Membrane Adsorber for the Fast Purification of a Monoclonal Antibody Using Protein A Chromatography

 ,
,
Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Cell Separation with Depth Filtration
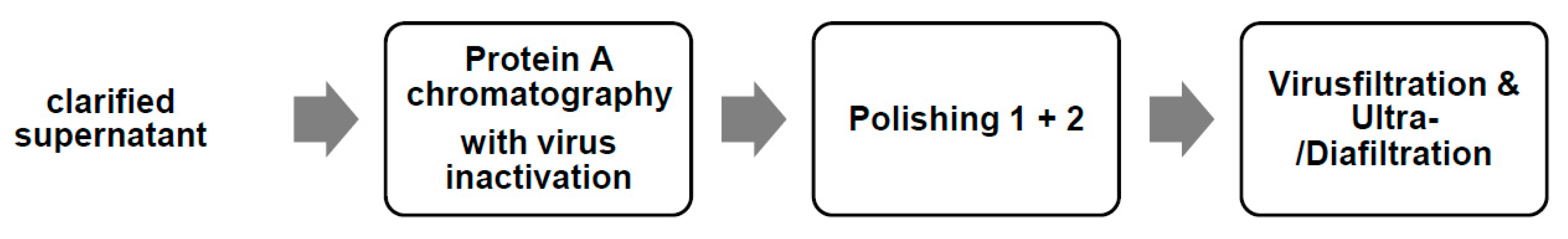
2.2.2. Purification of the Monoclonal Antibody
2.2.3. Stability Experiments
2.3. Analytical Methods
2.3.1. SEC-HPLC
2.3.2. SDS-PAGE
2.3.3. Intrinsic Protein Fluorescence
2.4. Equipment
2.4.1. Membrane Adsorber
2.4.2. Chromatographic System
3. Results
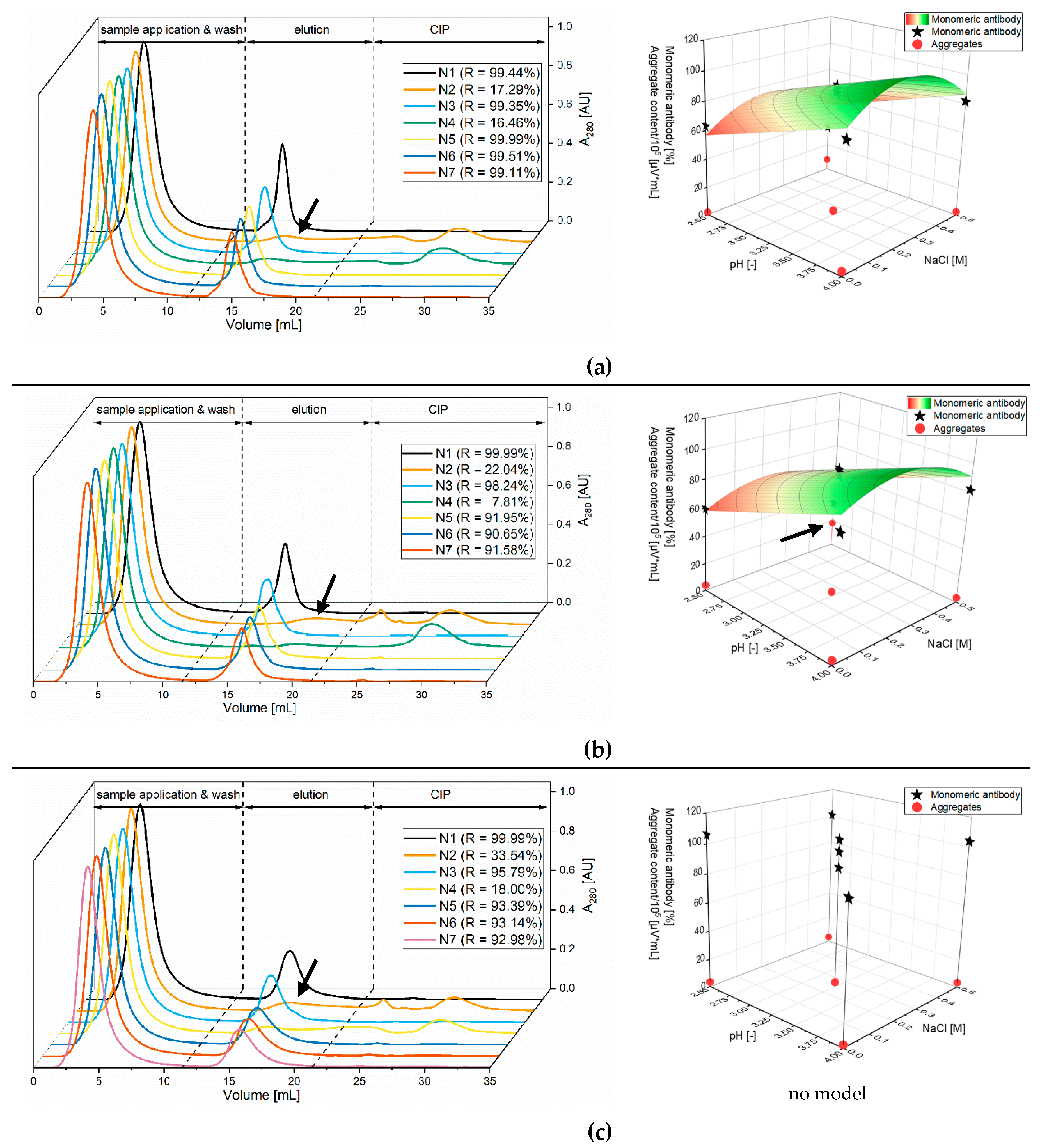
3.1. Optimization of Chromatography Buffers
3.1.1. Screening of the Elution Buffers with Stability Testing
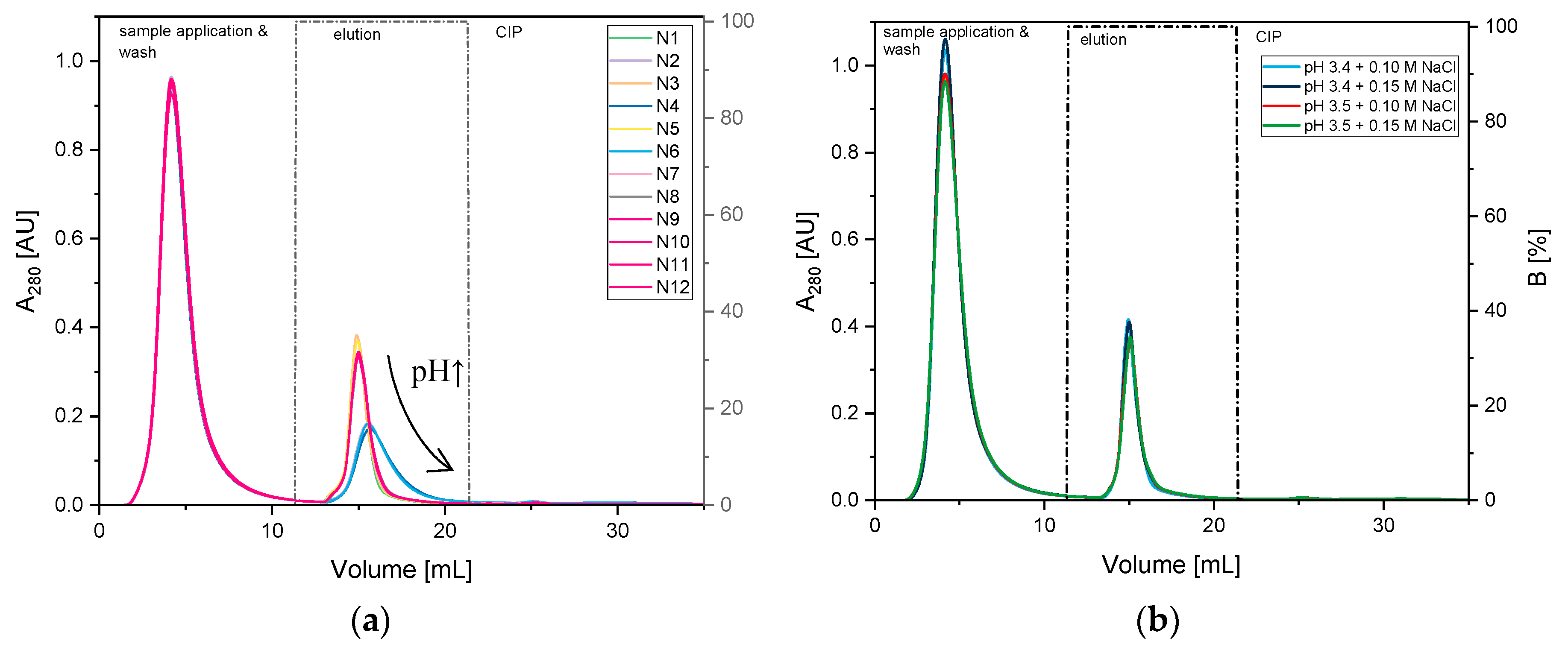
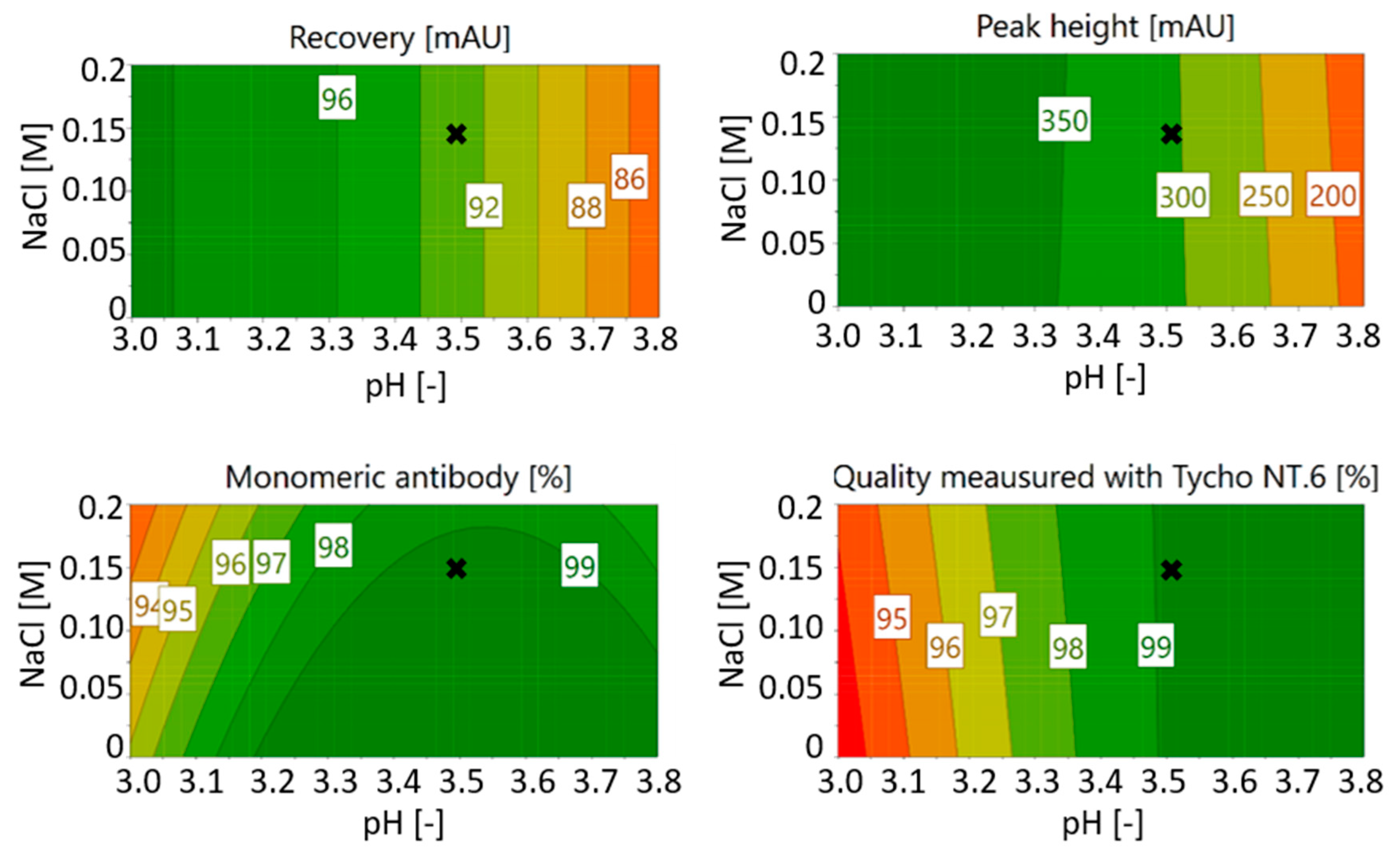
3.1.2. Optimization of Citrate Buffer
3.2. Continuous Membrane Purification of the Monoclonal Antibody
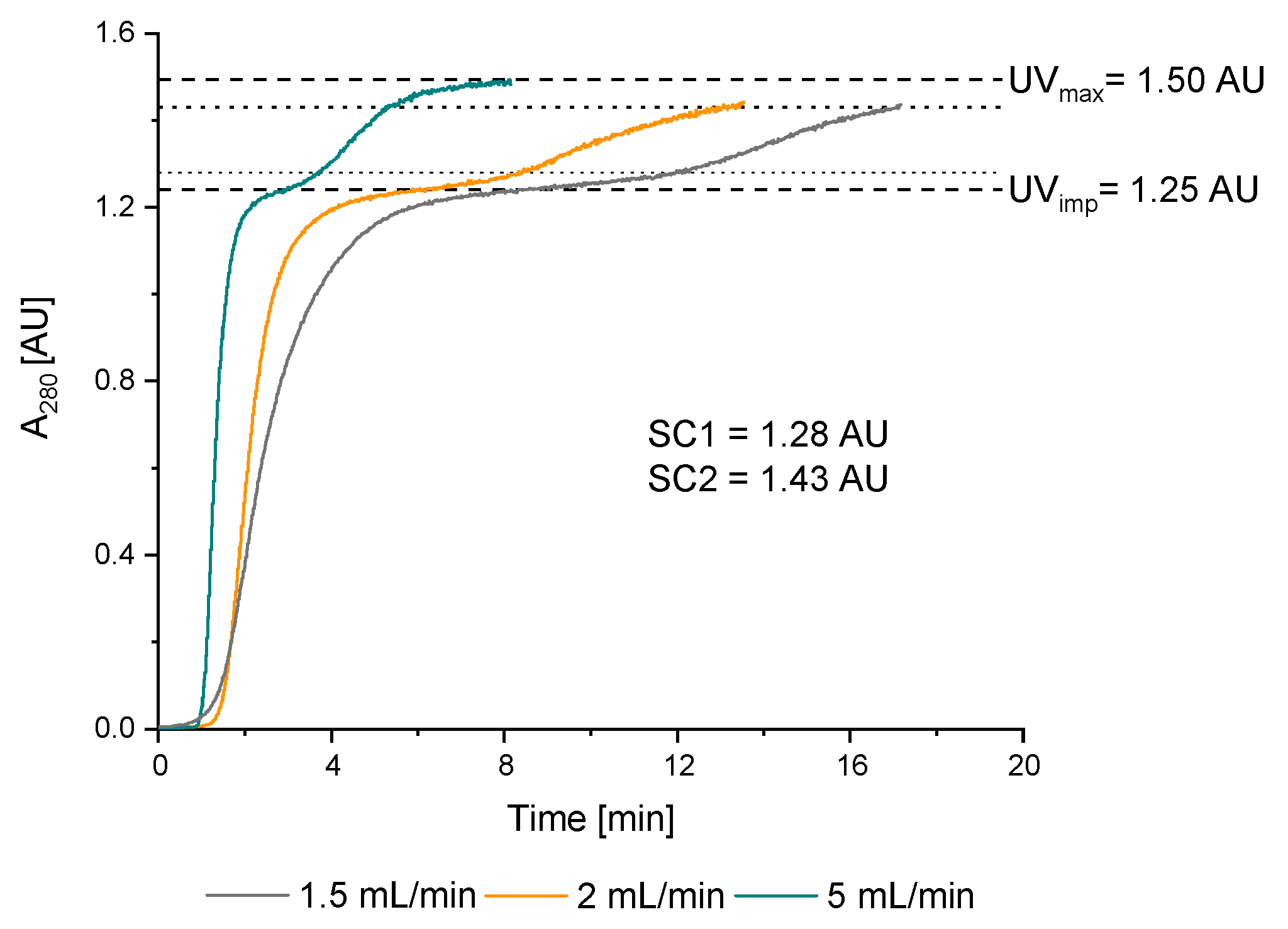
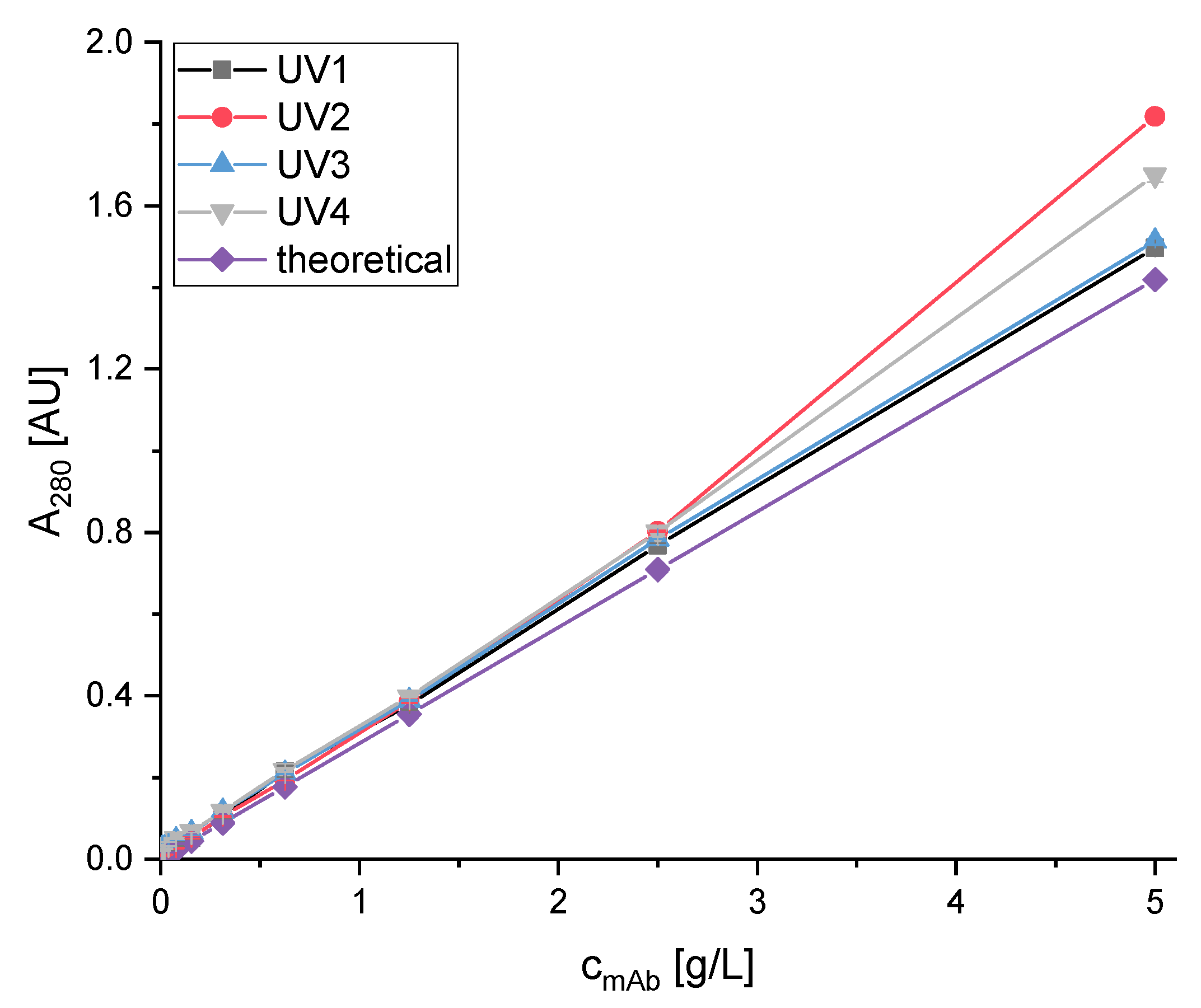
3.2.1. Double Breakthrough Curve
3.2.2. PCCC Application
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A



References
- Chames, P.; Van Regenmortel, M.; Weiss, E.; Baty, D. Therapeutic antibodies: successes, limitations and hopes for the future. Br. J. Pharmacol. 2009, 157, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Reichert, J.M. Antibodies to watch in 2017. MAbs 2017, 9, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Kaplon, H.; Reichert, J.M. Antibodies to watch in 2019. MAbs 2019, 11, 219–238. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.M.; Stryer, L.; Tymoczko, J.L. Stryer Biochemie; Springer: Berlin/Heidelberg, Germany, 2007. [Google Scholar]
- GE Healthcare. Affinity Chromatography Vol. 1: Antibodies; GE Healthcare: Uppsala, Sweden, 2014. [Google Scholar]
- Herschel, T.; El-Armouche, A.; Weber, S. Monoklonale Antikörper. DMW-Dtsch. Med. Wochenschr. 2016, 141, 1390–1394. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sack, U.; Emmrich, F. Monoklonale Antikörper. Internist 2008, 49, 919–928. [Google Scholar] [CrossRef]
- Schäffner, G.; Kabelitz, D. Monoklonale Antikörper zur Therapie. DMW-Dtsch. Med. Wochenschr. 2001, 126, 851–856. [Google Scholar] [CrossRef]
- Blum, H.E. Therapeutische monoklonale Antikörper. DMW-Dtsch. Med. Wochenschr. 2000, 125, 1501–1504. [Google Scholar]
- Ribatti, D. Edelman′s view on the discovery of antibodies. Immunol. Lett. 2015, 164, 72–75. [Google Scholar] [CrossRef]
- Kunert, R.; Reinhart, D. Advances in recombinant antibody manufacturing. Appl. Microbiol. Biotechnol. 2016, 100, 3451–3461. [Google Scholar] [CrossRef]
- Sommerfeld, S.; Strube, J. Challenges in biotechnology production—Generic processes and process optimization for monoclonal antibodies. Chem. Eng. Process. Process Intensif. 2005, 44, 1123–1137. [Google Scholar] [CrossRef]
- Chon, J.H.; Zarbis-Papastoitsis, G. Advances in the production and downstream processing of antibodies. New Biotechnol. 2011, 28, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Kelley, B. Industrialization of mAb production technology: The bioprocessing industry at a crossroads. MAbs 2009, 1, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Strube, J.; Grote, F.; Ditz, R. Bioprocess design and production technology for the future. Biopharm. Prod. Technol. 2012, 1, 657–705. [Google Scholar]
- Low, D.; O’Leary, R.; Pujar, N.S. Future of antibody purification. J. Chromatogr. B 2007, 848, 48–63. [Google Scholar] [CrossRef]
- Seewoester, T. Cell separation and product capture. Biotechnol. Bioprocess. Ser. 2005, 30, 417. [Google Scholar]
- Marichal-Gallardo, P.A.; Alvarez, M.M. State-of-the-art in downstream processing of monoclonal antibodies: process trends in design and validation. Biotechnol. Prog. 2012, 28, 899–916. [Google Scholar] [CrossRef]
- Mendiratta, S.K.; Bandyopadhyay, S.; Singh, A.K. Purification Process for Monoclonal Antibodies. U.S. Patent No. 9,708,365, 18 July 2017. [Google Scholar]
- Gottschalk, U. Process Scale Purification of Antibodies; no. 577.27 PRO; Wiley Online Library: Hoboken, NJ, USA, 2009. [Google Scholar]
- Li, R.; Dowd, V.; Stewart, D.J.; Burton, S.J.; Lowe, C.R. Design, synthesis, and application of a protein A mimetic. Nat. Biotechnol. 1998, 16, 190. [Google Scholar] [CrossRef]
- Ayyar, B.V.; Arora, S.; Murphy, C.; O’Kennedy, R. Affinity chromatography as a tool for antibody purification. Methods 2012, 56, 116–129. [Google Scholar] [CrossRef]
- Deisenhofer, J. Crystallographic refinement and atomic models of a human Fc fragment and its complex with fragment B of protein A from Staphylococcus aureus at 2.9- and 2.8-. ANG. resolution. Biochemistry 1981, 20, 2361–2370. [Google Scholar] [CrossRef]
- Gagnon, P. Purification Tools for Monoclonal Antibodies; Validated Biosystems Inc., 1996; Volume 196. [Google Scholar]
- Mazzer, A.R.; Perraud, X.; Halley, J.; O’Hara, J.; Bracewell, D.G. Protein A chromatography increases monoclonal antibody aggregation rate during subsequent low pH virus inactivation hold. J. Chromatogr. A 2015, 1415, 83–90. [Google Scholar] [CrossRef]
- Huse, K.; Böhme, H.-J.; Scholz, G.H. Purification of antibodies by affinity chromatography. J. Biochem. Biophys. Methods 2002, 51, 217–231. [Google Scholar] [CrossRef]
- Shukla, A.A.; Hubbard, B.; Tressel, T.; Guhan, S.; Low, D. Downstream processing of monoclonal antibodies—application of platform approaches. J. Chromatogr. B 2007, 848, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.A.; Hinckley, P.J.; Gupta, P.; Yigzaw, Y.; Hubbard, B. Strategies to address aggregation during protein A chromatography. BioProcess. Int. 2005, 3, 36–43. [Google Scholar]
- Seed, B. Affinity Purification Methods Involving Imidazole Elution. U.S. Patent No. 5,726,293, 10 March 1998. [Google Scholar]
- Gagnon, P.; Nian, R.; Leong, D.; Hoi, A. Transient conformational modification of immunoglobulin G during purification by protein A affinity chromatography. J. Chromatogr. A 2015, 1395, 136–142. [Google Scholar] [CrossRef]
- Arakawa, T.; Philo, J.S.; Tsumoto, K.; Yumioka, R.; Ejima, D. Elution of antibodies from a Protein-A column by aqueous arginine solutions. Protein Exp. Purif. 2004, 36, 244–248. [Google Scholar] [CrossRef]
- Müller, E.; Vajda, J. Routes to improve binding capacities of affinity resins demonstrated for protein A chromatography. J. Chromatogr. B 2016, 1021, 159–168. [Google Scholar] [CrossRef]
- Bickel, F.; Herold, E.M.; Signes, A.; Romeijn, S.; Jiskoot, W.; Kiefer, H. Reversible NaCl-induced aggregation of a monoclonal antibody at low pH: characterization of aggregates and factors affecting aggregation. Eur. J. Pharm. Biopharm. 2016, 107, 310–320. [Google Scholar] [CrossRef]
- Singla, A.; Bansal, R.; Joshi, V.; Rathore, A.S. Aggregation kinetics for IgG1-based monoclonal antibody therapeutics. AAPS J. 2016, 18, 689–702. [Google Scholar] [CrossRef]
- Rathore, A.S.; Kateja, N.; Agarwal, H. Continuous Downstream Processing for Production of Biotech Therapeutics. In Continuous Biomanufacturing: Innovative Technologies and Methods; Subramanian, G., Ed.; Wiley-VCH: Weinheim, Germany, 2017; pp. 261–288. [Google Scholar]
- Fraud, N.; Kuczewski, M.; Zarbis-Papastoitsis, G.; Hirai, M. Hydrophobic Membrane Adsorbers for Large-Scale Downstream Processing. Biopharm. Int. 2009, 22. [Google Scholar]
- Yu, D.; Ghosh, R. Purification of PEGylated Protein Using Membrane Chromatography. J. Pharm. Sci. 2010, 99, 3326–3333. [Google Scholar] [CrossRef]
- Zhou, J.X.; Tressel, T. Basic concepts in Q membrane chromatography for large-scale antibody production. Biotechnol. Prog. 2006, 22, 341–349. [Google Scholar] [CrossRef]
- Vogel, J.H.; Nguyen, H.; Giovannini, R.; Ignowski, J.; Garger, S.; Salgotra, A.; Tom, J. A new large-scale manufacturing platform for complex biopharmaceuticals. Biotechnol. Bioeng. 2012, 109, 3049–3058. [Google Scholar] [CrossRef]
- Boi, C. Membrane adsorbers as purification tools for monoclonal antibody purification. J. Chromatogr. B 2007, 848, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.F.; Ma, J.; Winter, C.; Bayer, R. Recovery and purification process development for monoclonal antibody production. MAbs 2010, 2, 480–499. [Google Scholar] [CrossRef] [PubMed]
- Kuczewski, M.; Schirmer, E.; Lain, B.; Zarbis-Papastoitsis, G. A single-use purification process for the production of a monoclonal antibody produced in a PER. C6 human cell line. Biotechnol. J. 2011, 6, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, H.L.; Fahrner, R.L.; Xu, Y.; Norling, L.A.; Blank, G.S. Membrane ion-exchange chromatography for process-scale antibody purification. J. Chromatogr. A 2001, 907, 145–154. [Google Scholar] [CrossRef]
- Mothes, B.; Pezzini, J.; Schroeder-Tittmann, K.; Villain, L. Accelerated, seamless antibody purification. Bioprocess. Int. 2016, 14, 5. [Google Scholar]
- Jungbauer, A. Continuous downstream processing of biopharmaceuticals. Trends Biotechnol. 2013, 31, 479–492. [Google Scholar] [CrossRef]
- Girard, V.; Hilbold, N.J.; Ng, C.K.; Pegon, L.; Chahim, W.; Rousset, F.; Monchois, V. Large-scale monoclonal antibody purification by continuous chromatography, from process design to scale-up. J. Biotechnol. 2015, 213, 65–73. [Google Scholar] [CrossRef]
- Gjoka, X.; Gantier, R.; Schofield, M. Transfer of a three step mAb chromatography process from batch to continuous: Optimizing productivity to minimize consumable requirements. J. Biotechnol. 2017, 242, 11–18. [Google Scholar] [CrossRef]
- De Fazekas, S.S.G.; Webster, R.G.; Datyner, A. Two new staining procedures for quantitative estimation of proteins on electrophoretic strips. Biochim. Biophys. Acta 1963, 71, 377–391. [Google Scholar]
- Wray, W.; Boulikas, T.; Wray, V.P.; Hancock, R. Silver staining of proteins in polyacrylamide gels. Anal. Biochem. 1981, 118, 197–203. [Google Scholar] [CrossRef]
- Breitsprecher, G.W.D.; Kern, B.; Gupta, A.J.; Knoll, K. Reproduzierbarkeit- Qualitätskontrolle von Proteinen durch thermische Entfaltungsprofile. BIOspektrum 2019, 60–62. Available online: https://www.biospektrum.de/blatt/d_bs_pdf&_id=1539789 (accessed on 27 November 2019).
- Brämer, C.; Schreiber, S.; Scheper, T.; Beutel, S. Continuous purification of Candida antarctica lipase B using 3-membrane adsorber periodic counter-current chromatography. Eng. Life Sci. 2018. [Google Scholar] [CrossRef]
- Mahajan, E.; George, A.; Wolk, B. Improving affinity chromatography resin efficiency using semi-continuous chromatography. J. Chromatogr. A 2012, 1227, 154–162. [Google Scholar] [CrossRef]
- Pollock, J.; Bolton, G.; Coffman, J.; Ho, S.V.; Bracewell, D.G.; Farid, S.S. Optimising the design and operation of semi-continuous affinity chromatography for clinical and commercial manufacture. J. Chromatogr. A 2013, 1284, 17–27. [Google Scholar] [CrossRef]
- Warikoo, V.; Godawat, R.; Brower, K.; Jain, S.; Cummings, D.; Simons, E.; Johnson, T.; Walther, J.; Yu, M.; Wright, B.; et al. Integrated continuous production of recombinant therapeutic proteins. Biotechnol. Bioeng. 2012, 109, 3018–3029. [Google Scholar] [CrossRef]
- Godawat, R.; Brower, K.; Jain, S.; Konstantinov, K.; Riske, F.; Warikoo, V. Periodic counter-current chromatography–design and operational considerations for integrated and continuous purification of proteins. Biotechnol. J. 2012, 7, 1496–1508. [Google Scholar] [CrossRef]
- Brämer, C.; Ekramzadeh, K.; Lammers, F.; Scheper, T.; Beutel, S. Optimization of continuous purification of recombinant patchoulol synthase from Escherichia coli with membrane adsorbers. Biotechnol. Prog. 2019, 35, e2812. [Google Scholar] [CrossRef]
- Shukla, A.A.; Wolfe, L.S.; Mostafa, S.S.; Norman, C. Evolving trends in mAb production processes. Bioeng. Transl. Med. 2017, 2, 58–69. [Google Scholar] [CrossRef]
- Skoglar, H.; Blom, H.; Mathiasson, L.; Akerblom, A.; Łącki, K. The use of dynamic control in periodic counter-current chromatography. Bioprocess. Int. 2015, 13, 29150261. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Field of Application | Ref. |
|---|---|
| Affinity chromatography | [40] |
| Ion exchange chromatography | [40,41,42,43] |
| Hydrophobic interaction chromatography | [41,42] |
| Accelerated, Seamless Antibody Purification (ASAP)-continuous method (Protein A chromatography, cation and anion exchange chromatography) | [44] |
| Screening 1 | Optimization 2 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Citrate/Glycine Buffer | Acetate Buffer | Citrate Buffer | |||||||||
| Exp. | Order | pH | NaCl [M] | Exp. | Order | pH | NaCl [M] | Exp. | Order | pH | NaCl [M] |
| N1 | 1 | 2.5 | 0 | N1 | 1 | 3.5 | 0 | N1 | 4 | 3 | 0 |
| N2 | 7 | 4 | 0 | N2 | 7 | 4 | 0 | N2 | 7 | 3.8 | 0 |
| N3 | 2 | 2.5 | 0.5 | N3 | 2 | 3.5 | 0.5 | N3 | 2 | 3 | 0.2 |
| N4 | 4 | 4 | 0.5 | N4 | 4 | 4 | 0.5 | N4 | 6 | 3.8 | 0.2 |
| N5 | 5 | 3.25 | 0.25 | N5 | 5 | 3.75 | 0.25 | N5 | 10 | 3 | 0.1 |
| N6 | 6 | 3.25 | 0.25 | N6 | 6 | 3.75 | 0.25 | N6 | 1 | 3.8 | 0.1 |
| N7 | 3 | 3.25 | 0.25 | N7 | 3 | 3.75 | 0.25 | N7 | 3 | 3.4 | 0 |
| N8 | 11 | 3.4 | 0.2 | ||||||||
| N9 | 5 | 3.4 | 0.1 | ||||||||
| N10 | 8 | 3.4 | 0.1 | ||||||||
| N11 | 9 | 3.4 | 0.1 | ||||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brämer, C.; Tünnermann, L.; Gonzalez Salcedo, A.; Reif, O.-W.; Solle, D.; Scheper, T.; Beutel, S. Membrane Adsorber for the Fast Purification of a Monoclonal Antibody Using Protein A Chromatography. Membranes 2019, 9, 159. https://doi.org/10.3390/membranes9120159
Brämer C, Tünnermann L, Gonzalez Salcedo A, Reif O-W, Solle D, Scheper T, Beutel S. Membrane Adsorber for the Fast Purification of a Monoclonal Antibody Using Protein A Chromatography. Membranes. 2019; 9(12):159. https://doi.org/10.3390/membranes9120159
Chicago/Turabian StyleBrämer, Chantal, Lisa Tünnermann, Alina Gonzalez Salcedo, Oscar-Werner Reif, Dörte Solle, Thomas Scheper, and Sascha Beutel. 2019. "Membrane Adsorber for the Fast Purification of a Monoclonal Antibody Using Protein A Chromatography" Membranes 9, no. 12: 159. https://doi.org/10.3390/membranes9120159
APA StyleBrämer, C., Tünnermann, L., Gonzalez Salcedo, A., Reif, O.-W., Solle, D., Scheper, T., & Beutel, S. (2019). Membrane Adsorber for the Fast Purification of a Monoclonal Antibody Using Protein A Chromatography. Membranes, 9(12), 159. https://doi.org/10.3390/membranes9120159