1. Introduction
Increasing interest has been directed over recent decades at the use of lignin, the most abundant natural source of aromatic polymers, as a feedstock for the production of renewable chemicals and fuels [
1]. Lignin is currently treated as a by-product in many different processes, such as the Kraft process, the soda process, the sulphite process, and the second-generation bioethanol bio-refinery process. The Kraft process is the dominant chemical pulping process used worldwide in the paper and pulp industries, NaOH and Na
2S being used in that context as chemicals for digesting lignocellullosic materials in order to obtain cellulose fibers. Around 130 million tons of Kraft pulp are generated annually [
2]. During the Kraft process, more than 90% of the lignin is degraded into fractions and is dissolved in alkali solutions. It thus represents one of the major constituent of Kraft black liquor. To date, the only use of Kraft lignin in black liquor is to be burned as an internal fuel for the recovery of pulping-reagents and for power generation [
3]. However, for a modern Kraft pulp mill using energy-efficient equipment, the high level of internal energy recovery obtained leads to a stream surplus that limits the capacity of the recovery boiler. The extraction of 25–50% of the lignin does not disturb milling operations, and it can provide the opportunity for increasing the milling capacity [
4]. Thus, the use of lignin obtained from black liquor as a raw material for producing valuable products is potentially as significant as the use of fibers for producing pulp and paper in modern Kraft mills [
5].
Two of the main approaches to extracting lignin from the Kraft black liquor are those of acid precipitation and ultrafiltration. The acid precipitation of lignin from the black liquor through use of sulfuric acid or some other acid and carbon dioxide is the most common and easiest process [
6,
7]. For example, the LignoBoost process has currently been a commercially available process for the production of lignin from the black liquor [
8]. The main disadvantage of acid precipitation is that the use of large amounts of acidification chemicals is required. In addition, the sodium and other impurity contents of lignin during the process are very high, so more water or acid is needed for washing it than would otherwise be required [
8]. Moreover, the use of sulfuric acid for acidification would disturb the balance between sodium and sulfur and increase the need to withdraw sulfates together with fly-ash from the recovery boiler fly-ash, which would seriously affect the recovery of cooking chemicals [
9]. In previous research, this was solved by membrane filtration of the solution so as to separate the lignin from the pulping chemicals prior to acidification [
10]. This was accomplished through the use of ultrafiltration or nanofiltration membranes for reducing the concentration of the pulping chemicals and increasing the concentration of lignin in the black liquor [
10]. The black liquor that was studied contained lignin with a molecular weight of less than 10 kDa, this resulting in the authors selecting membranes in the molecular weight cut-off (MWCO) region of 1000 to 200 Da. Lignin retentions of greater than 80% have been achieved with use of maximal initial fluxes of around 200 L/m
2h. The densest membranes had a high retention of ash, which led to the authors choosing the membrane with the highest MWCO levels for further studies. Other studies have shown that the separation of lignin into low and high molecular weight fractions is possible through use of ultrafiltration membranes [
11]. The fractions obtained had lignin that differed in molecular weight but had the same main structure, as evidenced by nuclear magnetic resonance (NMR) and Fourier-transform infrared spectroscopy (FTIR) results. Further results were obtained by Padilla et al. [
12], where successful fractionation of the lignin was achieved through use of a series of ultrafiltration membranes with varying MWCO (100, 30, 10, 5, and 1 kDa). The lignin obtained showed an increase in surface activity as the molecular weight increased due to the decreasing hydrophilic groups. The study also showed that the lignin of high molecular weight had a strong impact on the viscosity of the solution, which in turn could possibly affect the membrane filtration capacity. Kraft lignin was also fractionated in another study through use of sequential ultrafiltration membranes with different MWCO [
13]. The different fractions contained lignin that was more homogeneous in terms of molecular weight and polydispersity than that in non-filtrated specimens. The authors have also shown that the effects on the lignin of various functional groups, differing, for example, in the quantity of the hydroxyl groups that are present, is a function of the molecular weights. A fractionation of wheat straw and Sarkanda grass lignin in a soda pulp process was performed by Allegretti et al. [
14]. Their results showed a multi-step filtration process that included microfiltration and ultrafiltration to yield different lignin fractions of varying molecular weight. The high-molecular-weight fractions were proposed as being suitable as a starting material for further depolymerization of the lignin.
In addition to the structural complexity of lignin, which represents a common challenge in the utilization of lignin, the high level of condensation that occurs during the Kraft process increases the recalcitrance of Kraft lignin [
2]. To break down different lignin bonding motifs into small molecules, use of appropriate depolymerization approaches is essential. In recent years, efforts have been made to depolymerize Kraft lignin that has been extracted through acidification into low-molecular-mass compounds (LMMC). Mahmood et al. reported the depolymerization of softwood Kraft lignin at 250 °C for 45–90 min with use of NaOH as a catalyst, and this resulted in a yield of 80–90% depolymerized lignin compounds having a molecular weight ranging from 3 to 5 kDa [
15]. The LignoBoost Kraft lignin was depolymerized at 290–370 °C in a near-critical water medium with use of ZrO2/K2CO3 and phenol as catalysts, and 69–87% bio-oil, 5–11% water-soluble organics, and 16–22% char were obtained after depolymerization [
16]. During the lignin depolymerization process, repolymerization (or condensation) is a common and severe problem. The repolymerization mechanism can possibly be that of the electron-rich aromatic rings being easily attacked by benzyl carbocations, leading to C-C bond formation [
17]. The formation of high molecular weight insoluble structures and char is the consequence of the repolymerization that occurs. In order to suppress repolymerization, we have developed a novel continuous flow reactor (CFR) to depolymerize ‘Indulin AT’ (a commericial Kraft lignin that was precipitated from the black liquor of softwood pulp) at 170–240 °C for 1–4 min of residence time by using NaOH as a catalyst [
18]. We found that, under the experimental conditions, some of the Indulin AT macromolecules were depolymerized into LMMC.
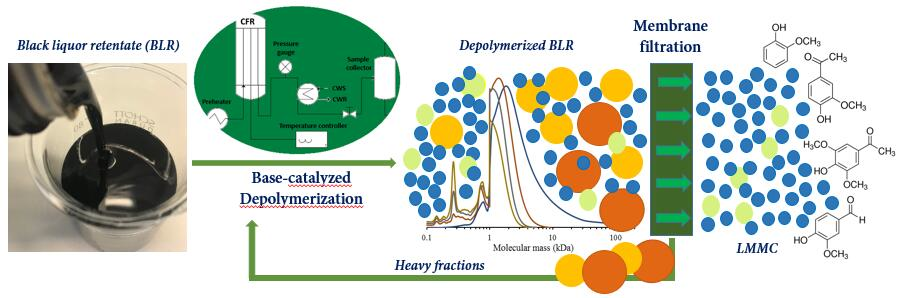
For Kraft lignin samples obtained from a membrane separation process conducted at a high pH level, the internal reagent NaOH represents a desirable catalyst for depolymerizing it to obtain LMMC. In our previous depolymerization study, most of the lignin macromolecules could not yet be depolymerized under these mild conditions. Increasing the temperature and the reaction time, or repeating depolymerization by recycling the product stream, could be effective in improving yields. At the same time, the repolymerization reactions obtained could be more severe. Using a suitable membrane to separate the LMMC from depolymerized lignin samples could be an effective option for further depolymerizing the lignin macromolecules and, at the same time, minimizing the repolymerization that occurs. To the best of our knowledge, few additional reports have been made regarding the continuous ultrafiltration and depolymerization of lignin for producing LMMC. The aim of this study was to develop a process that combined base-catalyzed continuous-flow depolymerization and membrane separation, so as to increase the yield of high-value aromatic compounds originating from the Kraft lignin. The feedstock employed in this study was black liquor retentate (BLR) obtained from a membrane filtration pilot plant. The depolymerization was performed within the temperature range of 170–210 °C, at a pressure level of 120–140 bar, and with a 2 min residence time. NaOH was the only catalyst employed in the study. The GR95PP membrane was used for the separation of LMMC after each depolymerization of the BLR samples. SEC, 2D HSQC NMR, and HPLC were used to analyze the lignin products that were obtained.
2. Materials and Methods
2.1. Materials
The BLR used in this work was obtained from a membrane filtration pilot plant situated in a pulp and paper mill in northern Sweden. The black liquor was a mixture from the softwood (70%) and hardwood (30%). The black liquor had been filtered continuously at 110 °C by a 1 kDa cut-off ceramic membrane. After membrane separation had taken place, BLR was obtained as a retentate containing 32.9% total solids, 22.4% total lignin, and 6.5% ash.
The total dry solid (TDS) content of each of the BLR samples was determined from the difference in weight before and after drying the BLR in a weighted porcelain crucible at 105 °C for 24 h. After drying, the samples were heated to 575 °C and were kept at that temperature for 3 h. The ash content of the samples was determined by weighing the residue again after it had been cooled to room temperature in a desiccator. The total amount of lignin was determined by measuring the UV absorbance at 280 nm by a spectrophotometer (UV-160, manufactured by the Shimadzu Corp., Kyoto, Japan). The absorption coefficient used was 24.6 g L
−1 cm
−1 [
10]. The samples were diluted by a 0.5% NaOH solution. The content of polysaccharides and of Klason lignin was determined after a standard two-step acid hydrolysis of the samples, in line with NREL procedures [
19]. The monomeric sugar concentration was measured using high-performance anion-exchange chromatography coupled with pulsed amperometric detection in an ICS-3000 chromatography system (Dionex Corp., Sunnyvale, USA), running at 30 °C. The sugars were separated using a CarboPac PA 1 column containing deionized water as an eluent, 200 mM of sodium hydroxide being added post-column at flow rates of 1.0 and 0.5 mL/min, respectively. The injection volume was 10 µL, and standard solutions of D-galactose, D-glucose, D-mannose, D-xylose, and L-arabinose (Fluka Chemie AG, Buchs, Swizerland) were employed. The amounts of the hemicelluloses involved were determined after anhydro-corrections of 0.9 and 0.88 for the hexoses and the pentoses respectively. Sodium hydroxide, hydrochloric acid (37%), DMSO-
d6, ethyl acetate and all the other chemicals employed were purchased from Sigma-Aldrich Sweden AB.
2.2. Depolymerization of BLR
The base-catalyzed BLR depolymerization experiments were performed in a continuous flow reactor (CFR) setup. The BLR was diluted 5 times by a 2 wt % NaOH solution to obtain a solution with 5 wt. % lignin, and was filtered prior to the depolymerization experiments. The experimental setup was described in greater detail in an earlier work [
18]. It is composed of an HPLC pump, a tubular CFR reactor, a filter, and a pressure control valve. A tubular preheater with a volume of 28 mL (out of which 6 mL is included in the reactor volume) was connected to the reactor (with a volume of 14 mL) beforehand to gradually preheat the sample to the required temperature. After the reaction had taken place, the sample stream was quickly condensed by a water bath that was connected to it, located just after the reactor.
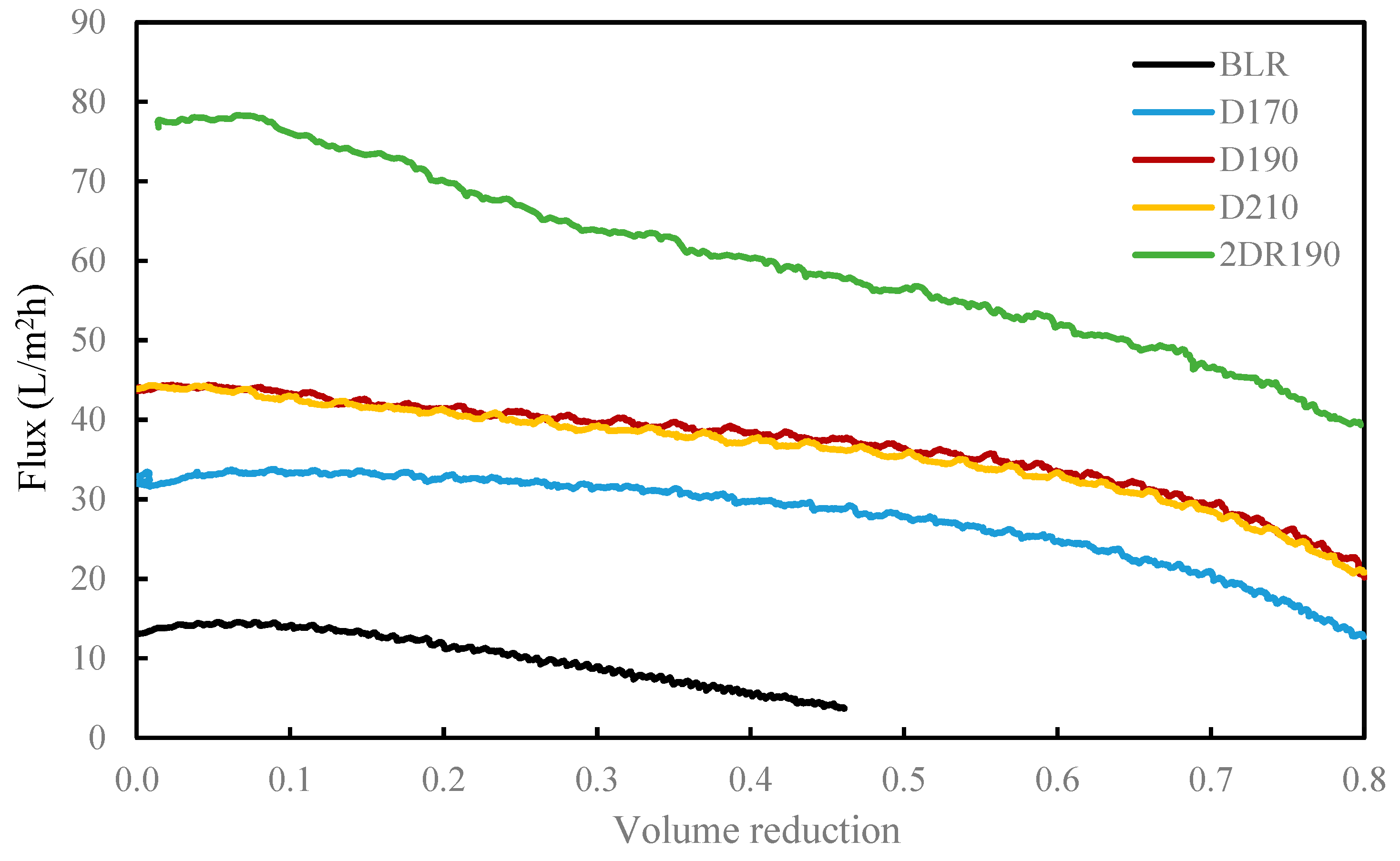
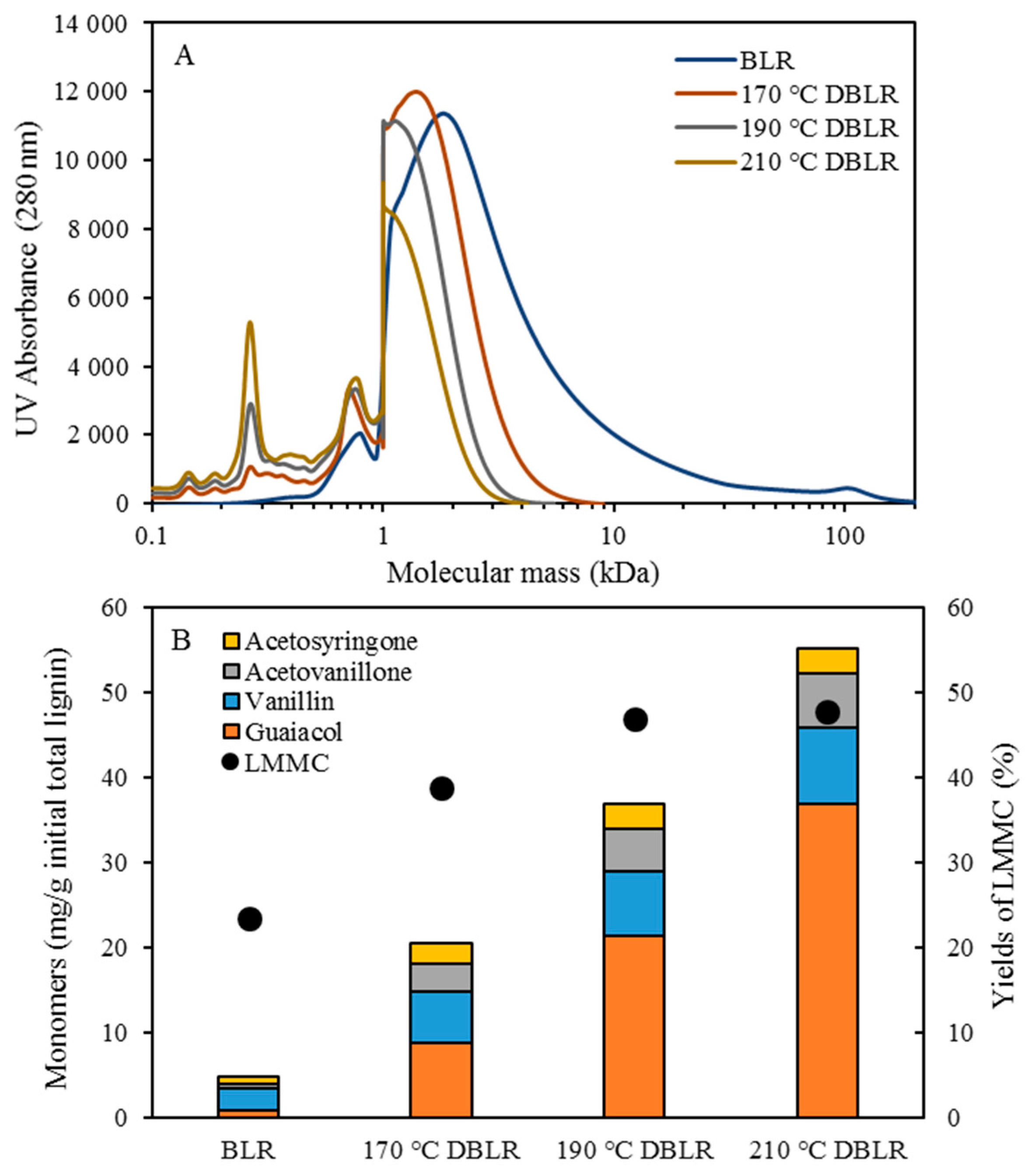
Prior to starting each depolymerization experiment, deionized water was used as the feedstock. When the system was heated up and was pressurized to operating conditions, the BLR sample was pumped continuously through the reactor at a flow rate of 10 ml/min. On the basis of the volume of interest in the preheater and the volume of the reactor, a flowrate of 10 mL/min corresponded to 2 min of residence time. In the present study, the experiments were performed at temperatures ranging from 170 to 210 °C, for 10 °C intervals, at pressures of around 130 bar. The experimental parameters and the conditions employed are summarized in
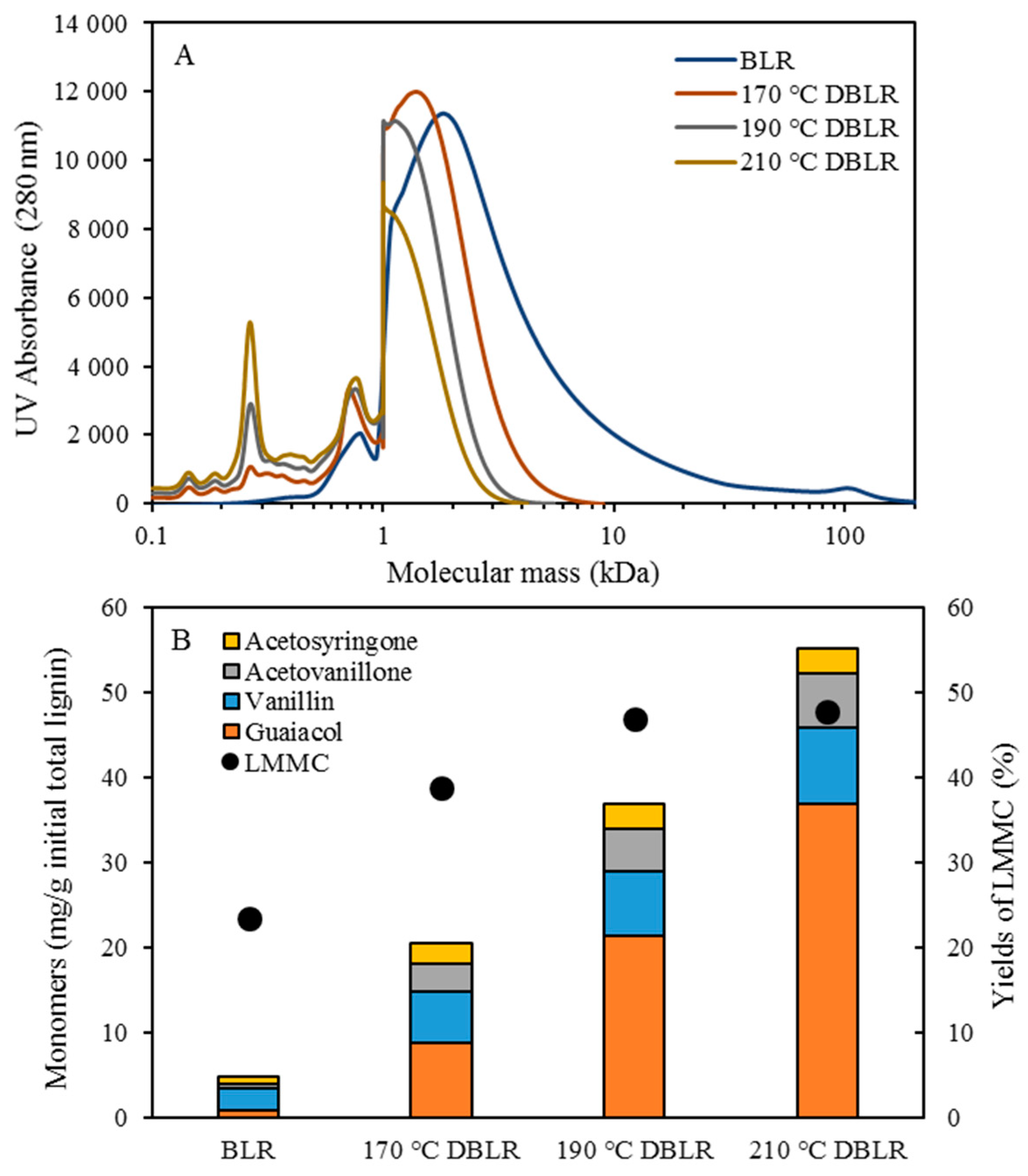
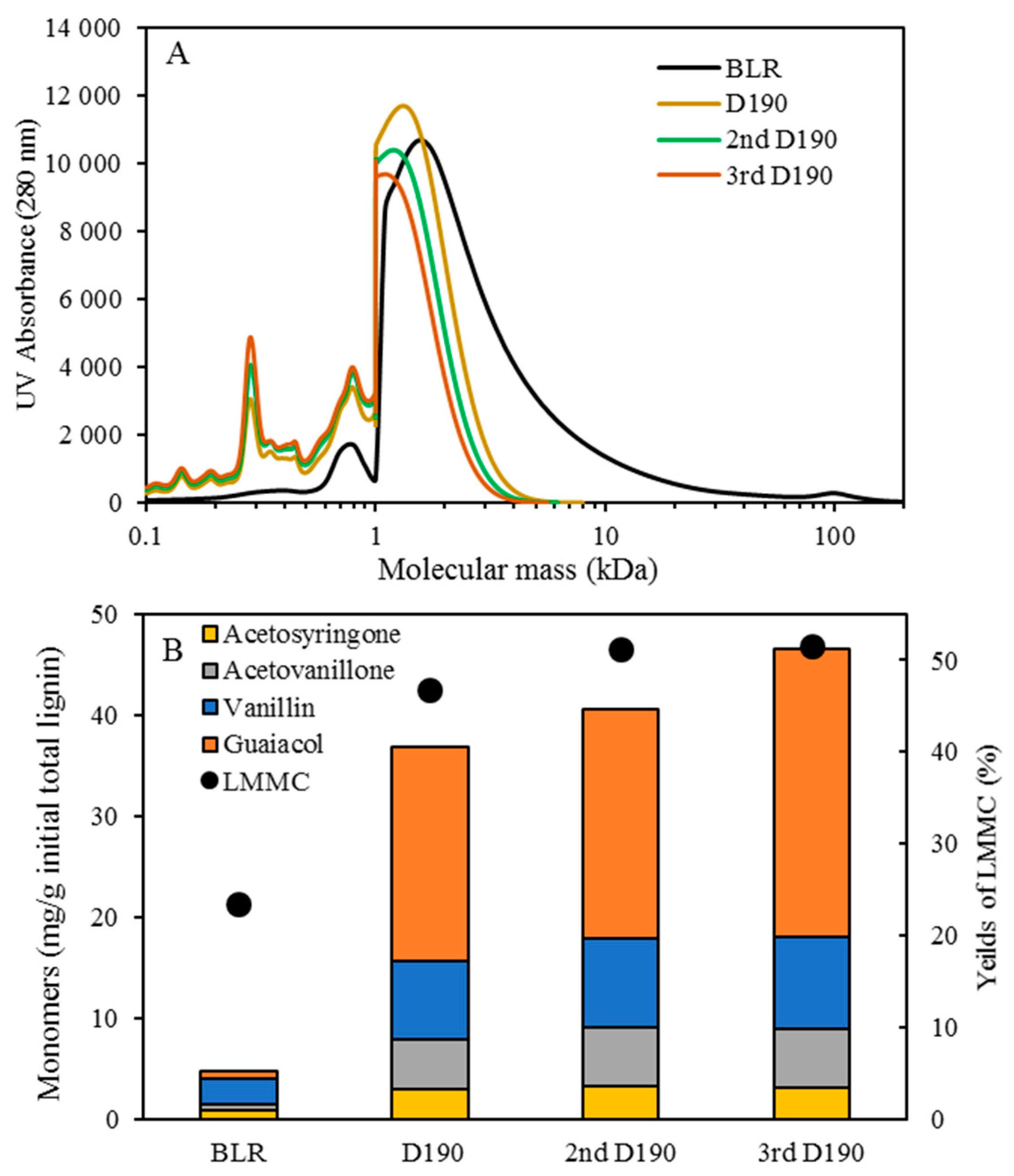
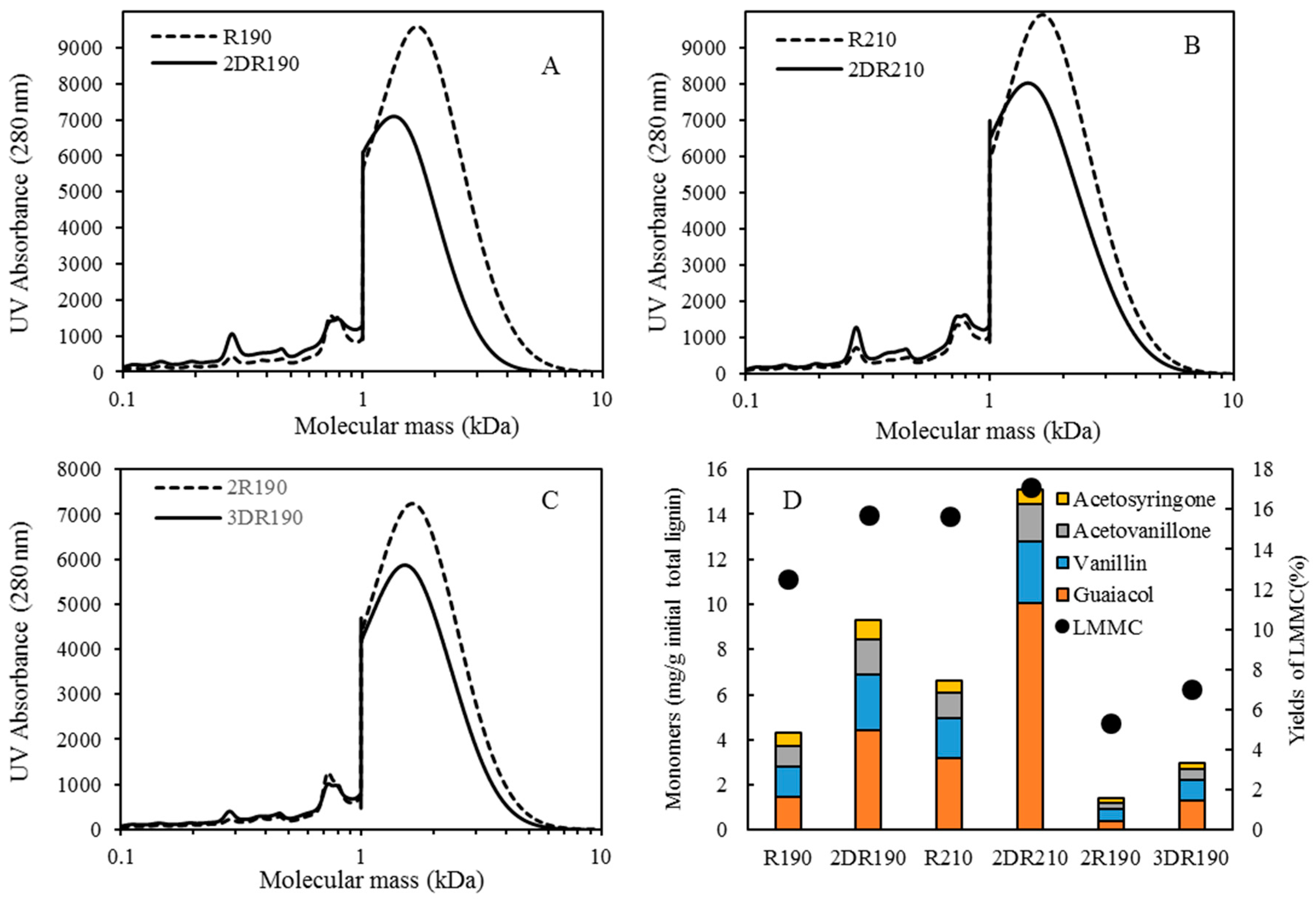
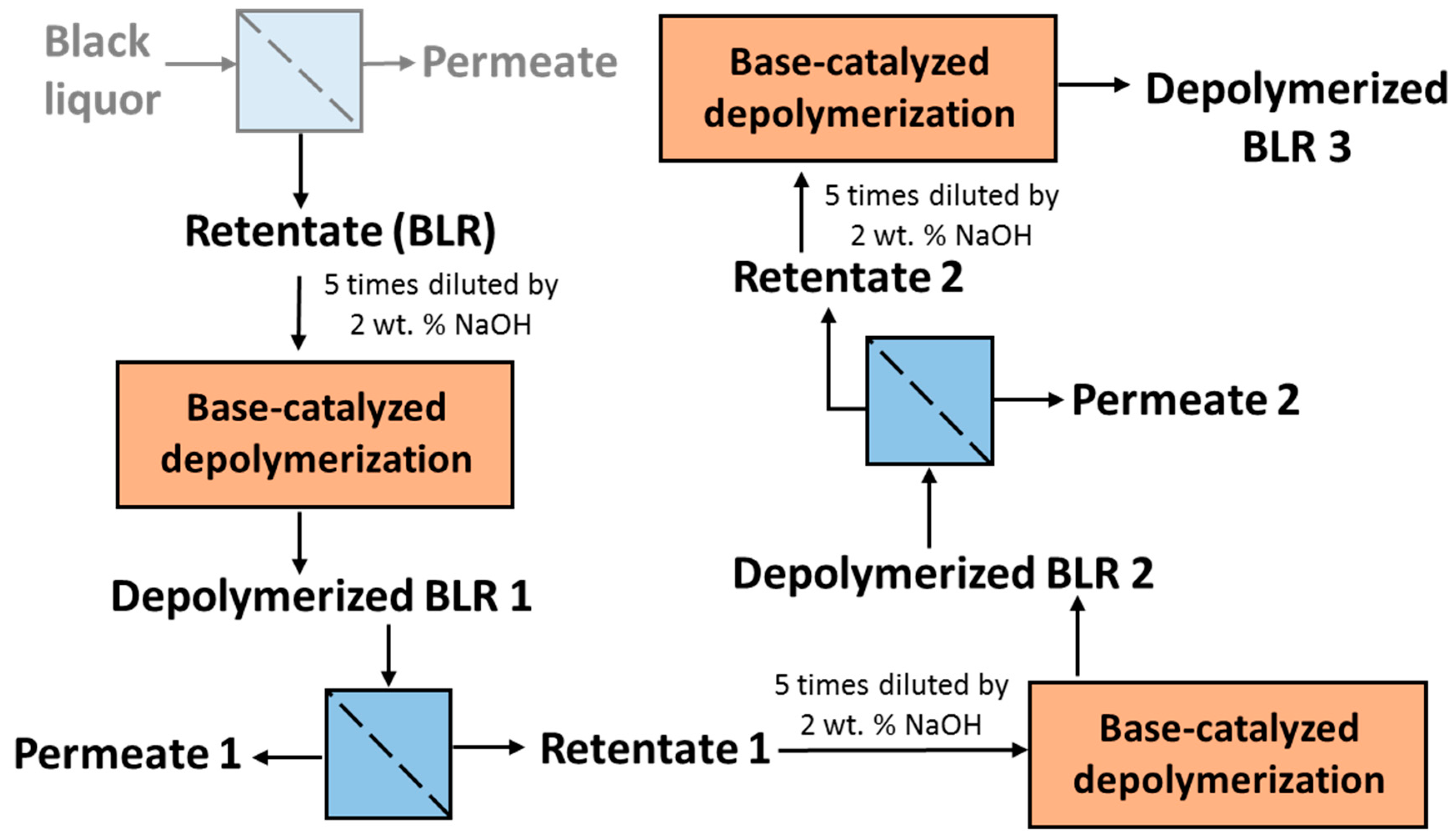
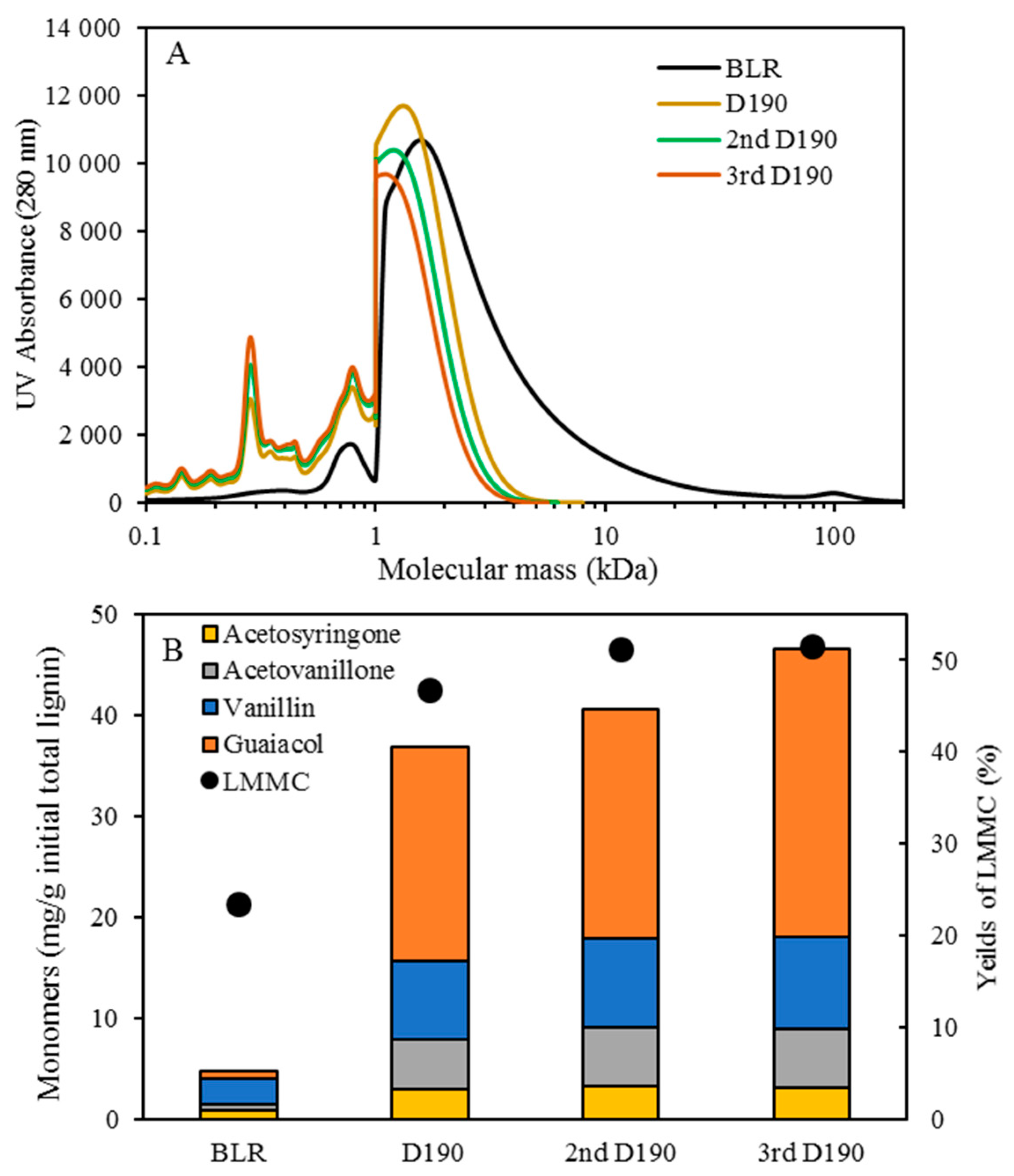
Table 1. After the reactions had taken place, the 170–210 °C depolymerized BLR products were collected continuously and were stored at 4 °C for analysis and for further use. The 190 °C depolymerized BLR samples were selected for a second and a third recycling depolymerization under the same conditions to check the effects of the number of depolymerization times on LMMC production. Samples D190, 2D190, and 3D190 were obtained.
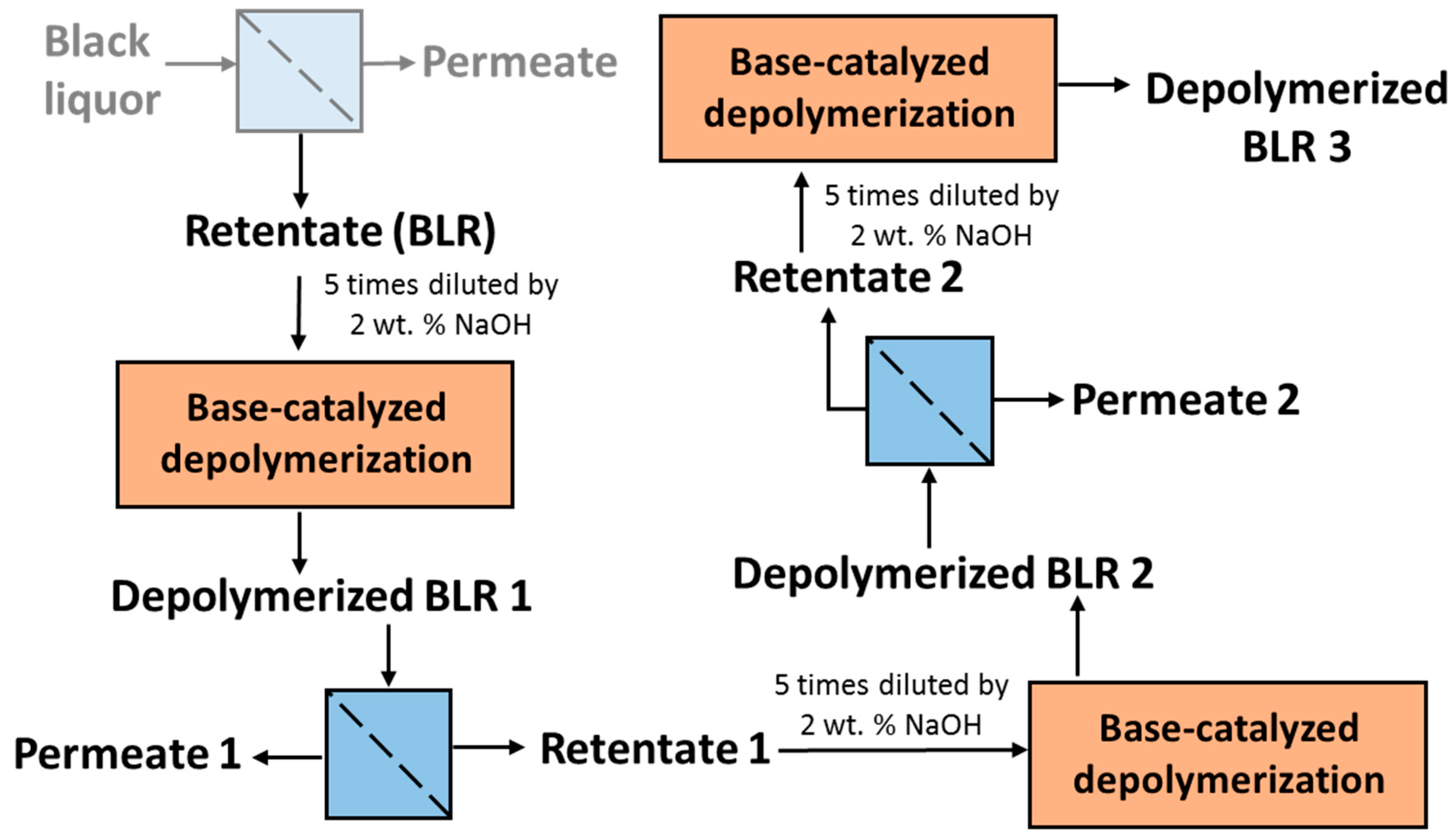
2.3. Membrane Filtration and the Depolymerization Recycling
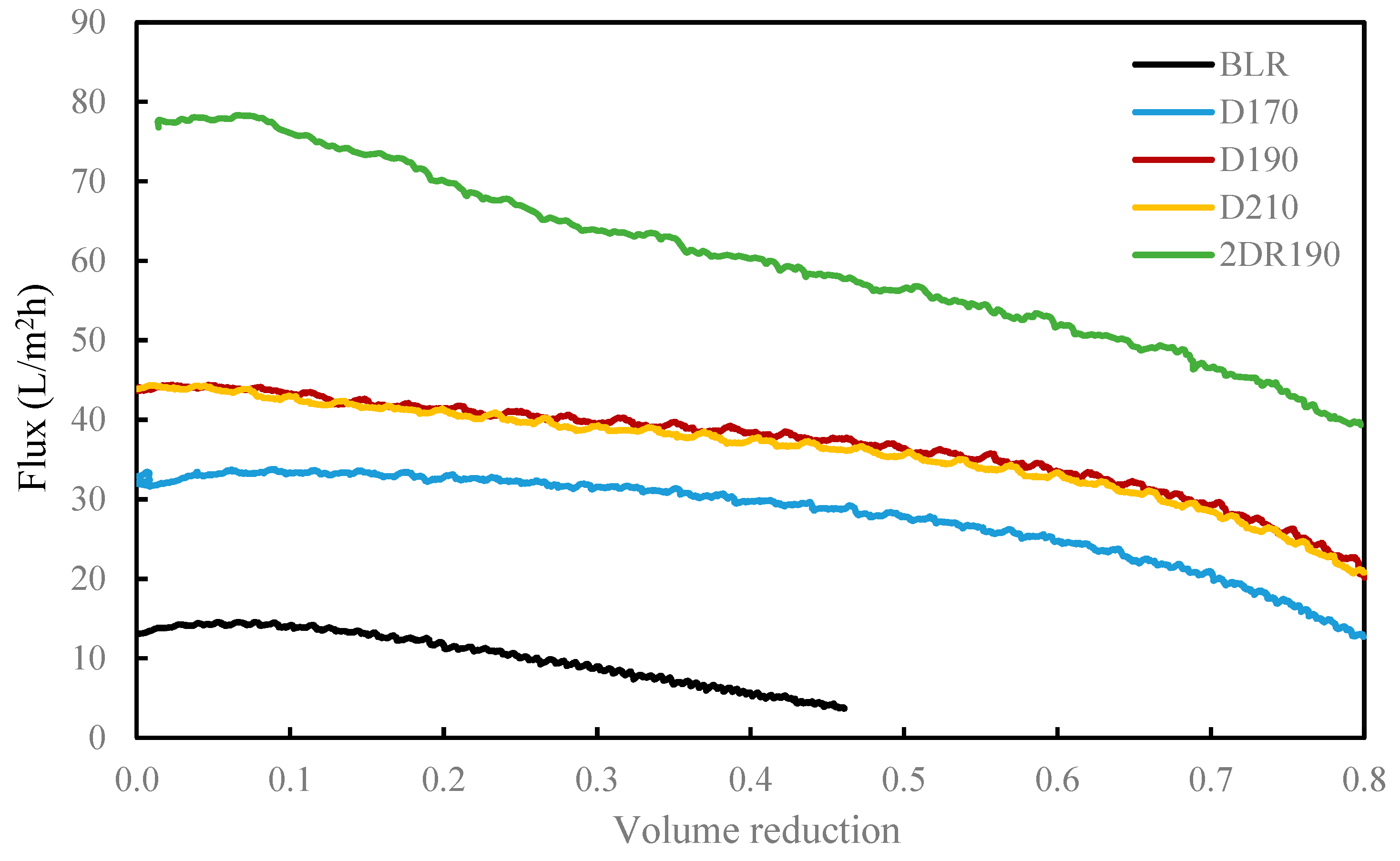
In order to separate the LMMC from the depolymerized BLR, use was made of a membrane filtration step. The type of membrane involved was an Alfa Laval membrane GR95PP, a polymeric membrane having an MWCO value of 2 kDa. The new membrane was washed at room temperature with a 4 g/L of sodium hydroxide solution prior to the diafiltration of BLR samples, in accordance with procedures described by Al-Rudainy et al. [
20]. The BLR lignin samples were diafiltrated at constant temperature, transmembrane pressure and diafiltration factor values of 50 °C, 5.5 bar, and 5 g/g in a stirred pressure vessel at a calculated cross-flow velocity of 0.5 m/s [
20].
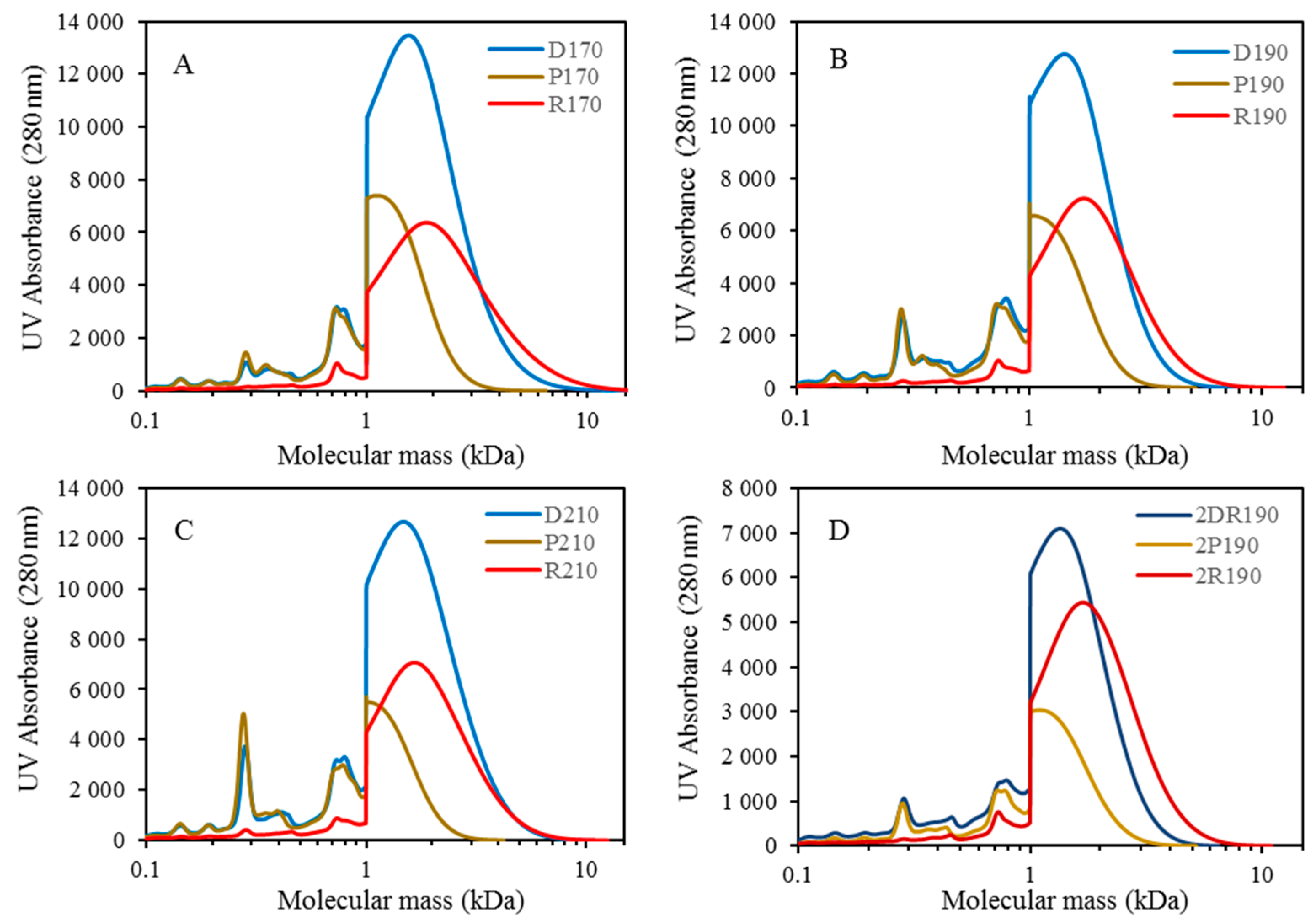
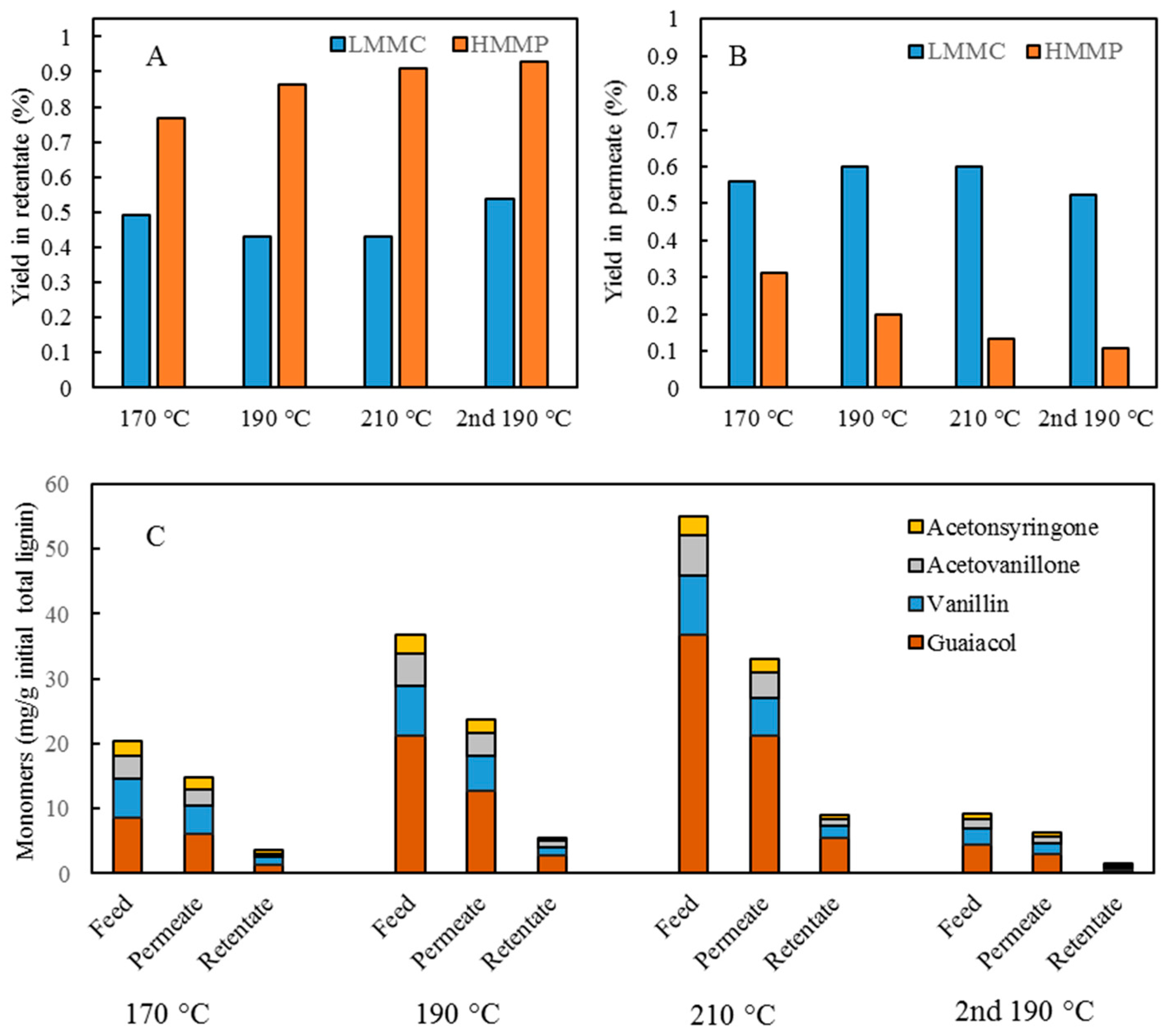
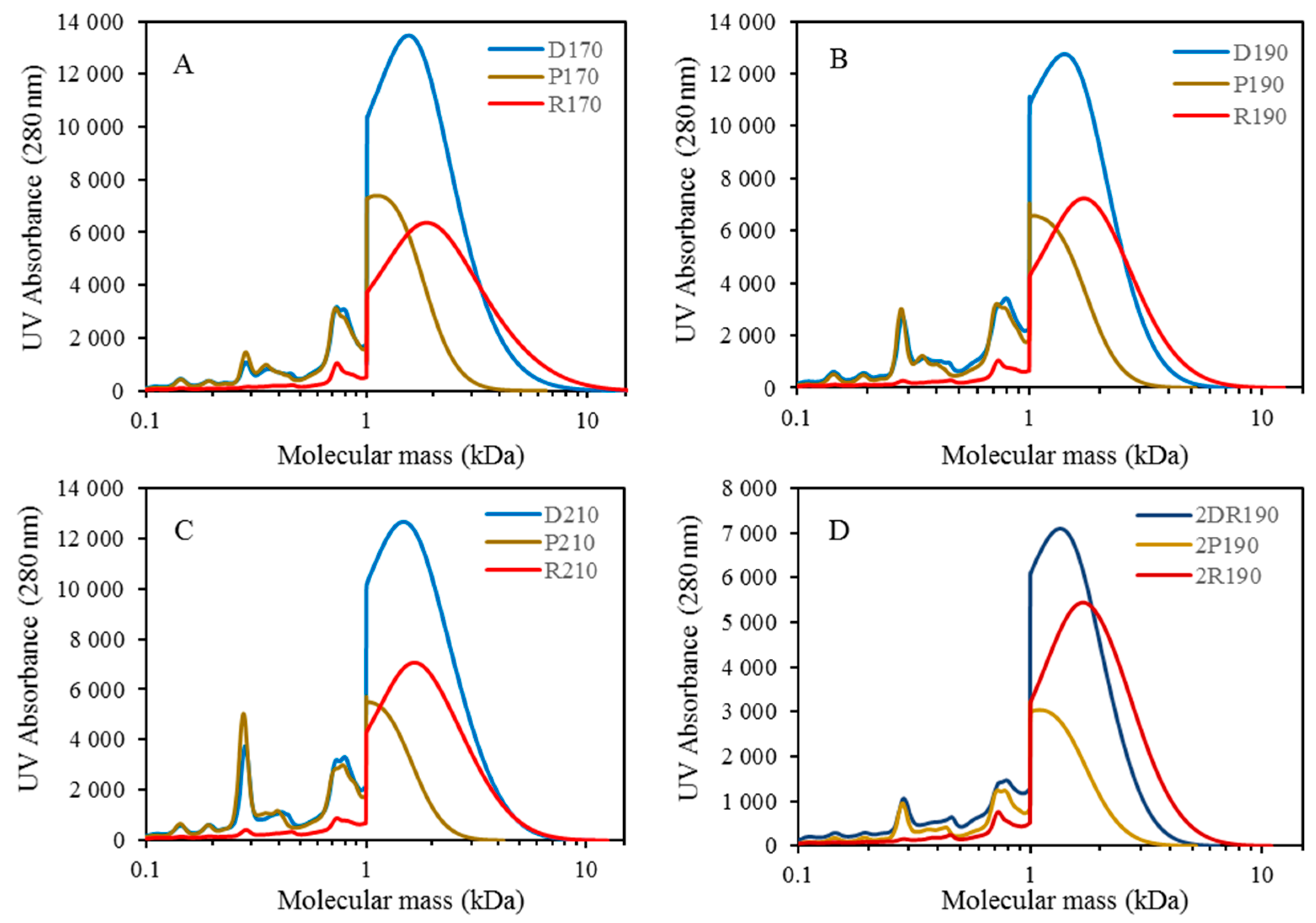
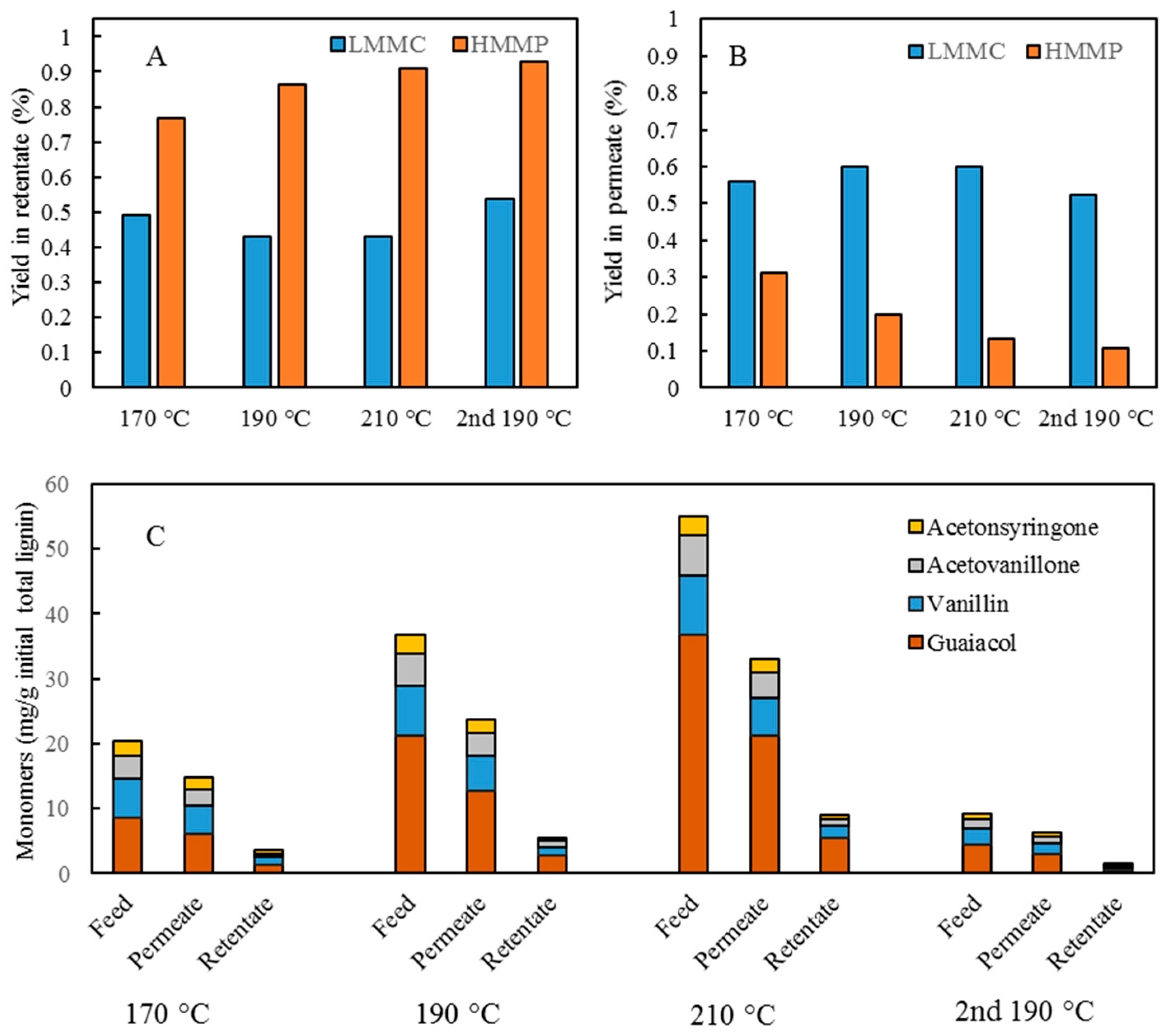
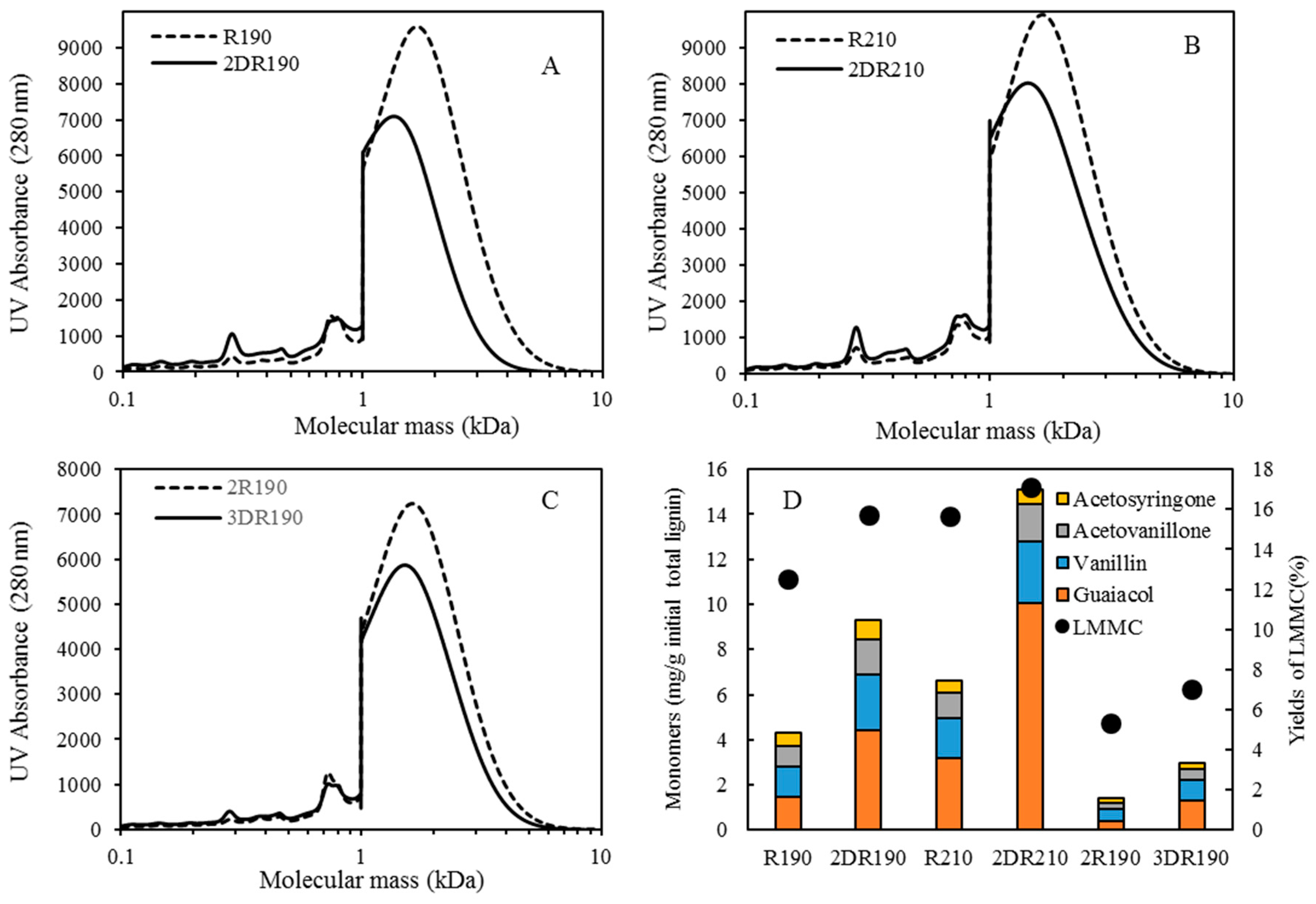
For combining depolymerization and membrane separation, the depolymerized BLR (DBLR) samples were filtered by a GR95PP membrane (2 kD MWCO) at three different temperatures, those of 170 °C, 190 °C, and 210 °C. After the membrane separation had taken place, three permeates (P170, P190, and P210) and three retentates (R170, R190, and R210) were collected for further analysis. The retentate R190 was diluted 5 times by 2 wt % of NaOH for the second round of depolymerization (with the resulting depolymerized retentate ‘2DR190’ being obtained). After the second depolymerization, another membrane filtration was performed to separate the LMMC from the 2DR190 sample, the resulting permeate (2P190) and retentate (2R190) being collected then. The retentate 2R190 was diluted 5 times again by a 2 wt % of NaOH and was depolymerized for a third time, 3DR190 being obtained. The entire process of the combined depolymerization and membrane filtration involved is shown in
Figure 1.
2.4. Molecule Weight Distribution
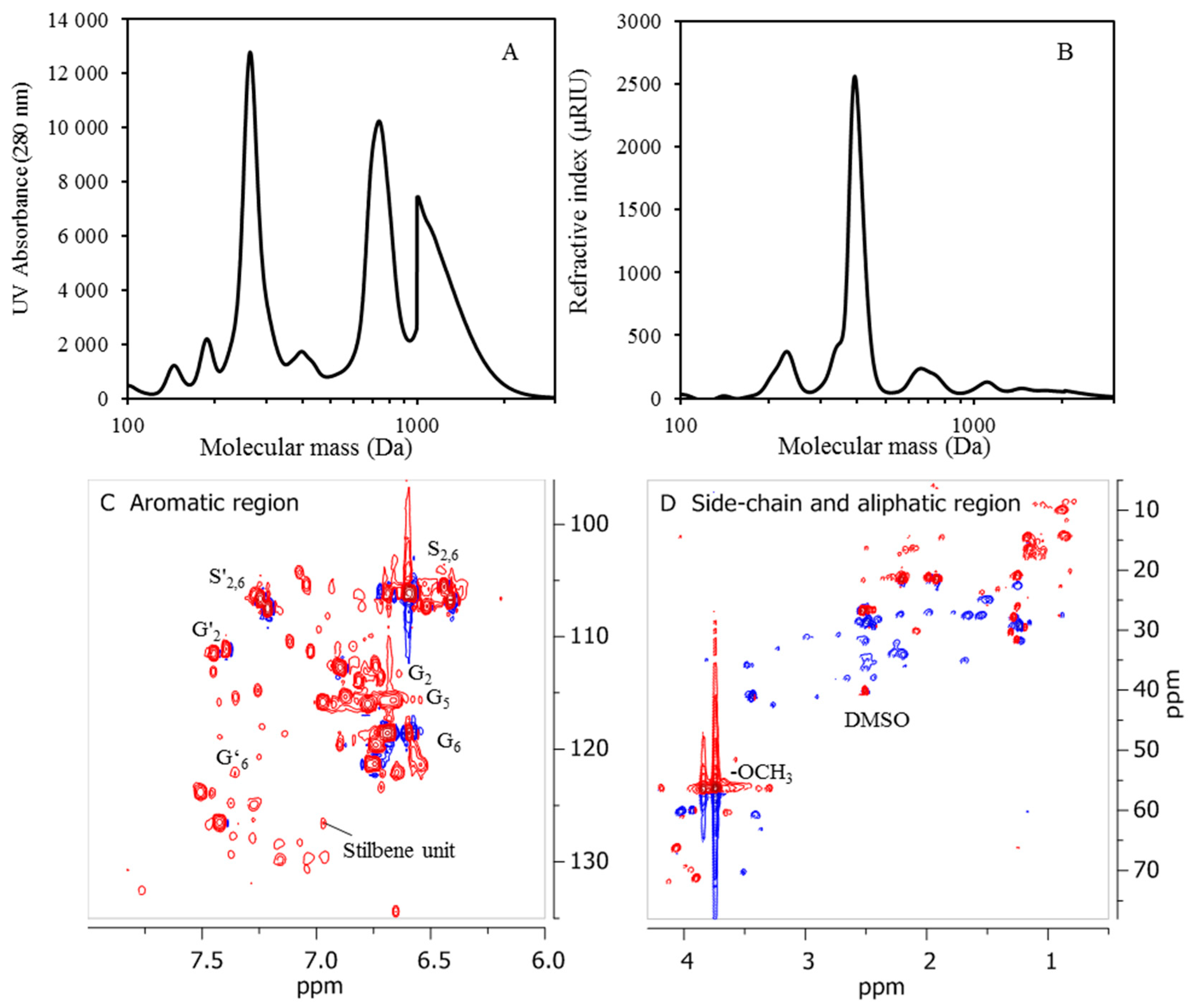
The molecular weight distributions of the BLR samples used in this work were determined by means of size exclusion chromatography (SEC), making use of an Azura HPLC system from the firm Knauer (Berlin, Germany) containing UVD 2.1L and RID 2.1L detectors, respectively, and a P 6.1L pump. ClarityChrom 6.1.0 software was used to control the system. Two different Superdx 10/300 GL columns (Peptide and 200 Increase) from GE Healthcare Bio-Sciences AB (Uppsala, Sweden) were combined to provide a high level of resolution over a large interval of molecular weights.
The column was operated at an ambient temperature and was eluted with a 0.1 M NaOH solution as the mobile phase. All the samples were diluted by the 0.1 M NaOH eluent to a concentration of 5–10 g/L lignin, and were then passed through a 0.2 µm filter (Schleicher and Schuell, Dassel, Germany) and were prepared then for a molecular weight distribution analysis. A 50 µL amount of the diluted sample was injected and elution was performed for a period of 100 min, using an aqueous solution containing 0.1 M NaOH. For each lignin sample, two analyses were performed, the one on the Peptide column and the other on the 200 Increase column. The data from the two columns was processed and then combined to provide a visualization of the molecular weight distribution of each detector signal.
2.5. Extraction of Lignin LMMC
Initial 10 mL amounts of BLR, depolymerized BLR, or membrane-filtrated samples were acidified by 6 M HCl to pH 1-2, 3-Ethoxy-4-hydroxybenzaldehyde (1 mg/mL, 2 mL) being added to the lignin sample prior to acidification as an internal standard. Following acidification, each sample was centrifuged to separate the supernatant and the precipitate. The precipitate was washed 3 times by acidic water, and was then freeze-dried and was stored thereafter for further analysis. The supernatant was mixed with the washing water, and was extracted with use of 10 mL ethyl acetate three times. The extract was collected in a sample tube of known weight. The solvent was then evaporated during a gentle flow of N2, the sample tube containing the oily LMMC product that was left then being weighed. The lignin oil (the extracted LMMC) was stored thereafter at −20 °C in a freezer until analysis was carried out. Taking into consideration the recovery obtained with use of an internal standard, the theoretical yield of the lignin oil was calculated, based on the ratio of the weight of the lignin oil to that of the initial total lignin content.
2.6. Lignin Aromatic Monomers Analysis
The lignin oil was dissolved in a 3 mL acetonitrile-water mixture (50:50 vol.), filtered through a 0.45 µm syringe filter prior to an HPLC analysis being performed. The HPLC analysis was carried out using an Agilent UHPLC system consisting of a degasser (G4225A, Agilent Technologies), a binary pump (G4220A, Agilent Technologies), a thermostated column compartment (G1316C, Agilent Technologies) and a diode array detector (G4212B, Agilent Technologies). An Agilent Eclipse Plus C18 column (2.1 mm × 100 mm, 1.8 µm) was used for this purpose. The flow rate was set at 0.3 mL/min. The mobile phase consisted of: A) water; and B) acetonitrile. The gradient started at 10% B and then ramped up to 30% within a period of 8 min. The amount of the acetonitrile was then increased to 90% within a period of 1 min. The gradient was then maintained at 90% acetonitrile for 3 min before being rapidly decreased during a period of 1 min to the starting composition for re-equilibration, in which it remained for 3 min. The column temperature was held constant at 45 °C. The sample volume that was injected was 5 μL. The separation was monitored with use of diode array detector (DAD) at 280 nm with use of 10 Hz as the sampling frequency. System control and data processing were performed using Agilent Chemstation software.
2.7. NMR Analysis
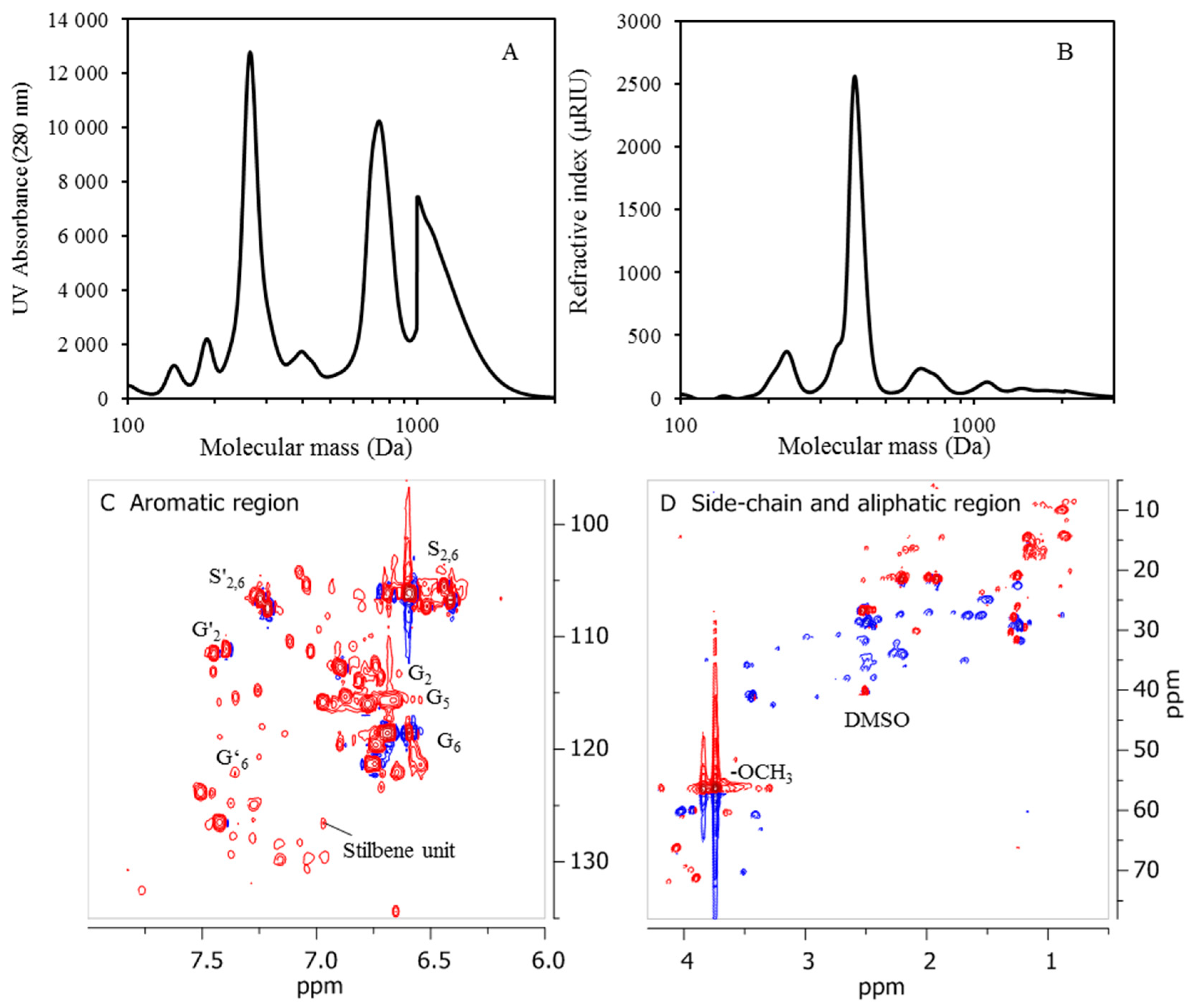
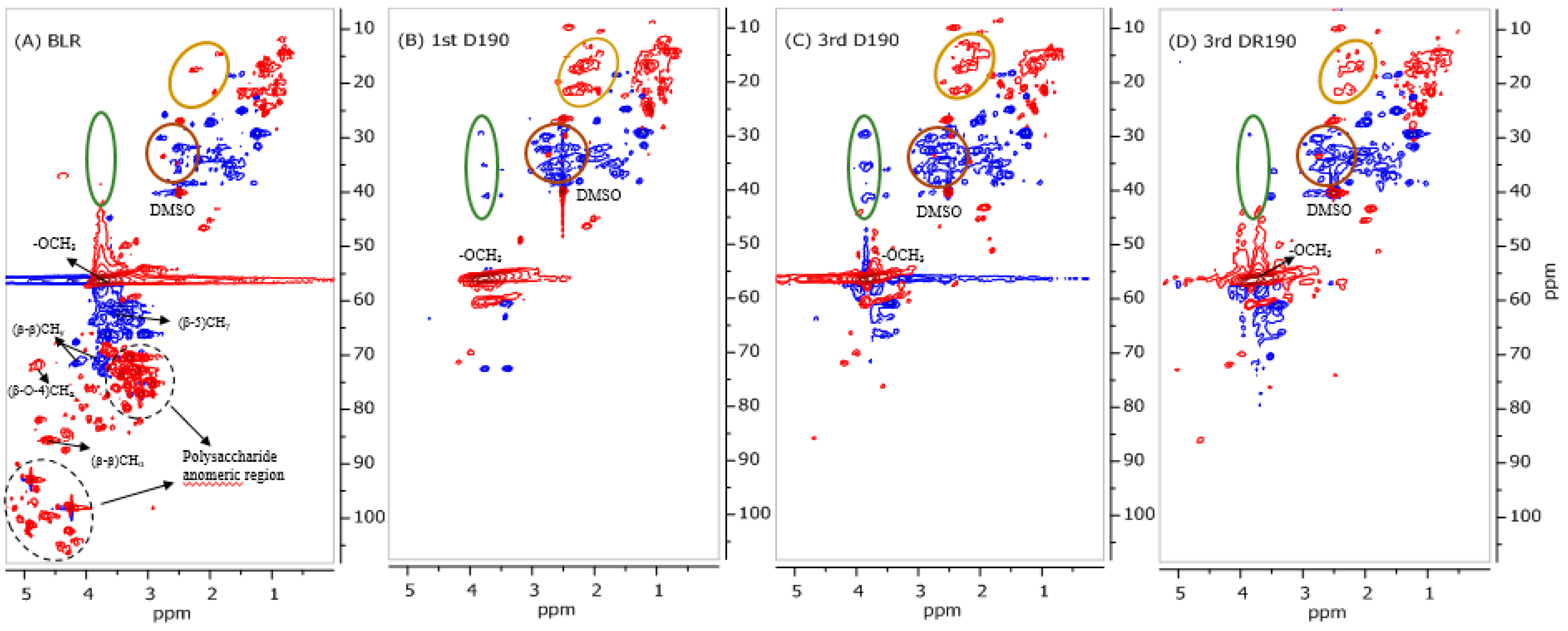
The NMR characterization carried out was performed on a Bruker Avance III HD at 500 MHz, 80 mg of lignin oil from D190 and 100 mg of freeze-dried precipitates from BLR, D190, 3D190, and 3DR190 then being dissolved in 0.5 mL of deuterated dimethyl sulfoxide-d6 (DMSO-d6). The 1H and 2D 1H-13C HSQC NMR spectra obtained were recorded using standard pulse sequences from Bruker. The spectra were processed using MestReNova software (version 9.0).

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}