Modification of Nanofiber Support Layer for Thin Film Composite Forward Osmosis Membranes via Layer-by-Layer Polyelectrolyte Deposition

 ,
,  ,
,  ,
, 


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
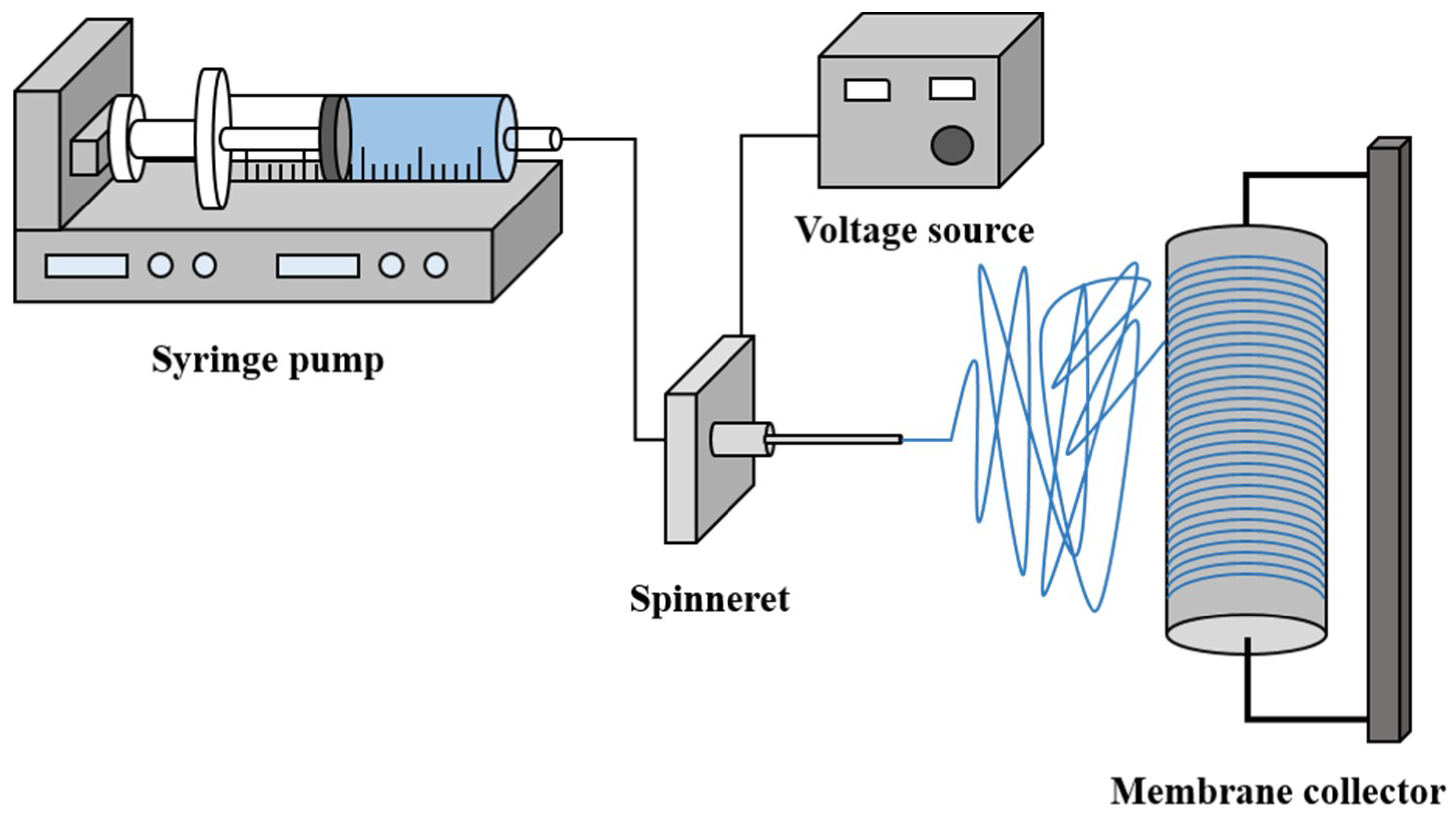
2.2. Preparation of Nanofiber Membrane Support via Electrospinning
2.2.1. Dope, Electrolyte, and Monomeric Solution Preparation
2.2.2. Electrospinning
2.3. Layer-by-Layer Polyelectrolyte Deposition
2.4. Interfacial Polymerization
2.5. Osmotic Performance
2.6. Determination of Membrane Parameters
2.7. Membrane Characterization
2.7.1. Surface and Cross-Section Morphology
2.7.2. Water Contact Angle
2.7.3. Pore Size and Porosity Determination
2.7.4. Membrane Mechanical Strength and Thickness
2.7.5. Surface Chemistry Characterization
3. Results and Discussion
3.1. Properties of Nanofiber PVDF Membrane Support
3.2. Molecular Layer-by-Layer Approach
3.3. Properties of the TFC Membranes
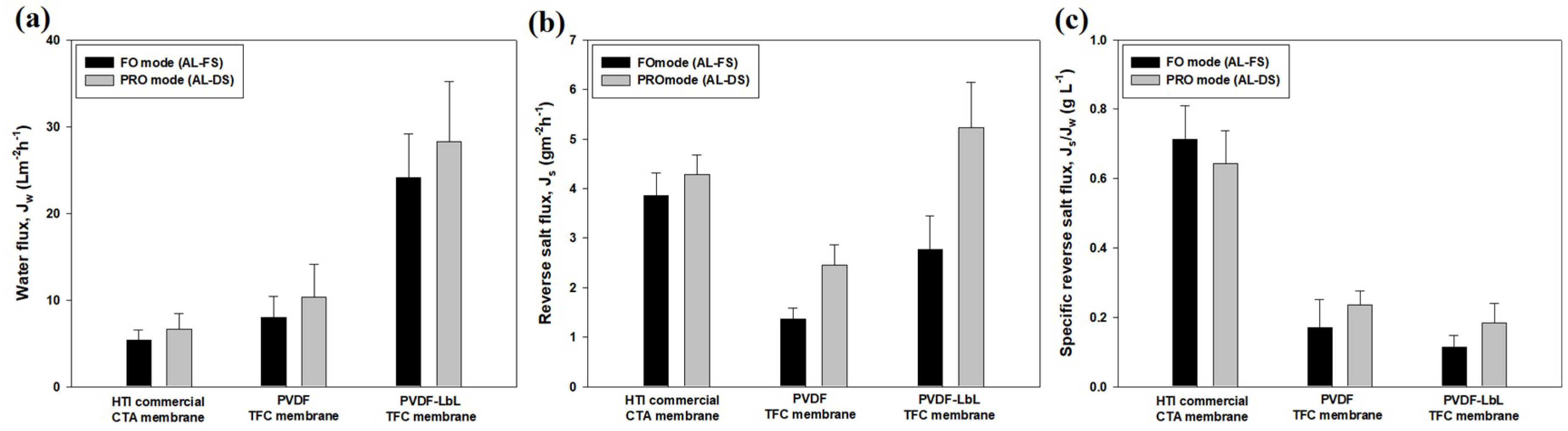
3.4. Membrane Performance
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cath, T.Y.; Childress, A.E.; Elimelech, M. Forward osmosis: Principles, applications, and recent developments. J. Membr. Sci. 2006, 281, 70–87. [Google Scholar] [CrossRef]
- Cutcheon, M.J.R.; Elimelech, M. Influence of concentrative and dilutive internal concentration polarization on flux behavior in forward osmosis. J. Membr. Sci. 2006, 284, 237–247. [Google Scholar] [CrossRef]
- Yip, N.Y.; Tiraferri, A.; Phillip, W.A.; Schiffman, J.D.; Elimelech, M. High performance thin-film composite forward osmosis membrane. Environ. Sci. Technol. 2010, 44, 3812–3818. [Google Scholar] [CrossRef] [PubMed]
- Tiraferri, A.; Yip, N.Y.; Phillip, W.A.; Schiffman, J.D.; Elimelech, M. Relating performance of thin-film composite forward osmosis membranes to support layer formation and structure. J. Membr. Sci. 2011, 367, 340–352. [Google Scholar] [CrossRef]
- Huang, L.; Arena, J.T.; Cutcheon, M.J.R. Surface modified PVDF nanofiber supported thin film composite membranes for forward osmosis. J. Membr. Sci. 2016, 499, 352–360. [Google Scholar] [CrossRef]
- Alsvik, I.L.; Hägg, M.B. Pressure retarded osmosis and forward osmosis membranes: Materials and methods. Polymers 2013, 5, 303–327. [Google Scholar] [CrossRef]
- Lee, K.L.; Baker, R.W.; Lonsdale, H.K. Membranes for power generation by pressure-retarded osmosis. J. Membr. Sci. 1981, 8, 141–171. [Google Scholar] [CrossRef]
- Cadotte, J.E.; Petersen, R.J.; Larson, R.E.; Erickson, E.E. A new thin-film composite seawater reverse osmosis membrane. Desalination 1980, 32, 25–31. [Google Scholar] [CrossRef]
- Peyki, A.; Rahimpour, A.; Jahanshahi, M. Preparation and characterization of thin film composite reverse osmosis membranes incorporated with hydrophilic SiO2 nanoparticles. Desalination 2015, 368, 152–158. [Google Scholar] [CrossRef]
- Wei, J.; Qiu, C.; Tang, C.Y.; Wang, R.; Fane, A.G. Synthesis and characterization of flat-sheet thin film composite forward osmosis membranes. J. Membr. Sci. 2011, 372, 292–302. [Google Scholar] [CrossRef]
- Klaysom, C.; Hermans, S.; Gahlaut, A.; Van Craenenbroeck, S.; Vankelecom, I.F.J. Polyamide/Polyacrylonitrile (PA/PAN) thin film composite osmosis membranes: Film optimization, characterization and performance evaluation. J. Membr. Sci. 2013, 445, 25–33. [Google Scholar] [CrossRef]
- Chung, T.S.; Li, X.; Ong, R.C.; Ge, Q.; Wang, H.; Han, G. Emerging forward osmosis (FO) technologies and challenges ahead for clean water and clean energy applications. Curr. Opin. Chem. Eng. 2012, 1, 246–257. [Google Scholar] [CrossRef]
- Han, G.; Zhang, S.; Li, X.; Chung, T.S. Progress in pressure retarded osmosis (PRO) membranes for osmotic power generation. Prog. Polym. Sci. 2015, 51, 1–27. [Google Scholar] [CrossRef]
- Li, G.; Li, X.M.; He, T.; Jiang, B.; Gao, C. Cellulose triacetate forward osmosis membranes: Preparation and characterization. Desalination Water Treat. 2013, 51, 2656–2665. [Google Scholar] [CrossRef]
- Cui, Y.; Liu, X.Y.; Chung, T.S. Enhanced osmotic energy generation from salinity gradients by modifying thin film composite membranes. Chem. Eng. J. 2014, 242, 195–203. [Google Scholar] [CrossRef]
- Park, M.J.; Phuntsho, S.; He, T.; Nisola, G.M.; Tijing, L.D.; Li, X.M.; Chen, G.; Chung, W.J.; Shon, H.K. Graphene oxide incorporated polysulfone substrate for the fabrication of flat-sheet thin-film composite forward osmosis membranes. J. Membr. Sci. 2015, 493, 496–507. [Google Scholar] [CrossRef]
- Choi, W.; Jeon, S.; Kwon, S.J.; Park, H.; Park, Y.I.; Nam, S.E.; Lee, P.S.; Lee, J.S.; Choi, J.; Hong, S.; et al. Thin film composite reverse osmosis membranes prepared via layered interfacial polymerization. J. Membr. Sci. 2017, 527, 121–128. [Google Scholar] [CrossRef]
- Song, X.; Liu, Z.; Sun, D.D. Energy recovery from concentrated seawater brine by thin-film nanofiber composite pressure retarded osmosis membranes with high power density. Energy Environ. Sci. 2013, 6, 1199–1210. [Google Scholar] [CrossRef]
- Bui, N.N.; Lind, M.L.; Hoek, E.M.V.; Cutcheon, M.J.R. Electrospun nanofiber supported thin film composite membranes for engineered osmosis. J. Membr. Sci. 2011, 385–386, 10–19. [Google Scholar] [CrossRef]
- Song, X.; Liu, Z.; Sun, D.D. Nano gives the answer: Breaking the bottleneck of internal concentration polarization with a nanofiber composite forward osmosis membrane for a higher water production rate. Adv. Mater. 2011, 23, 3256–3260. [Google Scholar] [CrossRef] [PubMed]
- Puguan, J.M.C.; Kim, H.S.; Lee, K.J.; Kim, H. Low internal concentration polarization in forward osmosis membranes with hydrophilic crosslinked PVA nanofibers as porous support layer. Desalination 2014, 336, 24–31. [Google Scholar] [CrossRef]
- Bui, N.N.; Cutcheon, M.J.R. Hydrophilic nanofibers as new supports for thin film composite membranes for engineered osmosis. Environ. Sci. Technol. 2013, 47, 1761–1769. [Google Scholar] [CrossRef] [PubMed]
- Bui, N.N.; Cutcheon, M.J.R. Nanofiber supported thin-film composite membrane for pressure-retarded osmosis. Environ. Sci. Technol. 2014, 48, 4129–4136. [Google Scholar] [CrossRef] [PubMed]
- Park, M.J.; Gonzales, R.R.; Wahab, A.A.; Phuntsho, S.; Shon, H.K. Hydrophilic polyvinyl alcohol coating on hydrophobic electrospun nanofiber membrane for high performance thin film composite forward osmosis membrane. Desalination 2018, 426, 50–59. [Google Scholar] [CrossRef]
- Tijing, L.D.; Woo, Y.C.; Johir, M.A.H.; Choi, J.S.; Shon, H.K. A novel dual-layer bicomponent electrospun nanofibrous membrane for desalination by direct contact membrane distillation. Chem. Eng. J. 2014, 256, 155–159. [Google Scholar] [CrossRef]
- Tian, E.L.; Zhou, H.; Ren, Y.W.; Mirza, Z.; Wang, X.Z.; Xiong, S.W. Novel design of hydrophobic/hydrophilic interpenetrating network composite nanofibers for the support layer of forward osmosis membrane. Desalination 2014, 347, 207–214. [Google Scholar] [CrossRef]
- Huang, L.; Manickam, S.S.; Cutcheon, M.J.R. Increasing strength of electrospun nanofiber membranes for water filtration using solvent vapor. J. Membr. Sci. 2013, 436, 213–220. [Google Scholar] [CrossRef]
- Huang, L.; Cutcheon, M.J.R. Hydrophilic nylon 6,6 nanofibers supported thin film composite membranes for engineered osmosis. J. Membr. Sci. 2014, 457, 162–169. [Google Scholar] [CrossRef]
- Tan, C.H.; Ng, H.Y. Modified models to predict flux behavior in forward osmosis in consideration of external and internal concentration polarizations. J. Membr. Sci. 2008, 324, 209–219. [Google Scholar] [CrossRef]
- Tan, C.H.; Ng, H.Y. Revised external and internal concentration polarization models to improve flux prediction in forward osmosis process. Desalination 2013, 309, 125–140. [Google Scholar] [CrossRef]
- Gerstandt, K.; Peinemann, K.V.; Skilhagen, S.E.; Thorsen, T.; Holt, T. Membrane processes in energy supply for an osmotic power plant. Desalination 2008, 224, 64–70. [Google Scholar] [CrossRef]
- Li, X.M.; He, T.; Dou, P.; Zhao, S. Forward Osmosis and Forward Osmosis Membranes. In Reference Module in Chemistry, Molecular Sciences and Chemical Engineering; Elsevier: Amsterdam, The Netherlands, 2010. [Google Scholar]
- Pan, Y.; Wang, W.; Peng, C.; Shi, K.; Luo, Y.; Ji, X. Novel hydrophobic polyvinyl alcohol-formaldehyde foams for organic solvents absorption and effective separation. RSC Adv. 2014, 4, 660–669. [Google Scholar] [CrossRef]
- Onuki, Y.; Nishikawa, M.; Morishita, M.; Takayama, K. Development of photocrosslinked polyacrylic acid hydrogel as an adhesive for dermatological patches: Involvement of formulation factors in physical properties and pharmacological effects. Int. J. Pharm. 2008, 349, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Sugama, T.; Kukacka, L.E.; Clayton, C.R.; Hua, H.C. Effects of polyacrylic acid primers on adhesion and durability of FPL-etched aluminum/polyurethane systems. J Adhes. Sci. Technol. 1987, 1, 265–280. [Google Scholar] [CrossRef]
- Choi, W.; Gu, J.E.; Park, S.H.; Kim, S.; Bang, J.; Baek, K.Y.; Park, B.; Lee, J.S.; Chan, E.P.; Lee, J.H. Tailor-made polyamide membranes for water desalination. ACS Nano 2015, 9, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Obaid, M.; Ghouri, Z.K.; Fadali, O.A.; Khalil, K.A.; Almajid, A.A.; Barakat, N.A. Amorphous SiO2 NP-Incorporated Poly(vinylidene fluoride) Electrospun Nanofiber Membrane for High Flux Forward Osmosis Desalination. ACS Appl. Mater. Interfaces 2016, 8, 4561–4574. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Yu, D.; He, Z.; Liu, J.; Xiao, F.X.; Zhang, Y.; Wang, R.; Bhattacharyya, D.; Tan, T.T. Graphene Oxide Quantum Dots Covalently Functionalized PVDF Membrane with Significantly-Enhanced Bactericidal and Antibiofouling Performances. Sci. Rep. 2016, 6, 20142. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Cheng, Z.L.; Chung, T.S. Thin-film composite (TFC) hollow fiber membrane with double-polyamide active layers for internal concentration polarization and fouling mitigation in osmotic processes. J. Membr. Sci. 2017, 523, 497–504. [Google Scholar] [CrossRef]
- Ren, J.; Cutcheon, M.J.R. A new commercial thin film composite membrane for forward osmosis. Desalination 2014, 343, 187–193. [Google Scholar] [CrossRef]
- Huang, L.; Bui, N.N.; Meyering, M.T.; Hamlin, T.J.; Cutcheon, M.J.R. Novel hydrophilic nylon 6,6 microfiltration membrane supported thin film composite membranes for engineered osmosis. J. Membr. Sci. 2013, 437, 141–149. [Google Scholar] [CrossRef]
- Obaid, M.; Mohamed, H.O.; Yasin, A.S.; Fadali, O.A.; Khalil, K.A.; Kim, T.; Barakat, N.A.M. A novel strategy for enhancing the electrospun PVDF support layer of thin-film composite forward osmosis membranes. RSC Adv. 2016, 6, 102762–102772. [Google Scholar] [CrossRef]
- Shibuya, M.; Park, M.J.; Lim, S.; Phuntsho, S.; Matsuyama, H.; Shon, H.K. Novel CA/PVDF nanofiber supports strategically designed via coaxial electrospinning for high performance thin-film composite forward osmosis membranes for desalination. Desalination 2018, 445, 63–74. [Google Scholar] [CrossRef]
- Tian, M.; Qiu, C.; Liao, Y.; Chou, S.; Wang, R. Preparation of polyamide thin film composite forward osmosis membranes using electrospun polyvinylidene fluoride (PVDF) nanofibers as substrates. Sep. Purif. Technol. 2013, 118, 727–736. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | PVDF Nanofiber | PVDF-PAA Nanofiber | PVDF-Lbl |
|---|---|---|---|
| Tensile Strength (MPa) | 7.14 ± 0.61 | 8.51 ± 0.38 | 8.89 ± 0.71 |
| Elongation (%) | 138.12 ± 37.88 | 177.21 ± 18.03 | 191.16 ± 15.34 |
| Young’s Modulus (MPa) | 36.19 ± 11.14 | 68.31 ± 6.18 | 72.15 ± 10.14 |
| Porosity (%) | 79.21 ± 1.37 | 71.05 ± 0.68 | 72.18 ± 1.19 |
| Water Uptake (%) | 4.29 ± 0.45 | 138.21 ± 8.41 | 127.18 ± 5.88 |
| Membrane | A (L m−2 h−1 bar−1) | B (L m−2 h−1) | R (%) | S (µm) |
|---|---|---|---|---|
| HTI CTA | 0.64 | 0.57 | 92.18 | 721 |
| PVDF TFC | 1.88 | 0.43 | 95.17 | 482 |
| PVDF-LbL TFC | 4.12 | 0.38 | 96.43 | 221 |
| Membrane | Draw Solution | Jw (L m−2 h−1) | Js (g m−2 h−1) | Js/Jw (g L−1) | Reference |
|---|---|---|---|---|---|
| Nylon 6,6-modified PVDF | 0.5 M NaCl | 16.0 | 2.7 | 0.17 | [5] |
| PVA a | 0.5 M NaCl | 27.7 | - | - | [21] |
| PET b-supported CA c/PAN d | 1.5 M NaCl | 27.6 | 3.9 | 0.14 | [22] |
| PVDF-PVA | 0.5 M NaCl | 24.8 | 3.3 | 0.13 | [24] |
| PET/PVA (1:4) | 0.5 M NaCl | 47.2 | 9.5 | 0.20 | [26] |
| Nylon 6,6 | 1.0 M NaCl | 21.0 | 5.2 | 0.24 | [41] |
| TEA e-modified PVDF | 2.0 M NaCl | 68.0 | 2.0 | 0.03 | [42] |
| PVDF | 0.5 M NaCl | 18.5 | 2.7 | 0.14 | [43] |
| PVDF/CA composite | 0.5 M NaCl | 20.2 | 2.1 | 0.10 | |
| PVDF/CA blend | 0.5 M NaCl | 31.3 | 0.8 | 0.03 | |
| PVDF | 1.0 M NaCl | 28.0 | 12.9 | 0.46 | [44] |
| PVDF-LbL | 0.5 M NaCl | 24.1 | 2.8 | 0.12 | This work |
| 1.0 M NaCl | 32.4 | 3.9 | 0.12 | ||
| 1.5 M NaCl | 37.8 | 4.5 | 0.12 | ||
| 2.0 M NaCl | 45.2 | 4.9 | 0.11 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzales, R.R.; Park, M.J.; Tijing, L.; Han, D.S.; Phuntsho, S.; Shon, H.K. Modification of Nanofiber Support Layer for Thin Film Composite Forward Osmosis Membranes via Layer-by-Layer Polyelectrolyte Deposition. Membranes 2018, 8, 70. https://doi.org/10.3390/membranes8030070
Gonzales RR, Park MJ, Tijing L, Han DS, Phuntsho S, Shon HK. Modification of Nanofiber Support Layer for Thin Film Composite Forward Osmosis Membranes via Layer-by-Layer Polyelectrolyte Deposition. Membranes. 2018; 8(3):70. https://doi.org/10.3390/membranes8030070
Chicago/Turabian StyleGonzales, Ralph Rolly, Myoung Jun Park, Leonard Tijing, Dong Suk Han, Sherub Phuntsho, and Ho Kyong Shon. 2018. "Modification of Nanofiber Support Layer for Thin Film Composite Forward Osmosis Membranes via Layer-by-Layer Polyelectrolyte Deposition" Membranes 8, no. 3: 70. https://doi.org/10.3390/membranes8030070
APA StyleGonzales, R. R., Park, M. J., Tijing, L., Han, D. S., Phuntsho, S., & Shon, H. K. (2018). Modification of Nanofiber Support Layer for Thin Film Composite Forward Osmosis Membranes via Layer-by-Layer Polyelectrolyte Deposition. Membranes, 8(3), 70. https://doi.org/10.3390/membranes8030070