile Myasthenia Gravis: A Twenty-Year Experience. J. Clin. Neuromuscul. Dis. 2013, 14, 95–102. [Google Scholar] [CrossRef]
- Barraud, C.; Desguerre, I.; Barnerias, C.; Gitiaux, C.; Boulay, C.; Chabrol, B. Clinical Features and Evolution of Juvenile Myasthenia Gravis in a French Cohort. Muscle Nerve 2018, 57, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Parr, J.R.; Jayawant, S. Childhood Myasthenia: Clinical Subtypes and Practical Management. Dev. Med. Child. Neurol. 2007, 49, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Lindner, A.; Schalke, B.; Toyka, K.V. Outcome in Juvenile-Onset Myasthenia Gravis: A Retrospective Study with Long-Term Follow-up of 79 Patients. J. Neurol. 1997, 244, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Vanikieti, K.; Lowwongngam, K.; Padungkiatsagul, T.; Visudtibhan, A.; Poonyathalang, A. Juvenile Ocular Myasthenia Gravis: Presentation and Outcome of a Large Cohort. Pediatr. Neurol. 2018, 87, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Gui, M.; Luo, X.; Lin, J.; Li, Y.; Zhang, M.; Zhang, X.; Yang, M.; Wang, W.; Bu, B. Long-Term Outcome of 424 Childhood-Onset Myasthenia Gravis Patients. J. Neurol. 2015, 262, 823–830. [Google Scholar] [CrossRef]
- Pineles, S.L.; Avery, R.A.; Moss, H.E.; Finkel, R.; Blinman, T.; Kaiser, L.; Liu, G.T. Visual and Systemic Outcomes in Pediatric Ocular Myasthenia Gravis. Am. J. Ophthalmol. 2010, 150, 453–459.e3. [Google Scholar] [CrossRef] [PubMed]
- Anlar, B.; Senbil, N.; Köse, G.; Değerliyurt, A. Serological Follow-up in Juvenile Myasthenia: Clinical and Acetylcholine Receptor Antibody Status of Patients Followed for at Least 2 Years. Neuromuscul. Disord. 2005, 15, 355–357. [Google Scholar] [CrossRef]
- Skjei, K.L.; Lennon, V.A.; Kuntz, N.L. Muscle Specific Kinase Autoimmune Myasthenia Gravis in Children: A Case Series. Neuromuscul. Disord. 2013, 23, 874–882. [Google Scholar] [CrossRef]
- Zisimopoulou, P.; Evangelakou, P.; Tzartos, J.; Lazaridis, K.; Zouvelou, V.; Mantegazza, R.; Antozzi, C.; Andreetta, F.; Evoli, A.; Deymeer, F.; et al. A Comprehensive Analysis of the Epidemiology and Clinical Characteristics of Anti-LRP4 in Myasthenia Gravis. J. Autoimmun. 2014, 52, 139–145. [Google Scholar] [CrossRef]
- Li, M.; Han, J.; Zhang, Y.; Lv, J.; Zhang, J.; Zhao, X.; Ren, L.; Fang, H.; Yang, J.; Zhang, Y.; et al. Clinical Analysis of Chinese Anti-Low-Density-Lipoprotein-Receptor-Associated Protein 4 Antibodies in Patients with Myasthenia Gravis. Eur. J. Neurol. 2019, 26, 1296-e84. [Google Scholar] [CrossRef] [PubMed]
- Namba, T.; Brown, S.B.; Grob, D. Neonatal Myasthenia Gravis: Report of Two Cases and Review of the Literature. Pediatrics 1970, 45, 488–504. [Google Scholar] [PubMed]
- Vernet-der Garabedian, B.; Lacokova, M.; Eymard, B.; Morel, E.; Faltin, M.; Zajac, J.; Sadovsky, O.; Dommergues, M.; Tripon, P.; Bach, J.F. Association of Neonatal Myasthenia Gravis with Antibodies against the Fetal Acetylcholine Receptor. J. Clin. Investig. 1994, 94, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Hacohen, Y.; Jacobson, L.W.; Byrne, S.; Norwood, F.; Lall, A.; Robb, S.; Dilena, R.; Fumagalli, M.; Born, A.P.; Clarke, D.; et al. Fetal Acetylcholine Receptor Inactivation Syndrome: A Myopathy Due to Maternal Antibodies. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e57. [Google Scholar] [CrossRef]
- Parr, J.R.; Andrew, M.J.; Finnis, M.; Beeson, D.; Vincent, A.; Jayawant, S. How Common Is Childhood Myasthenia? The UK Incidence and Prevalence of Autoimmune and Congenital Myasthenia. Arch. Dis. Child. 2014, 99, 539–542. [Google Scholar] [CrossRef]
- Koneczny, I.; Herbst, R. Myasthenia Gravis: Pathogenic Effects of Autoantibodies on Neuromuscular Architecture. Cells 2019, 8, 671. [Google Scholar] [CrossRef]
- Ruff, R.L.; Lisak, R.P. Nature and Action of Antibodies in Myasthenia Gravis. Neurol. Clin. 2018, 36, 275–291. [Google Scholar] [CrossRef]
- Ruff, R.L.; Lennon, V.A. How Myasthenia Gravis Alters the Safety Factor for Neuromuscular Transmission. J. Neuroimmunol. 2008, 201–202, 13–20. [Google Scholar] [CrossRef]
- Lee, J.; Stathopoulos, P.; Gupta, S.; Bannock, J.M.; Barohn, R.J.; Cotzomi, E.; Dimachkie, M.M.; Jacobson, L.; Lee, C.S.; Morbach, H.; et al. Compromised Fidelity of B-cell Tolerance Checkpoints in AChR and MuSK Myasthenia Gravis. Ann. Clin. Transl. Neurol. 2016, 3, 443–454. [Google Scholar] [CrossRef]
- Vander Heiden, J.A.; Stathopoulos, P.; Zhou, J.Q.; Chen, L.; Gilbert, T.J.; Bolen, C.R.; Barohn, R.J.; Dimachkie, M.M.; Ciafaloni, E.; Broering, T.J.; et al. Dysregulation of B Cell Repertoire Formation in Myasthenia Gravis Patients Revealed through Deep Sequencing. J. Immunol. 2017, 198, 1460–1473. [Google Scholar] [CrossRef] [PubMed]
- Takaba, H.; Takayanagi, H. The Mechanisms of T Cell Selection in the Thymus. Trends Immunol. 2017, 38, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Poëa-Guyon, S.; Christadoss, P.; Le Panse, R.; Guyon, T.; De Baets, M.; Wakkach, A.; Bidault, J.; Tzartos, S.; Berrih-Aknin, S. Effects of Cytokines on Acetylcholine Receptor Expression: Implications for Myasthenia Gravis. J. Immunol. 2005, 174, 5941–5949. [Google Scholar] [CrossRef] [PubMed]
- Dragin, N.; Bismuth, J.; Cizeron-Clairac, G.; Biferi, M.G.; Berthault, C.; Serraf, A.; Nottin, R.; Klatzmann, D.; Cumano, A.; Barkats, M.; et al. Estrogen-Mediated Downregulation of AIRE Influences Sexual Dimorphism in Autoimmune Diseases. J. Clin. Investig. 2016, 126, 1525–1537. [Google Scholar] [CrossRef] [PubMed]
- Truffault, F.; de Montpreville, V.; Eymard, B.; Sharshar, T.; Le Panse, R.; Berrih-Aknin, S. Thymic Germinal Centers and Corticosteroids in Myasthenia Gravis: An Immunopathological Study in 1035 Cases and a Critical Review. Clin. Rev. Allerg. Immunol. 2017, 52, 108–124. [Google Scholar] [CrossRef] [PubMed]
- Vrolix, K.; Fraussen, J.; Losen, M.; Stevens, J.; Lazaridis, K.; Molenaar, P.C.; Somers, V.; Bracho, M.A.; Le Panse, R.; Stinissen, P.; et al. Clonal Heterogeneity of Thymic B Cells from Early-Onset Myasthenia Gravis Patients with Antibodies against the Acetylcholine Receptor. J. Autoimmun. 2014, 52, 101–112. [Google Scholar] [CrossRef]
- Vinuesa, C.G.; Linterman, M.A.; Yu, D.; MacLennan, I.C.M. Follicular Helper T Cells. Annu. Rev. Immunol. 2016, 34, 335–368. [Google Scholar] [CrossRef]
- Gradolatto, A.; Nazzal, D.; Truffault, F.; Bismuth, J.; Fadel, E.; Foti, M.; Berrih-Aknin, S. Both Treg Cells and Tconv Cells Are Defective in the Myasthenia Gravis Thymus: Roles of IL-17 and TNF-α. J. Autoimmun. 2014, 52, 53–63. [Google Scholar] [CrossRef]
- Ashida, S.; Ochi, H.; Hamatani, M.; Fujii, C.; Kimura, K.; Okada, Y.; Hashi, Y.; Kawamura, K.; Ueno, H.; Takahashi, R.; et al. Immune Skew of Circulating Follicular Helper T Cells Associates With Myasthenia Gravis Severity. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8. [Google Scholar] [CrossRef]
- Kaul, R.; Shenoy, M.; Goluszko, E.; Christadoss, P. Major histocompatibility complex class II gene disruption prevents experimental autoimmune myasthenia gravis. J. Immunol. 1994, 152, 3152–3157. [Google Scholar]
- Christadoss, P.; Dauphinee, M.J. Immunotherapy for myasthenia gravis: A murine model. J. Immunol. 1986, 136, 2437–2440. [Google Scholar] [PubMed]
- Wu, B.; Deng, C.; Goluszko, E.; Christadoss, P. Tolerance to a dominant T cell epitope in the acetylcholine receptor molecule induces epitope spread and suppresses murine myasthenia gravis. J. Immunol. 1997, 159, 3016–3023. [Google Scholar] [PubMed]
- Balasa, B.; Sarvetnick, N. Is pathogenic humoral autoimmunity a Th1 response? Lessons from (for) myasthenia gravis. Immunol. Today 2000, 21, 19–23. [Google Scholar] [CrossRef]
- Link, J.; Söderström, M.; Ljungdahl, Å.; Höjeberg, B.; Olsson, T.; Xu, Z.; Fredrikson, S.; Wang, Z.-Y.; Link, H. Organ-specific autoantigens induce interferon-γ and interleukin-4 mRNA expression in mononuclear cells in multiple sclerosis and myasthenia gravis. Neurology 1994, 44, 728. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Moriyama, M.; Nakashima, H.; Miyake, K.; Hayashida, J.-N.; Maehara, T.; Shinozaki, S.; Kubo, Y.; Nakamura, S. Th2 and regulatory immune reactions contribute to IgG4 production and the initiation of Mikulicz disease. Arthritis Rheum. 2011, 64, 254–263. [Google Scholar] [CrossRef] [PubMed]
- Stevens, T.L.; Bossie, A.; Sanders, V.M.; Fernandez-Botran, R.; Coffman, R.L.; Mosmann, T.R.; Vitetta, E.S. Regulation of antibody isotype secretion by subsets of antigen-specific helper T cells. Nat. Cell Biol. 1988, 334, 255–258. [Google Scholar] [CrossRef]
- Çebi, M.; Durmus, H.; Aysal, F.; Özkan, B.; Gül, G.E.; Çakar, A.; Hocaoglu, M.; Mercan, M.; Yentür, S.P.; Tütüncü, M.; et al. CD4+ T Cells of Myasthenia Gravis Patients Are Characterized by Increased IL-21, IL-4, and IL-17A Productions and Higher Presence of PD-1 and ICOS. Front. Immunol. 2020, 11, 809. [Google Scholar] [CrossRef]
- Uzawa, A.; Kuwabara, S.; Suzuki, S.; Imai, T.; Murai, H.; Ozawa, Y.; Yasuda, M.; Nagane, Y.; Utsugisawa, K. Roles of cytokines and T cells in the pathogenesis of myasthenia gravis. Clin. Exp. Immunol. 2021, 203, 366–374. [Google Scholar] [CrossRef]
- Xie, Y.; Li, H.-F.; Jiang, B.; Li, Y.; Kaminski, H.J.; Kusner, L.L. Elevated plasma interleukin-17A in a subgroup of Myasthenia Gravis patients. Cytokine 2016, 78, 44–46. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, W.; Chen, Y.; Wei, D. T Helper Type 17 Cells Expand in Patients with Myasthenia-Associated Thymoma. Scand. J. Immunol. 2012, 76, 54–61. [Google Scholar] [CrossRef]
- Cao, Y.; Amezquita, R.A.; Kleinstein, S.H.; Stathopoulos, P.; Nowak, R.J.; O’Connor, K.C. Autoreactive T Cells from Patients with Myasthenia Gravis Are Characterized by Elevated IL-17, IFN-γ, and GM-CSF and Diminished IL-10 Production. J. Immunol. 2016, 196, 2075–2084. [Google Scholar] [CrossRef]
- Huan, X.; Luo, S.; Zhong, H.; Zheng, X.; Song, J.; Zhou, L.; Lu, J.; Wang, Y.; Xu, Y.; Xi, J.; et al. In-depth peripheral CD4 + T profile correlates with myasthenic crisis. Ann. Clin. Transl. Neurol. 2021, 8, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Guidon, A.; Sparks, S.; Osborne, R.; Juel, V.; Massey, J.; Sanders, D.; Weinhold, K.; Guptill, J. Characterization of CD4 and CD8 T cell responses in MuSK myasthenia gravis. J. Autoimmun. 2014, 52, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Villegas, J.A.; Van Wassenhove, J.; Le Panse, R.; Berrih-Aknin, S.; Dragin, N. An imbalance between regulatory T cells and T helper 17 cells in acetylcholine receptor-positive myasthenia gravis patients. Ann. N. Y. Acad. Sci. 2018, 1413, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Balandina, A.; Lécart, S.; Dartevelle, P.; Saoudi, A.; Berrih-Aknin, S. Functional defect of regulatory CD4+CD25+ T cells in the thymus of patients with autoimmune myasthenia gravis. Blood 2005, 105, 735–741. [Google Scholar] [CrossRef]
- Battaglia, M.; Stabilini, A.; Roncarolo, M.-G. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood 2005, 105, 4743–4748. [Google Scholar] [CrossRef]
- Matsui, N.; Nakane, S.; Saito, F.; Ohigashi, I.; Nakagawa, Y.; Kurobe, H.; Takizawa, H.; Mitsui, T.; Kondo, K.; Kitagawa, T.; et al. Undiminished regulatory T cells in the thymus of patients with myasthenia gravis. Neurology 2010, 74, 816–820. [Google Scholar] [CrossRef]
- Thiruppathi, M.; Rowin, J.; Ganesh, B.; Sheng, J.R.; Prabhakar, B.S.; Meriggioli, M.N. Impaired regulatory function in circulating CD4+CD25highCD127low/− T cells in patients with myasthenia gravis. Clin. Immunol. 2012, 145, 209–223. [Google Scholar] [CrossRef]
- Mauri, C.; Menon, M. Human regulatory B cells in health and disease: Therapeutic potential. J. Clin. Investig. 2017, 127, 772–779. [Google Scholar] [CrossRef]
- Sheng, J.R.; Rezania, K.; Soliven, B. Impaired regulatory B cells in myasthenia gravis. J. Neuroimmunol. 2016, 297, 38–45. [Google Scholar] [CrossRef]
- Sun, F.; Ladha, S.S.; Yang, L.; Liu, Q.; Bs, S.X.S.; Su, N.; Bomprezzi, R.; Shi, S.X.-Y. Interleukin-10 producing-B cells and their association with responsiveness to rituximab in myasthenia gravis. Muscle Nerve 2013, 49, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.S.; Russo, M.A.; Massey, J.M.; Juel, V.; Hobson-Webb, L.D.; Gable, K.; Raja, S.M.; Balderson, K.; Weinhold, K.J.; Guptill, J.T. B10 Cell Frequencies and Suppressive Capacity in Myasthenia Gravis Are Associated with Disease Severity. Front. Neurol. 2017, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Karim, R.; Zhang, H.-Y.; Yuan, J.; Sun, Q.; Wang, Y.-F. Regulatory B Cells in Seropositive Myasthenia Gravis versus Healthy Controls. Front. Neurol. 2017, 8, 43. [Google Scholar] [CrossRef]
- Thompson, J.S.; Bixler, S.A.; Qian, F.; Vora, K.; Scott, M.L.; Cachero, T.G.; Hession, C.; Schneider, P.; Sizing, I.D.; Mullen, C.; et al. BAFF-R, a Newly Identified TNF Receptor That Specifically Interacts with BAFF. Science 2001, 293, 2108–2111. [Google Scholar] [CrossRef]
- Ragheb, S.; Lisak, R.P. B-Cell-Activating Factor and Autoimmune Myasthenia Gravis. Autoimmune Dis. 2011, 2011, 1–10. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kang, S.-Y.; Kang, C.-H.; Lee, K.-H. B-cell-activating factor is elevated in serum of patients with myasthenia gravis. Muscle Nerve 2016, 54, 1030–1033. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Xiao, B.-G.; Xi, J.-Y.; Lu, C.-Z.; Lu, J.-H. Decrease of CD4+CD25highFoxp3+ regulatory T cells and elevation of CD19+BAFF-R+ B cells and soluble ICAM-1 in myasthenia gravis. Clin. Immunol. 2008, 126, 180–188. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dresser, L.; Wlodarski, R.; Rezania, K.; Soliven, B. Myasthenia Gravis: Epidemiology, Pathophysiology and Clinical Manifestations. J. Clin. Med. 2021, 10, 2235. https://doi.org/10.3390/jcm10112235
Dresser L, Wlodarski R, Rezania K, Soliven B. Myasthenia Gravis: Epidemiology, Pathophysiology and Clinical Manifestations. Journal of Clinical Medicine. 2021; 10(11):2235. https://doi.org/10.3390/jcm10112235
Chicago/Turabian StyleDresser, Laura, Richard Wlodarski, Kourosh Rezania, and Betty Soliven. 2021. "Myasthenia Gravis: Epidemiology, Pathophysiology and Clinical Manifestations" Journal of Clinical Medicine 10, no. 11: 2235. https://doi.org/10.3390/jcm10112235
APA StyleDresser, L., Wlodarski, R., Rezania, K., & Soliven, B. (2021). Myasthenia Gravis: Epidemiology, Pathophysiology and Clinical Manifestations. Journal of Clinical Medicine, 10(11), 2235. https://doi.org/10.3390/jcm10112235