
Diagnosis of Myasthenia Gravis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
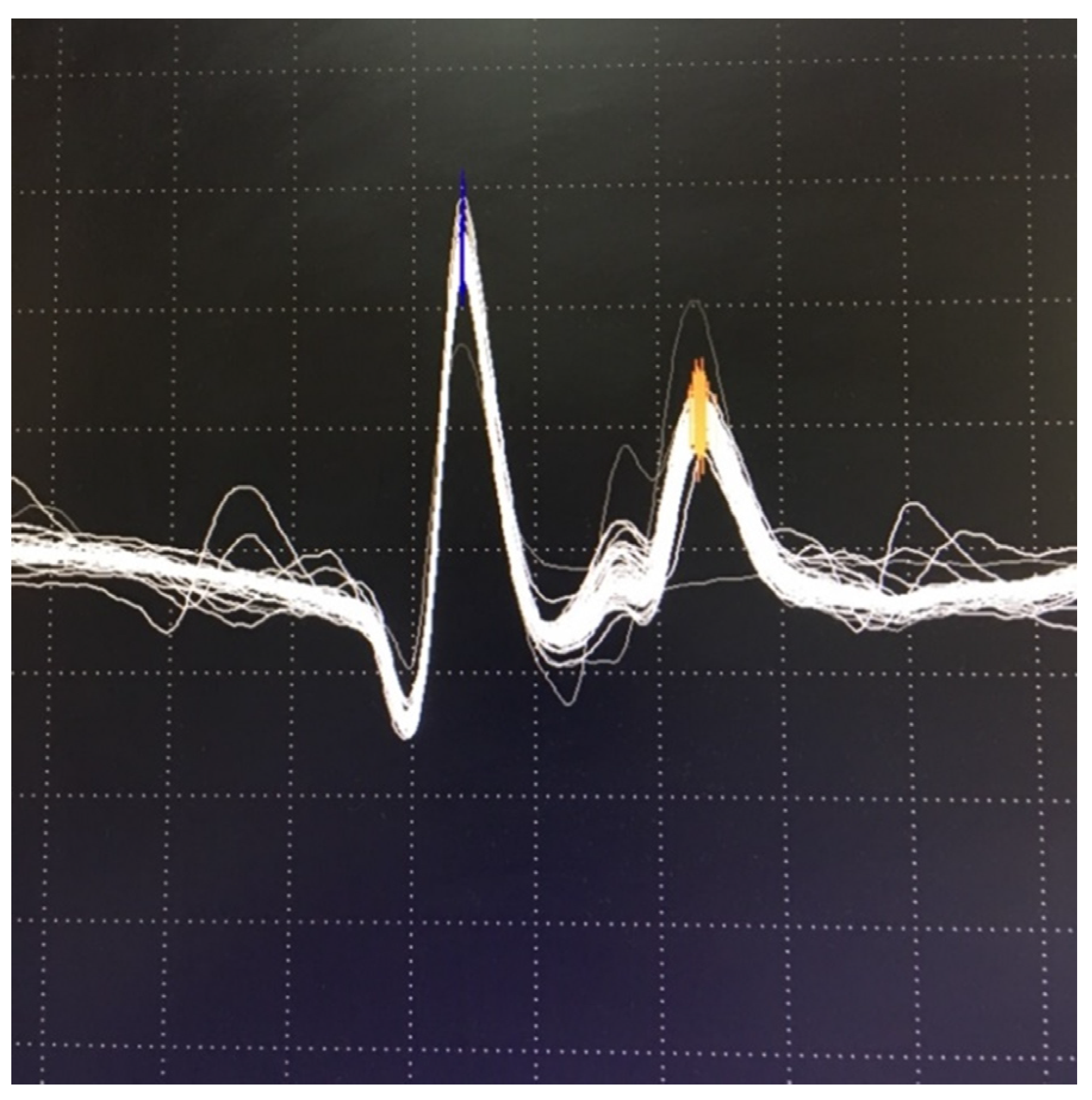{kind=link}
{kind=link}
{kind=link}
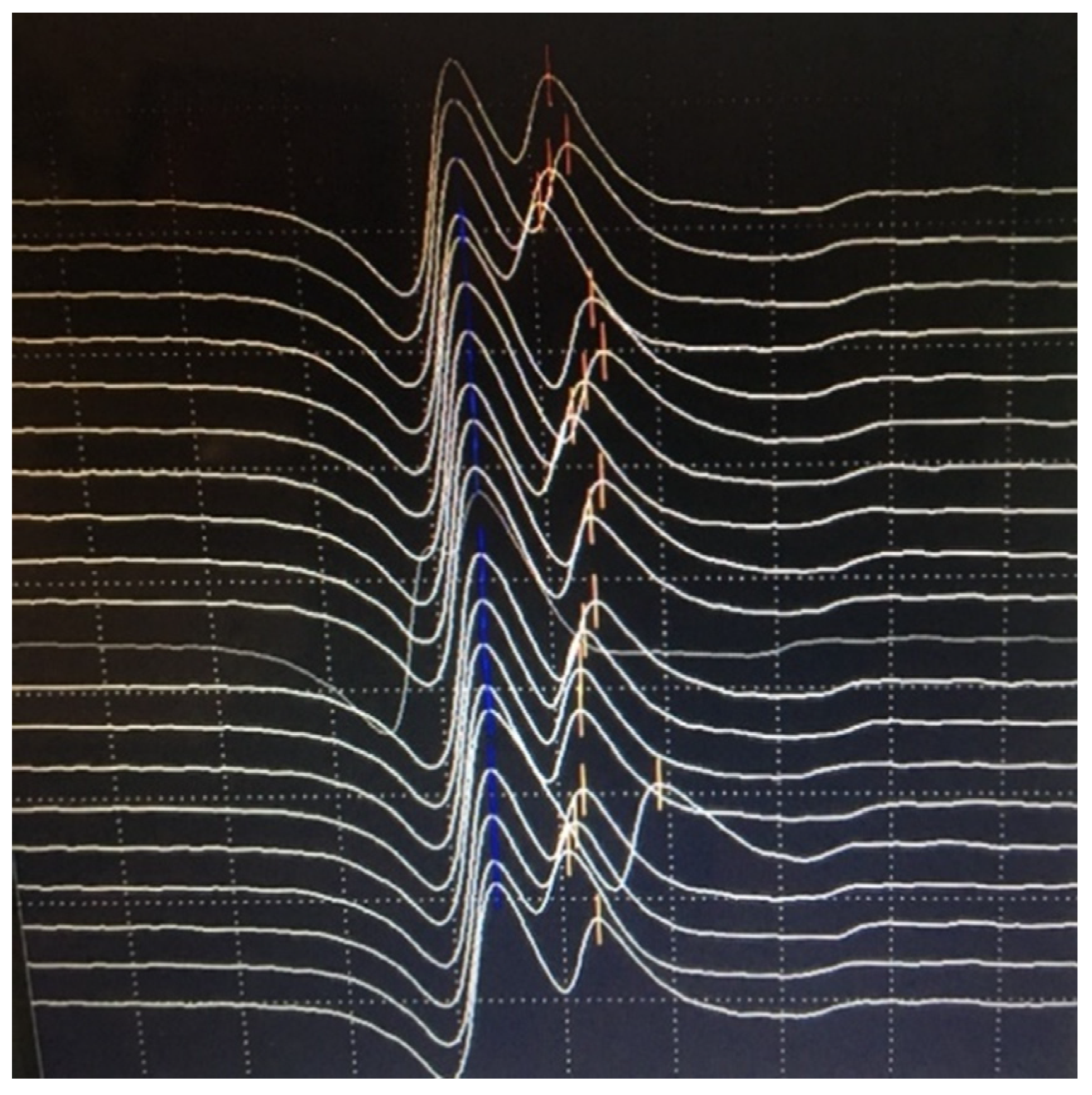{kind=link}
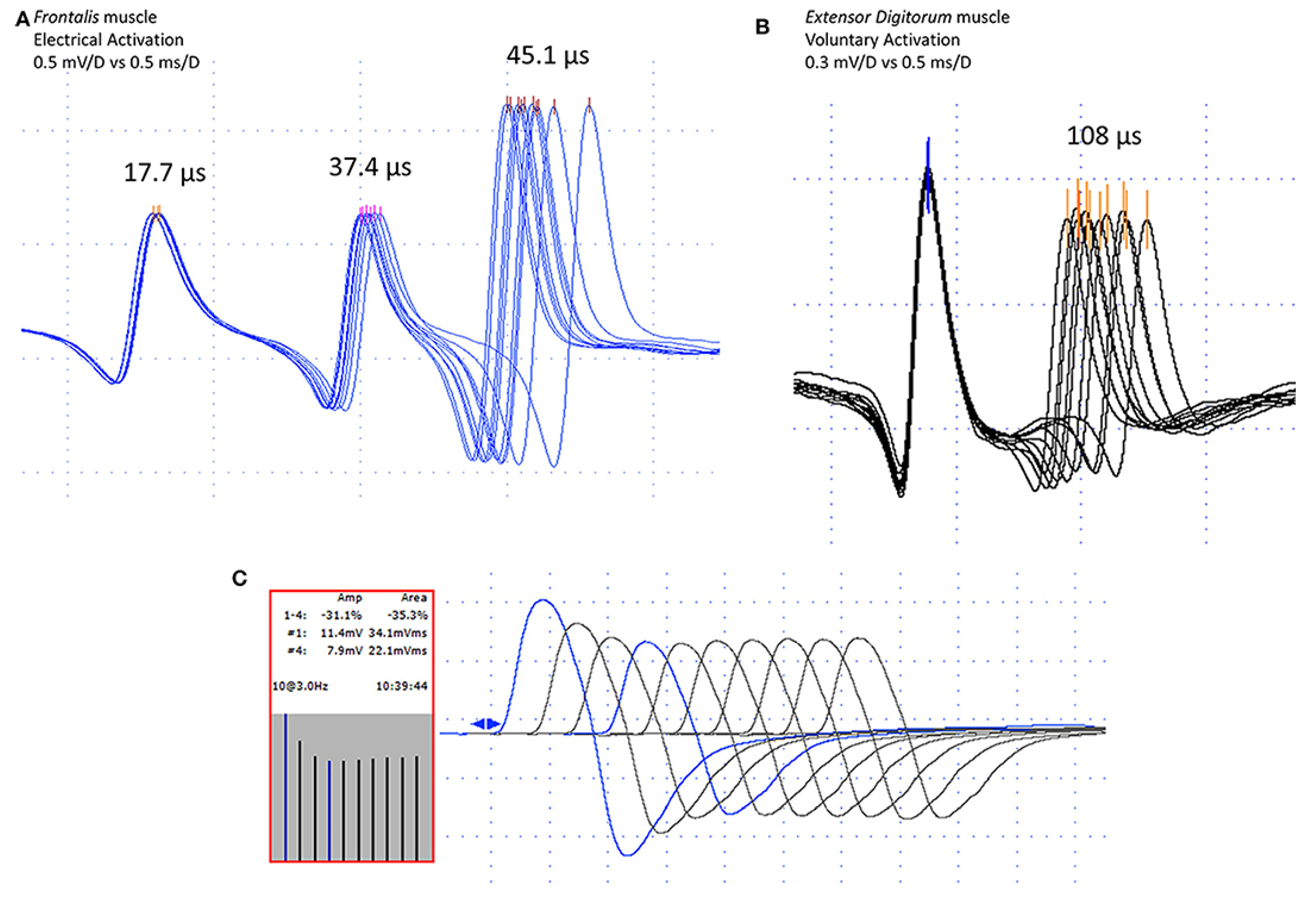{kind=link}
Abstract
1. Introduction
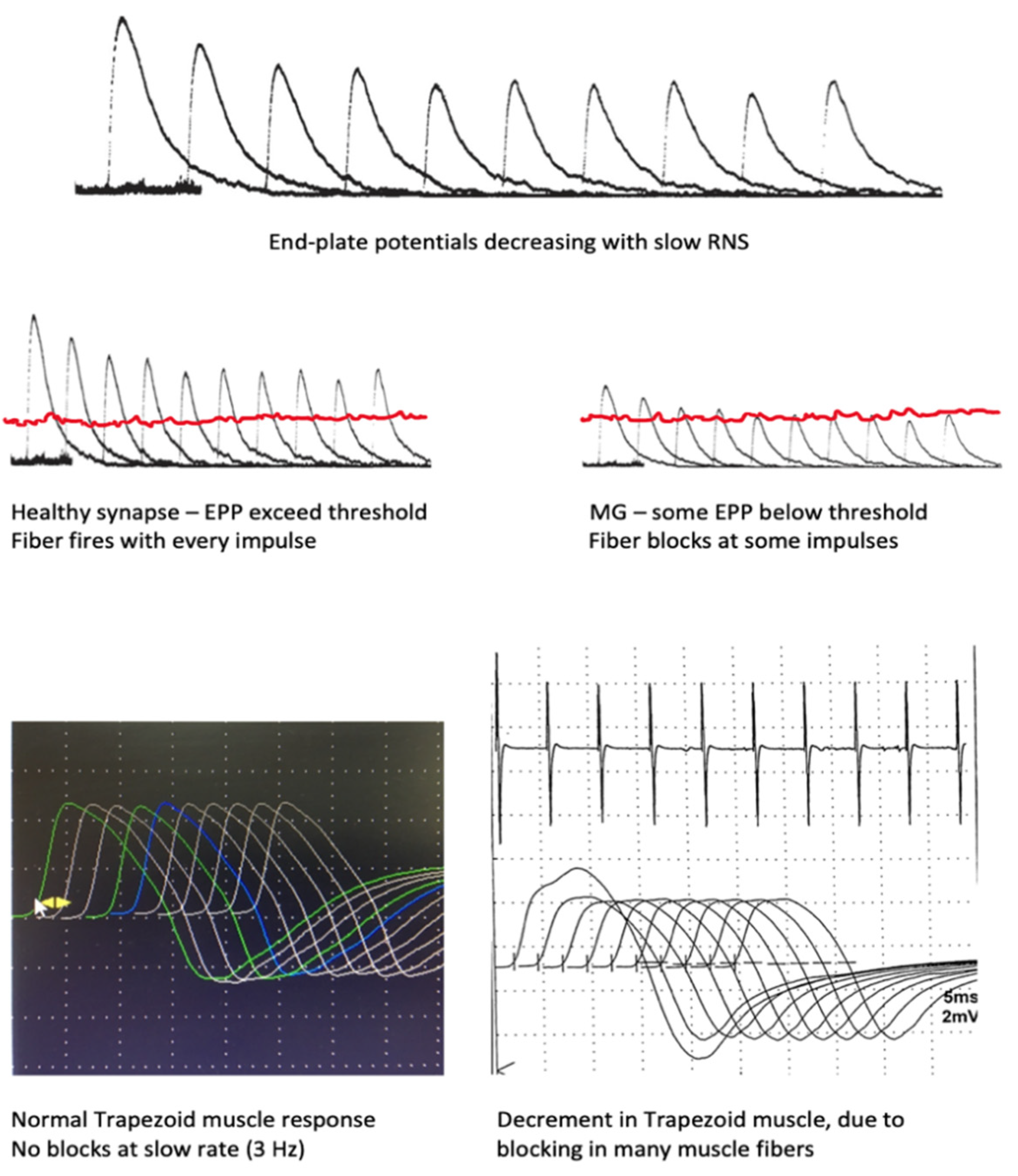
2. Neuromuscular Transmission
3. Antibodies in Diagnosis of Myasthenia Gravis
3.1. Anti-Acetylcholine Receptor Antibodies
3.2. Antibodies to MuSK
3.3. Antibodies to Anti-LRP4
3.4. Double-Seropositive Myasthenia Gravis
4. Pharmacologic Tests
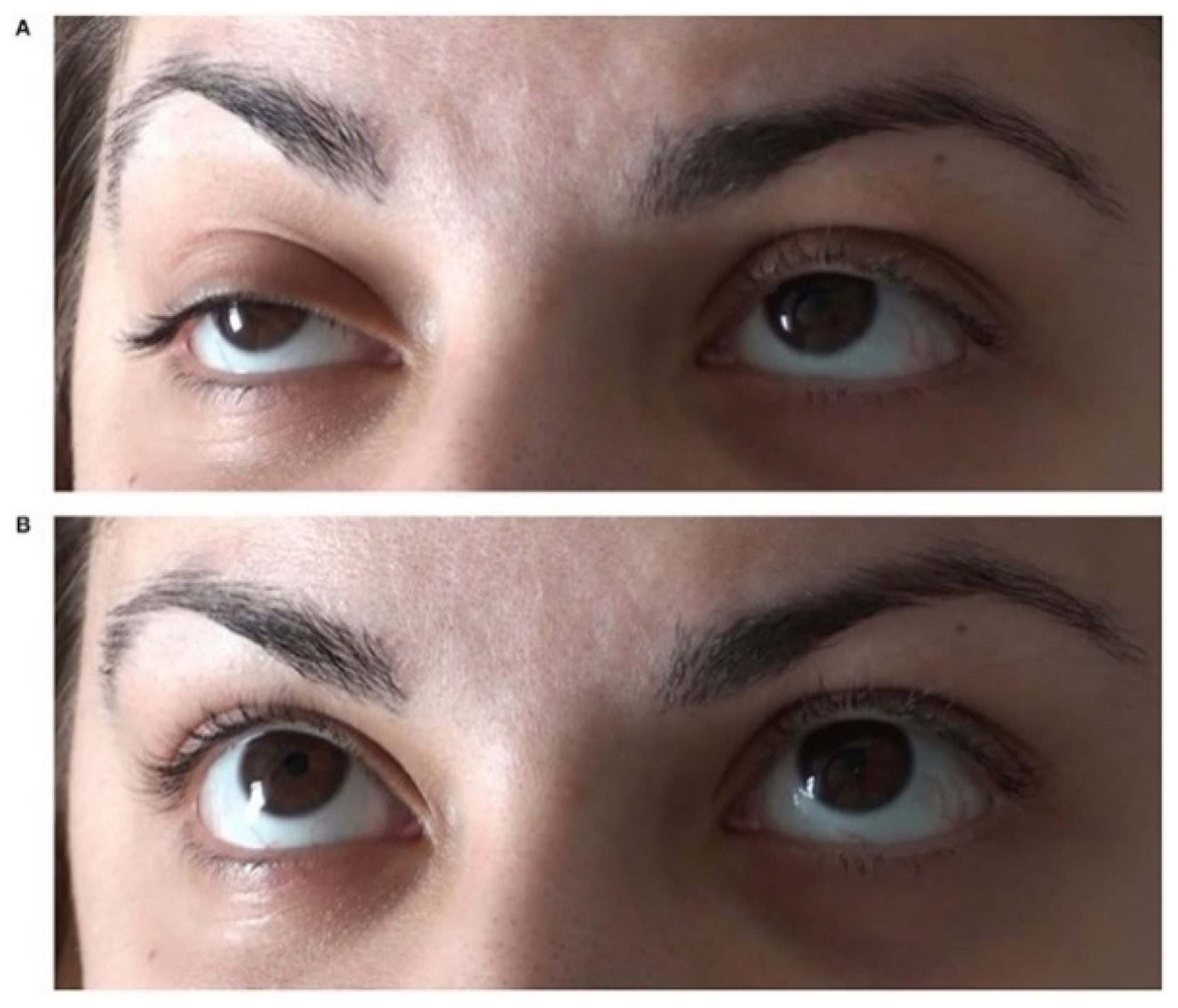
5. The Ice-Pack Test
6. Electrodiagnostic Studies
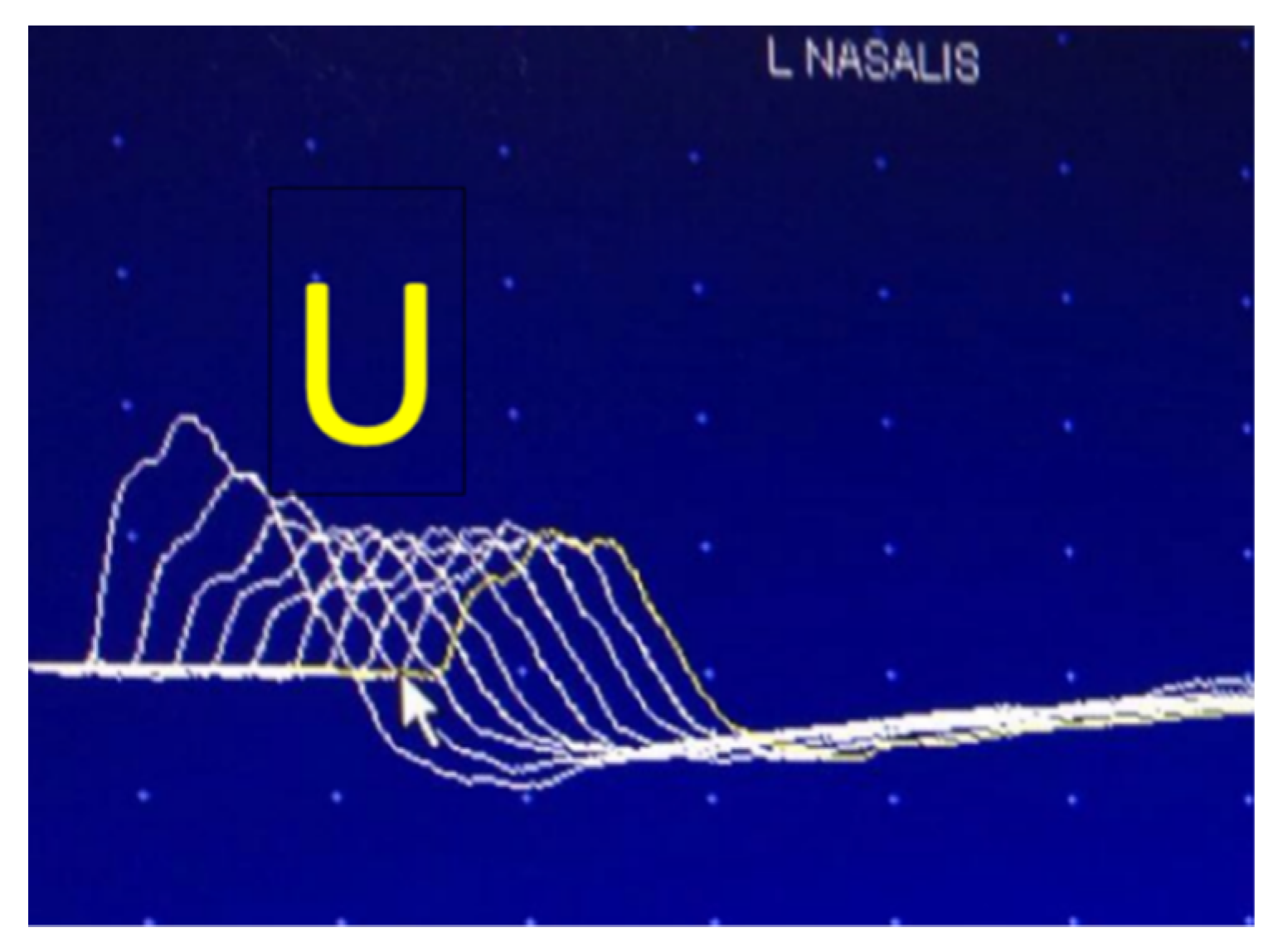
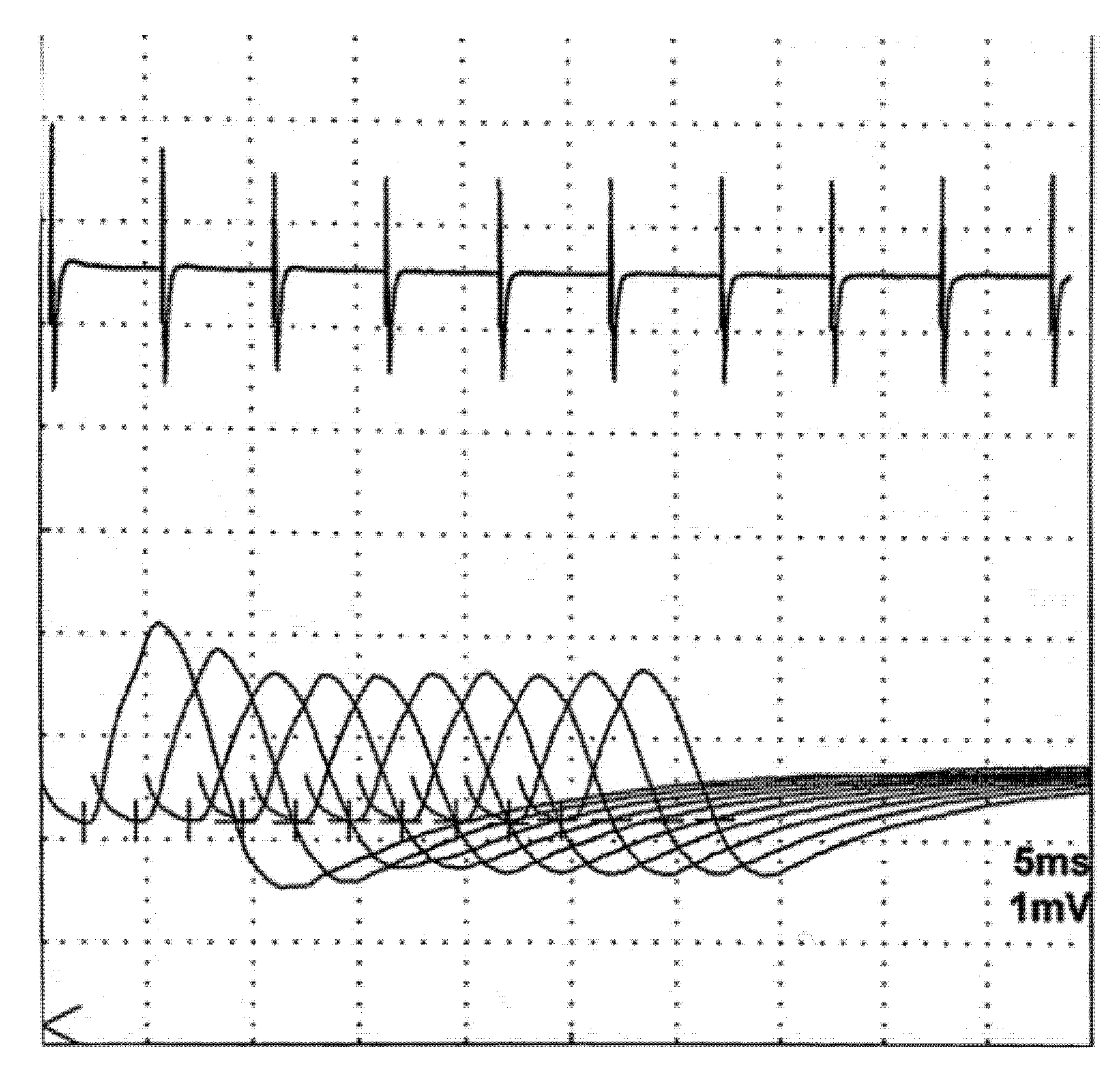
6.1. Repetitive Nerve Stimulation
6.2. Neuromuscular Jitter Study
7. Thymus Imaging
8. Diagnostic Approach
9. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vincent, A.; Huda, S.; Cao, M.; Cetin, H.; Koneczny, I.; Cruz, P.M.R.; Jacobson, L.; Viegas, S.; Jacob, S.; Woodhall, M.; et al. Serological and experimental studies in different forms of myasthenia gravis. Ann. N. Y. Acad. Sci. 2018, 1413, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Lazaridis, K.; Tzartos, S.J. Autoantibody Specificities in Myasthenia Gravis; Implications for Improved Diagnostics and Therapeutics. Front. Immunol. 2020, 11, 212. [Google Scholar] [CrossRef]
- Plomp, J.J.; Morsch, M.; Phillips, W.D.; Verschuuren, J.J. Electrophysiological analysis of neuromuscular synaptic function in myasthenia gravis patients and animal models. Exp. Neurol. 2015, 270, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Juel, V.C. Clinical neurophysiology of neuromuscular junction disease. Neurocutaneous Syndr. 2019, 161, 291–303. [Google Scholar] [CrossRef]
- Pasnoor, M.; Dimachkie, M.M.; Farmakidis, C.; Barohn, R.J. Diagnosis of Myasthenia Gravis. Neurol. Clin. 2018, 36, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Gilhus, N.E.; Tzartos, S.; Evoli, A.; Palace, J.; Burns, T.M.; Verschuuren, J.J.G.M. Myasthenia gravis. Nat. Rev. Dis. Prim. 2019, 5, 30. [Google Scholar] [CrossRef]
- Plomp, J.J. Neuromuscular junction physiology and pathophysiology. In Myasthenia Gravis and Related Disorders, 3rd ed.; Kaminski, H.J., Kushner, L.L., Eds.; Springer International: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Hoch, W.; McConville, J.; Helms, S.; Newsom-Davis, J.; Melms, A.; Vincent, A. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat. Med. 2001, 7, 365–368. [Google Scholar] [CrossRef]
- Higuchi, O.; Hamuro, J.; Motomura, M.; Yamanashi, Y. Autoantibodies to low-density lipoprotein receptor-related protein 4 in myasthenia gravis. Ann. Neurol. 2011, 69, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Meriggioli, M.N.; Sanders, D.B. Myasthenia Gravis: Diagnosis. Semin. Neurol. 2004, 24, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Rajput, R.; Sachdev, A.; Din, N.; Damato, E.M.; Murray, A. False positive acetylcholine receptor antibodies in a case of unilateral chronic progressive external ophthalmoplegia: Case report and review of literature. Orbit 2018, 37, 385–388. [Google Scholar] [CrossRef]
- Apiwattanakul, M.; McKeon, A.; Pittock, S.J.; Kryzer, T.J.; Lennon, V.A. Eliminating false-positive results in serum tests for neuromuscular autoimmunity. Muscle Nerve 2010, 41, 702–704. [Google Scholar] [CrossRef] [PubMed]
- Strijbos, E.; Verschuuren, J.J.; Kuks, J.B. Serum Acetylcholine Receptor Antibodies Before the Clinical Onset of Myasthenia Gravis. J. Neuromuscul. Dis. 2018, 5, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Fortin, E.; Cestari, D.M.; Weinberg, D.H. Ocular myasthenia gravis. Curr. Opin. Ophthalmol. 2018, 29, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Shelly, S.; Paul, P.; Bi, H.; Dubey, D.; Milone, M.; Sorenson, E.J.; Crum, B.A.; Laughlin, R.S.; Liewluck, T.; Mandrekar, J.; et al. Improving accuracy of myasthenia gravis autoantibody testing by reflex algorithm. Neurology 2020, 95, e3002–e3011. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.H.; Lachance, D.H.; Harper, C.M.; Lennon, V.A. Frequency of seronegativity in adult-acquired generalized myasthenia gravis. Muscle Nerve 2007, 36, 651–658. [Google Scholar] [CrossRef]
- Trakas, N.; Tzartos, S.J. Immunostick ELISA for rapid and easy diagnosis of myasthenia gravis. J. Immunol. Methods 2018, 460, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Bokoliya, S.; Patil, S.; Nagappa, M.; Taly, A. A Simple, Rapid and Non-Radiolabeled Immune Assay to Detect Anti-AChR Antibodies in Myasthenia Gravis. Lab. Med. 2019, 50, 229–235. [Google Scholar] [CrossRef]
- Devic, P.; Petiot, P.; Simonet, T.; Stojkovic, T.; Delmont, E.; Franques, J.; Magot, A.; Vial, C.; Lagrange, É.; Nicot, A.S.; et al. Antibodies to clustered acetylcholine receptor: Expanding the phenotype. Eur. J. Neurol. 2013, 21, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Koneczny, I.; Vincent, A. Myasthenia Gravis with Antibodies Against Muscle Specific Kinase: An Update on Clinical Features, Pathophysiology and Treatment. Front. Mol. Neurosci. 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Rodolico, C.; Bonanno, C.; Toscano, A.; Vita, G. MuSK-Associated Myasthenia Gravis: Clinical Features and Management. Front. Neurol. 2020, 11, 660. [Google Scholar] [CrossRef] [PubMed]
- Huda, S.; Waters, P.; Woodhall, M.; Leite, M.I.; Jacobson, L.; De Rosa, A.; Maestri, M.; Ricciardi, R.; Heckmann, J.M.; Maniaol, A.; et al. IgG-specific cell-based assay detects potentially pathogenic MuSK-Abs in seronegative MG. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e357. [Google Scholar] [CrossRef] [PubMed]
- Zisimopoulou, P.; Evangelakou, P.; Tzartos, J.; Lazaridis, K.; Zouvelou, V.; Mantegazza, R.; Antozzi, C.; Andreetta, F.; Evoli, A.; Deymeer, F.; et al. A comprehensive analysis of the epidemiology and clinical characteristics of anti-LRP4 in myasthenia gravis. J. Autoimmun. 2014, 52, 139–145. [Google Scholar] [CrossRef]
- Frykman, H.; Kumar, P.; Oger, J. Immunopathology of Autoimmune Myasthenia Gravis: Implications for Improved Testing Algorithms and Treatment Strategies. Front. Neurol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, Y.; Chen, J.; Huang, X.; Wang, H.; Li, Y.; Liu, W.; Feng, H. AChRAb and MuSKAb double-seropositive myasthenia gravis: A distinct subtype? Neurol. Sci. 2021, 42, 863–869. [Google Scholar] [CrossRef]
- Bokoliya, S.C.; Kumar, V.P.; Nashi, S.; Polavarapu, K.; Nalini, A.; Patil, S.A. Anti-AChR, MuSK, and LRP4 antibodies coexistence: A rare and distinct subtype of myasthenia gravis from Indian subcontinent. Clin. Chim. Acta 2018, 486, 34–35. [Google Scholar] [CrossRef] [PubMed]
- Gilhus, N.E.; Verschuuren, J.J. Myasthenia gravis: Subgroup classification and therapeutic strategies. Lancet Neurol. 2015, 14, 1023–1036. [Google Scholar] [CrossRef]
- Katz, B.; Miledi, R. The binding of acetylcholine to receptors and its removal from the synaptic cleft. J. Physiol. 1973, 231, 549–574. [Google Scholar] [CrossRef] [PubMed]
- Osserman, K.E.; Kaplan, L.I. Rapid diagnostic test for myasthenia gravis: Increased muscle strength, without fasciculations, after intravenous administration of edrophonium (tensilon) chloride. J. Am. Med Assoc. 1952, 150, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.; Rogozinski, S.; Puppe, W.; Framme, C.; Höglinger, G.; Hufendiek, K.; Wegner, F. Postinfectious Onset of Myasthenia Gravis in a COVID-19 Patient. Front. Neurol. 2020, 11, 576153. [Google Scholar] [CrossRef]
- Ing, E.B.; Ing, S.Y.; Ing, T.; Ramocki, J.A. The complication rate of edrophonium testing for suspected myasthenia gravis. Can. J. Ophthalmol. 2000, 35, 141–145. [Google Scholar] [CrossRef]
- Gould, L.; Zahir, M.; Gomprecht, R.F. Cardiac arrest during edrophonium administration. Am. Hear. J. 1971, 81, 437–438. [Google Scholar] [CrossRef]
- Evoli, A.; Padua, L. Diagnosis and therapy of myasthenia gravis with antibodies to muscle-specific kinase. Autoimmun. Rev. 2013, 12, 931–935. [Google Scholar] [CrossRef]
- Benatar, M. A systematic review of diagnostic studies in myasthenia gravis. Neuromuscul. Disord. 2006, 16, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Patil, S.A.; Bokoliya, S.C.; Nagappa, M.; Taly, A.B. Diagnosis of myasthenia gravis: Comparison of anti-nicotinic acetyl choline receptor antibodies, repetitive nerve stimulation and Neostigmine tests at a tertiary neuro care centre in India, a ten year study. J. Neuroimmunol. 2016, 292, 81–84. [Google Scholar] [CrossRef]
- Mittal, M.K.; Barohn, R.J.; Pasnoor, M.; McVey, A.; Herbelin, L.; Whittaker, T.; Dimachkie, M. Ocular Myasthenia Gravis in an Academic Neuro-Ophthalmology Clinic: Clinical Features and Therapeutic Response. J. Clin. Neuromuscul. Dis. 2011, 13, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Naji, A.; Owens, M.L. Edrophonium; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Evoli, A.; Antonini, G.; Antozzi, C.; DiMuzio, A.; Habetswallner, F.; Iani, C.; Inghilleri, M.; Liguori, R.; Mantegazza, R.; Massa, R.; et al. Italian recommendations for the diagnosis and treatment of myasthenia gravis. Neurol. Sci. 2019, 40, 1111–1124. [Google Scholar] [CrossRef] [PubMed]
- Sussman, J.; Farrugia, M.E.; Maddison, P.; Hill, M.; Leite, M.I.; Hilton-Jones, D. Myasthenia gravis: Association of British Neurologists’ management guidelines. Pr. Neurol. 2015, 15, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Borenstein, S.; Desmedt, J. Temperature and weather correlates of myasthenic fatigue. Lancet 1974, 304, 63–66. [Google Scholar] [CrossRef]
- Sethi, K.D.; Rivner, M.H.; Swift, T.R. Ice pack test for myasthenia gravis. Neurology 1987, 37, 1383. [Google Scholar] [CrossRef]
- Giannoccaro, M.P.; Paolucci, M.; Zenesini, C.; Di Stasi, V.; Donadio, V.; Avoni, P.; Liguori, R. Comparison of ice-pack test and single fiber EMG diagnostic accuracy in patients referred for myasthenic ptosis. Neurology 2020, 95. [Google Scholar] [CrossRef]
- Marinos, E.; Buzzard, K.; Fraser, C.L.; Reddel, S. Evaluating the temperature effects of ice and heat tests on ptosis due to Myasthenia Gravis. Eye 2018, 32, 1387–1391. [Google Scholar] [CrossRef] [PubMed]
- Pascuzzi, R.M. The History of Myasthenia Gravis. Neurol. Clin. 1994, 12, 231–242. [Google Scholar] [CrossRef]
- Wood, S.J.; Slater, C.R. Safety factor at the neuromuscular junction. Prog. Neurobiol. 2001, 64, 393–429. [Google Scholar] [CrossRef]
- Ruff, R.L.; Lennon, V.A. How myasthenia gravis alters the safety factor for neuromuscular transmission. J. Neuroimmunol. 2008, 201–202, 13–20. [Google Scholar] [CrossRef]
- Baslo, M.B.; Deymeer, F.; Serdaroglu, P.; Parman, Y.; Ozdemir, C.; Cuttini, M. Decrement pattern in Lambert–Eaton myasthenic syndrome is different from myasthenia gravis. Neuromuscul. Disord. 2006, 16, 454–458. [Google Scholar] [CrossRef]
- Preston, D.C.; Shapiro, B.E. Repetitive Nerve Stimulation. In Electromyography and Neuromuscular Disorders, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 52–61. [Google Scholar]
- Kimura, J. Repetitive Nerve Stimulation and Exercise Tests. In Electrodiagnosis in Diseases of Nerve and Muscle: Principles and Practice, 4th ed.; Oxford University Press (OUP): Oxford, UK, 2013; pp. 449–474. [Google Scholar]
- Oh, S.J. Distinguishing Features of the Repetitive Nerve Stimulation Test Between Lambert–Eaton Myasthenic Syndrome and Myasthenia Gravis, 50-Year Reappraisal. J. Clin. Neuromuscul. Dis. 2017, 19, 66–75. [Google Scholar] [CrossRef]
- Rubin, D.I.; Hentschel, K. Is exercise necessary with repetitive nerve stimulation in evaluating patients with suspected myasthenia gravis? Muscle Nerve 2006, 35, 103–106. [Google Scholar] [CrossRef]
- AAEM Quality Assurance Committee; American Association of Electrodiagnostic Medicine. Literature review of the usefulness of repetitive nerve stimulation and single fiber EMG in the electrodiagnostic evaluation of patients with suspected myasthenia gravis or Lambert-Eaton myasthenic syndrome. Muscle Nerve 2001, 24, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Chiou-Tan, F.Y.; Gilchrist, J.M. Repetitive nerve stimulation and single-fiber electromyography in the evaluation of patients with suspected myasthenia gravis or Lambert-Eaton myasthenic syndrome: Review of recent literature. Muscle Nerve 2015, 52, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.B.; Salort-Campana, E.; Grapperon, A.M.; Gallard, J.; Franques, J.; Sevy, A.; Delmont, E.; Verschueren, A.; Pouget, J.; Attarian, S. New strategy for improving the diagnostic sensitivity of repetitive nerve stimulation in myasthenia gravis. Muscle Nerve 2017, 55, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.-L.; Yin, H.-X.; Liu, M.-S.; Cui, L.-Y. Study on variation trend of repetitive nerve stimulation waveform in amyotrophic lateral sclerosis. Chin. Med. J. 2019, 132, 542–550. [Google Scholar] [CrossRef] [PubMed]
- Schumm, F.; Stöhr, M. Accessory nerve stimulation in the assessment of myasthenia gravis. Muscle Nerve 1984, 7, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Costa, J.; Evangelista, T.; Conceição, I.; de Carvalho, M. Repetitive nerve stimulation in myasthenia gravis—relative sensitivity of different muscles. Clin. Neurophysiol. 2004, 115, 2776–2782. [Google Scholar] [CrossRef] [PubMed]
- Rubin, D.I.; Harper, C.M.; Auger, R.G. Trigeminal nerve repetitive stimulation in myasthenia gravis. Muscle Nerve 2004, 29, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Mier, A.; Brophy, C.; Moxham, J.; Green, M. Repetitive stimulation of phrenic nerves in myasthenia gravis. Thorax 1992, 47, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Lamb, C.J.; Rubin, D.I. Sensitivity and specificity of repetitive nerve stimulation with lower cutoffs for abnormal decrement in myasthenia gravis. Muscle Nerve 2020, 62, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Abraham, A.; Breiner, A.; Barnett, C.; Katzberg, H.D.; Lovblom, L.E.; Rt, M.N.; Bril, V. Electrophysiological testing is correlated with myasthenia gravis severity. Muscle Nerve 2017, 56, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.H.; Kwon, S.B.; Kim, B.J.; Kim, B.J.; Kim, S.H.; Kim, J.K.; Park, K.S.; Park, K.J.; Sung, J.J.; Sohn, E.H.; et al. Prognosis of ocular myasthenia in Korea: A retrospective multicenter analysis of 202 patients. J. Neurol. Sci. 2008, 273, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Zhao, S.; Ren, K.; Dang, D.; Li, H.; Wu, F.; Zhang, M.; Li, Z.; Guo, J. Prediction of generalization of ocular myasthenia gravis under immunosuppressive therapy in Northwest China. BMC Neurol. 2020, 20, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ekstedt, J.E. Early struggles with single-fiber electromyography. Muscle Nerve 2002, 999, S5–S14. [Google Scholar] [CrossRef]
- Sanders, D.B. Clinical impact of single-fiber electromyography. Muscle Nerve 2002, 999, S15–S20. [Google Scholar] [CrossRef]
- Ertaş, M.; Baslo, M.B.; Yildiz, N.; Yazici, J.; Öge, A.E. Concentric needle electrode for neuromuscular jitter analysis. Muscle Nerve 2000, 23, 715–719. [Google Scholar] [CrossRef]
- Stålberg, E. Jitter analysis with concentric needle electrodes. Ann. N. Y. Acad. Sci. 2012, 1274, 77–85. [Google Scholar] [CrossRef]
- Juel, V.C. Single fiber electromyography. Neurocutaneous Syndr. 2019, 160, 303–310. [Google Scholar] [CrossRef]
- Gilchrist, J.M.; Massey, J.M.; Sanders, D.B. Single fiber EMG and repetitive stimulation of the same muscle in myasthenia gravis. Muscle Nerve 1994, 17, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Sonoo, M.; Uesugi, H.; Mochizuki, A.; Hatanaka, Y.; Shimizu, T. Single fiber EMG and repetitive nerve stimulation of the same extensor digitorum communis muscle in myasthenia gravis. Clin. Neurophysiol. 2001, 112, 300–303. [Google Scholar] [CrossRef]
- Padua, L.; Caliandro, P.; Di Iasi, G.; Pazzaglia, C.; Ciaraffa, F.; Evoli, A. Reliability of SFEMG in diagnosing myasthenia gravis: Sensitivity and specificity calculated on 100 prospective cases. Clin. Neurophysiol. 2014, 125, 1270–1273. [Google Scholar] [CrossRef]
- Caliandro, P.; Evoli, A.; Stålberg, E.; Granata, G.; Tonali, P.; Padua, L. The difficulty in confirming clinical diagnosis of myasthenia gravis in a seronegative patient: A possible neurophysiological approach. Neuromuscul. Disord. 2009, 19, 825–827. [Google Scholar] [CrossRef]
- Kouyoumdjian, J.A.; Graça, C.R.; Oliveira, F.N. Jitter Evaluation in Distant and Adjacent Muscles after Botulinum Neurotoxin Type A Injection in 78 Cases. Toxins 2020, 12, 549. [Google Scholar] [CrossRef] [PubMed]
- Mercelis, R. Abnormal Single-Fiber Electromyography in Patients Not Having Myasthenia: Risk for diagnostic confusion? Ann. N. Y. Acad. Sci. 2003, 998, 509–511. [Google Scholar] [CrossRef] [PubMed]
- Al-Hashel, J.Y.; Rousseff, R.T.; Khuraibet, A.J.; Tzvetanov, P. Single-Fiber Electromyography of Facial and Limb Muscles in Diabetic Patients with or without Neuropathy. J. Clin. Neurophysiol. 2014, 31, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Sanders, D.B.; Arimura, K.; Cui, L.; Ertaş, M.; Farrugia, M.E.; Gilchrist, J.; Kouyoumdjian, J.A.; Padua, L.; Pitt, M.; Stålberg, E. Guidelines for single fiber EMG. Clin. Neurophysiol. 2019, 130, 1417–1439. [Google Scholar] [CrossRef] [PubMed]
- Pitt, M.C. Use of stimulated electromyography in the analysis of the neuromuscular junction in children. Muscle Nerve 2017, 56, 841–847. [Google Scholar] [CrossRef]
- Priola, A. Imaging of thymus in myasthenia gravis: From thymic hyperplasia to thymic tumor. Clin. Radiol. 2014, 69, e230–e245. [Google Scholar] [CrossRef]
- Marx, A.; Pfister, F.; Schalke, B.; Saruhan-Direskeneli, G.; Melms, A.; Ströbel, P. The different roles of the thymus in the pathogenesis of the various myasthenia gravis subtypes. Autoimmun. Rev. 2013, 12, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Nacu, A.; Andersen, J.B.; Lisnic, V.; Owe, J.F.; Gilhus, N.E. Complicating autoimmune diseases in myasthenia gravis: A review. Autoimmunity 2015, 48, 362–368. [Google Scholar] [CrossRef] [PubMed]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rousseff, R.T. Diagnosis of Myasthenia Gravis. J. Clin. Med. 2021, 10, 1736. https://doi.org/10.3390/jcm10081736
Rousseff RT. Diagnosis of Myasthenia Gravis. Journal of Clinical Medicine. 2021; 10(8):1736. https://doi.org/10.3390/jcm10081736
Chicago/Turabian StyleRousseff, Rossen T. 2021. "Diagnosis of Myasthenia Gravis" Journal of Clinical Medicine 10, no. 8: 1736. https://doi.org/10.3390/jcm10081736
APA StyleRousseff, R. T. (2021). Diagnosis of Myasthenia Gravis. Journal of Clinical Medicine, 10(8), 1736. https://doi.org/10.3390/jcm10081736