Thin Hydrogel Films for Optical Biosensor Applications

Abstract
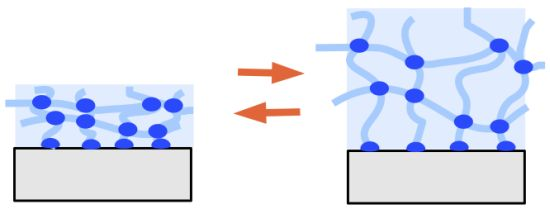
:
1. Introduction
2. Classification of Hydrogel Systems
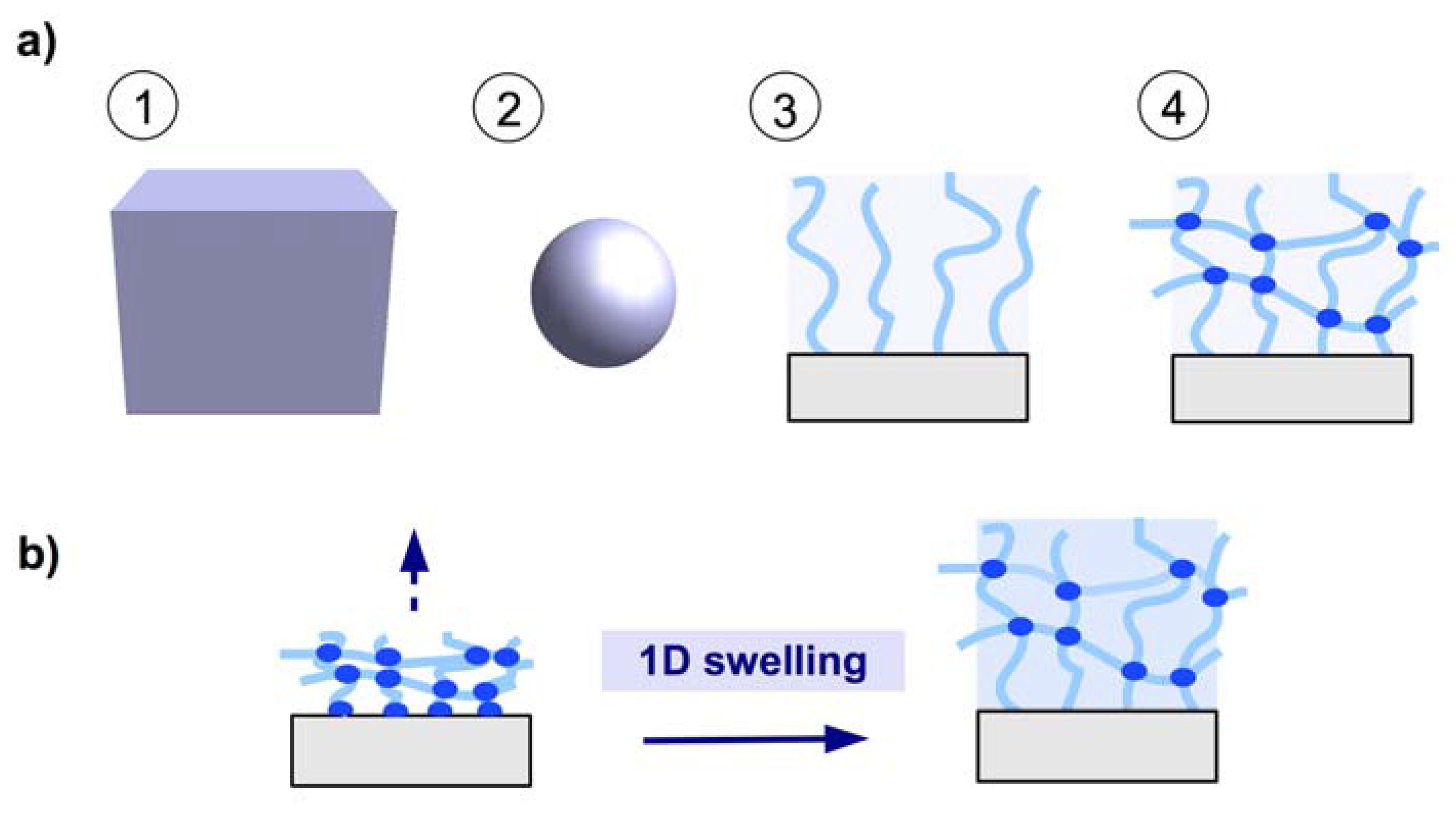
2.1. Chemical Structures of Polymer-Based Hydrogels
2.1.1. Main Monomer
2.1.1.1 Monomers and Polymers for Hydrogel Films
2.1.2. Functional Groups
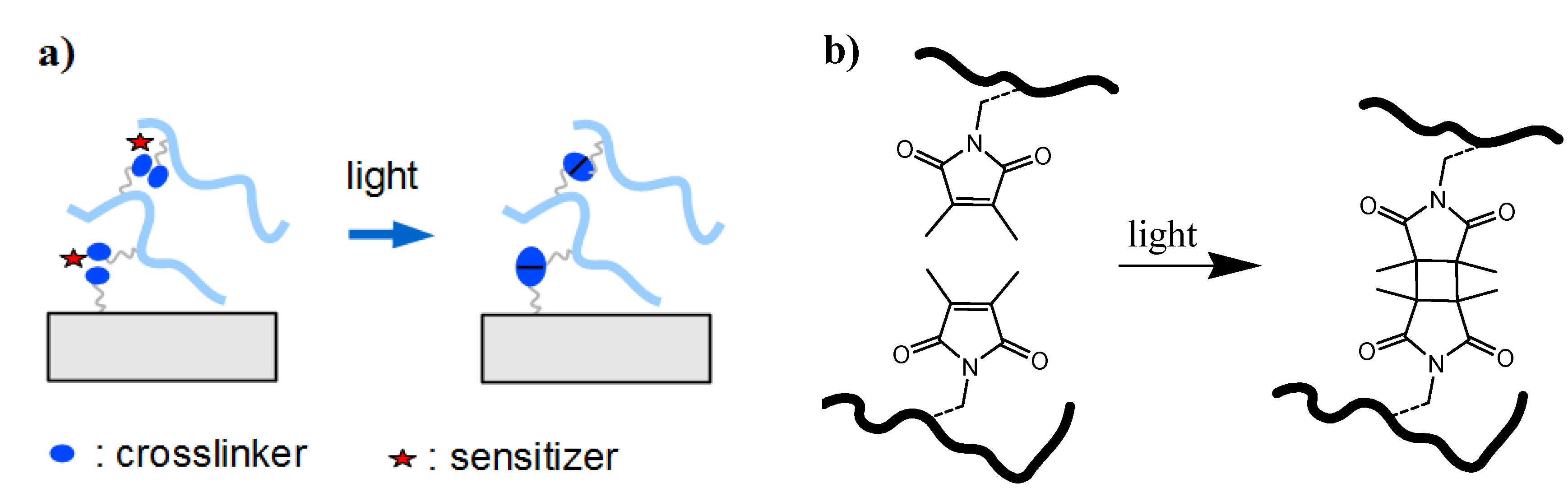
2.1.3. Crosslinkers
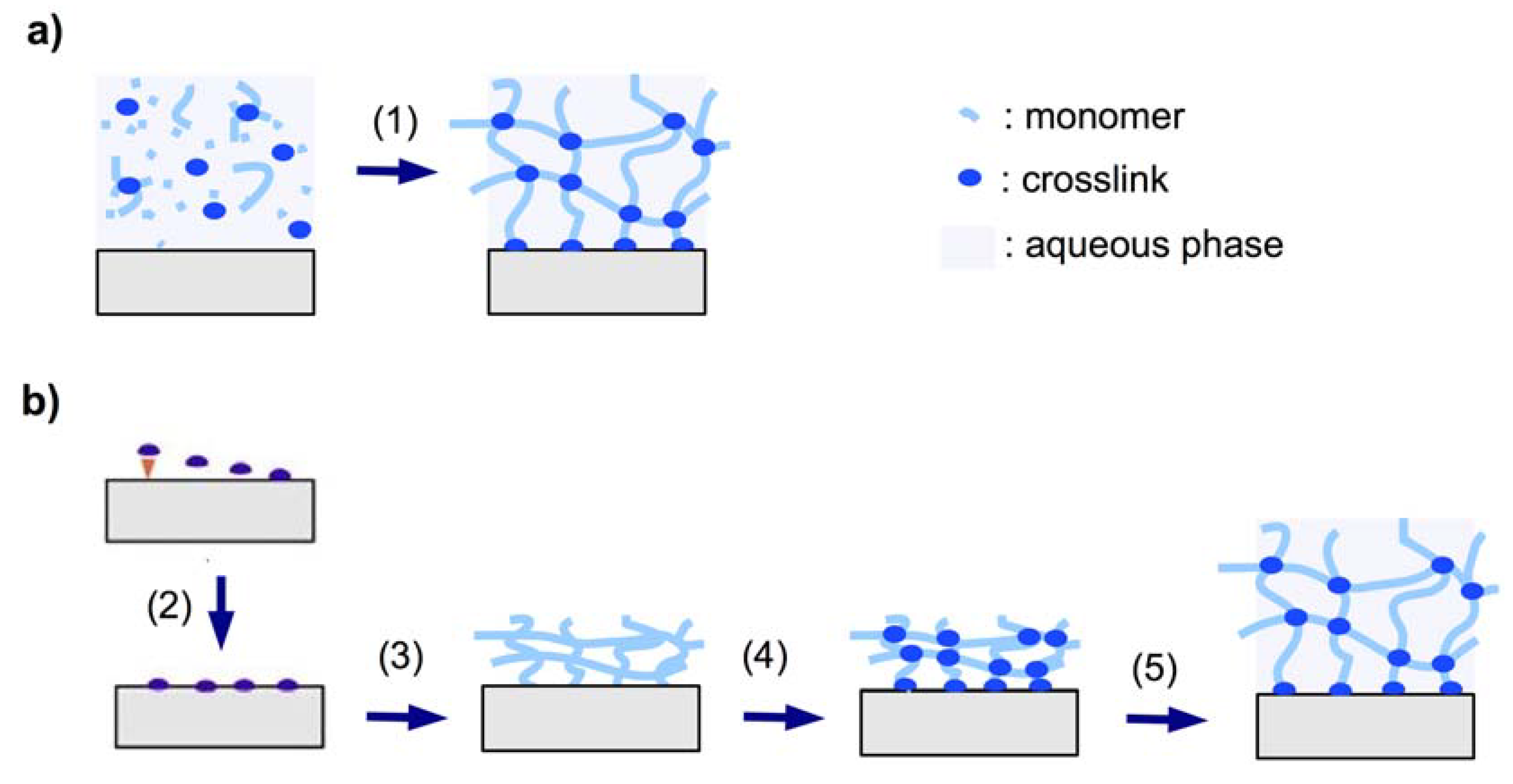
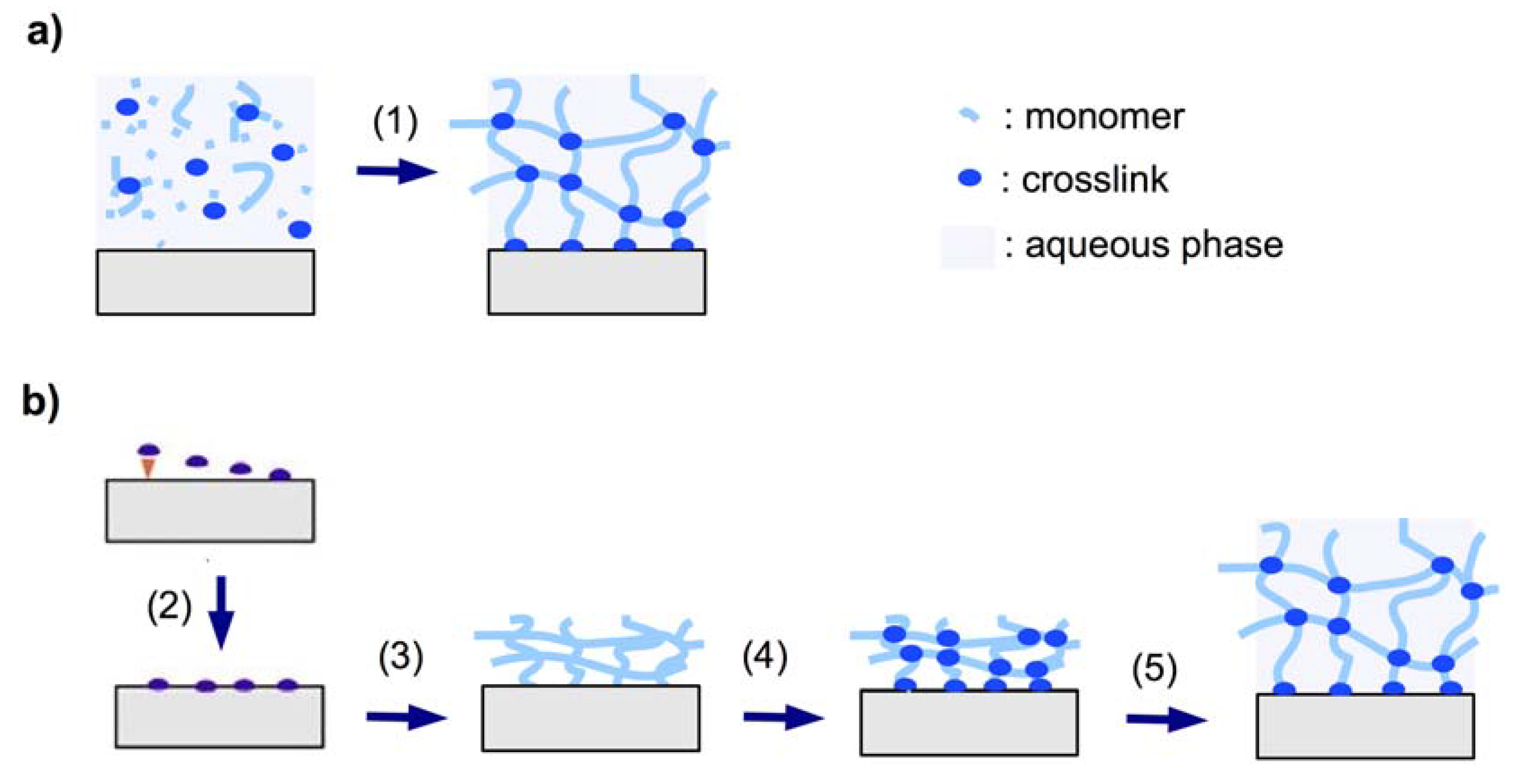
2.1.3.1. In Situ Crosslinking
2.1.3.2. Post-Synthetic Crosslinking
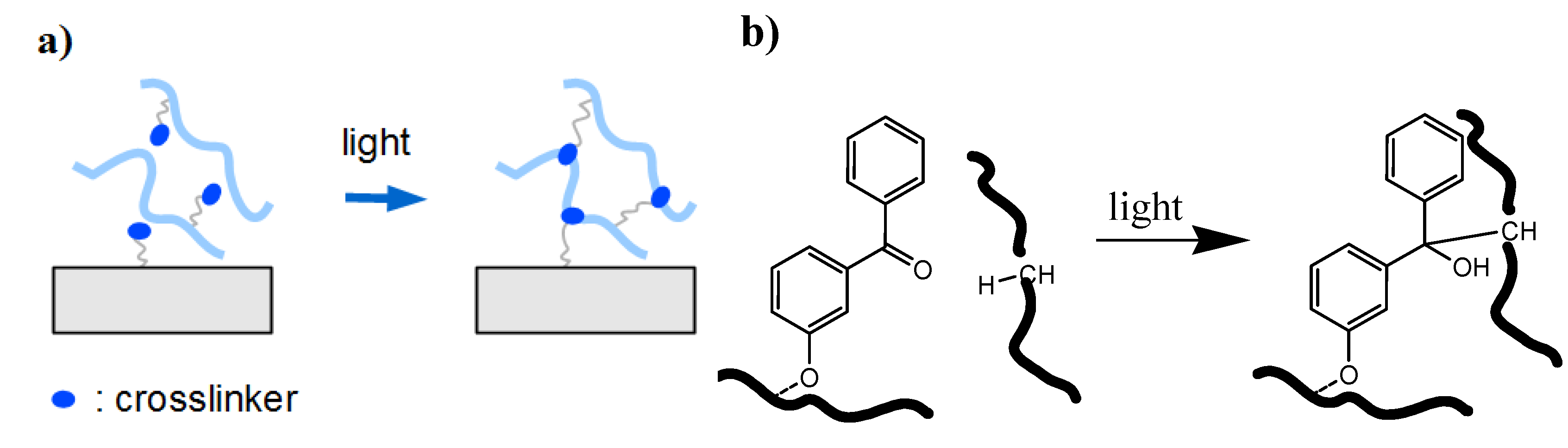
2.2. Surface Attachment Strategies
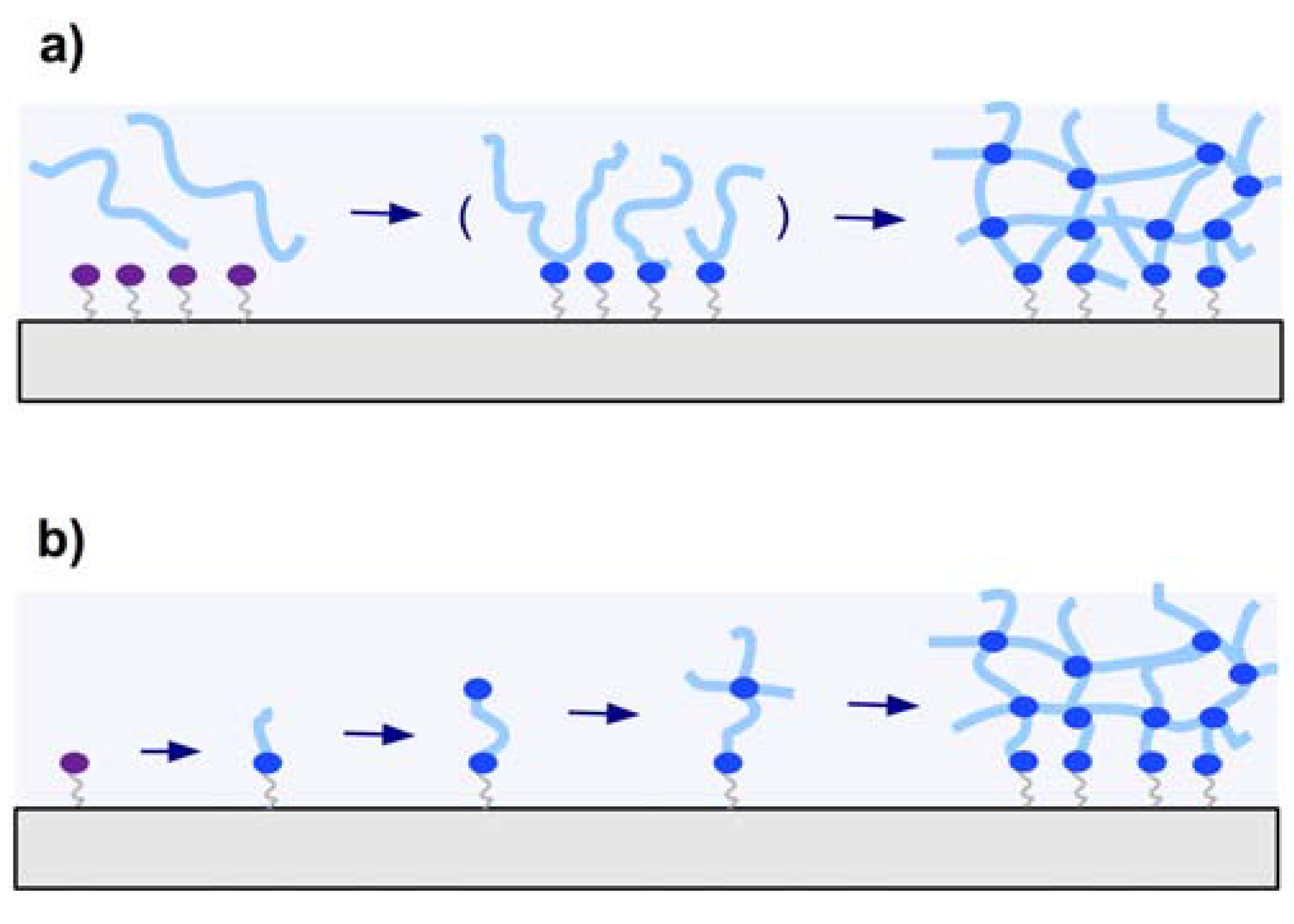
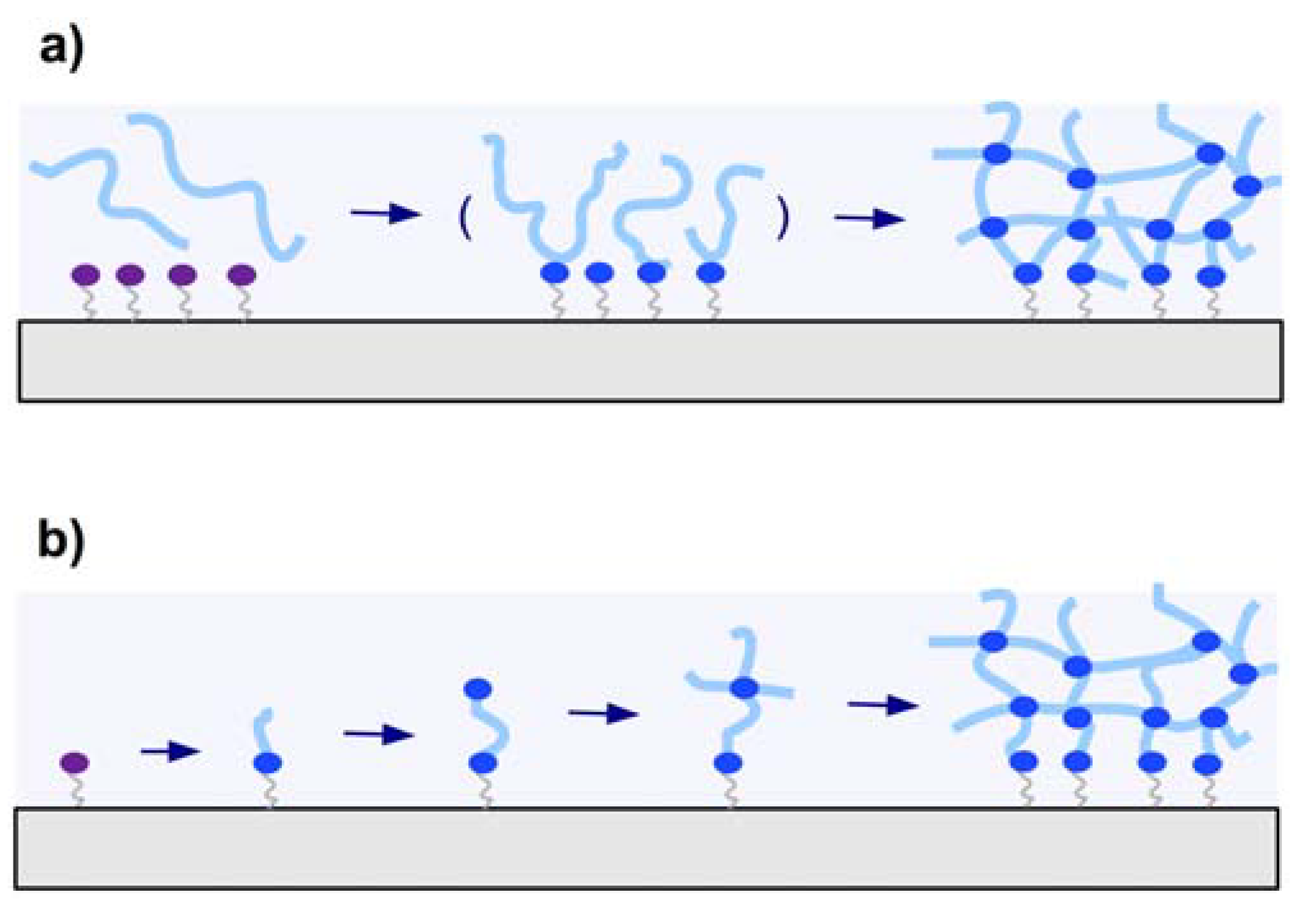
2.2.1. “Grafting to” Method
2.2.1.1. Chemical “Grafting to”
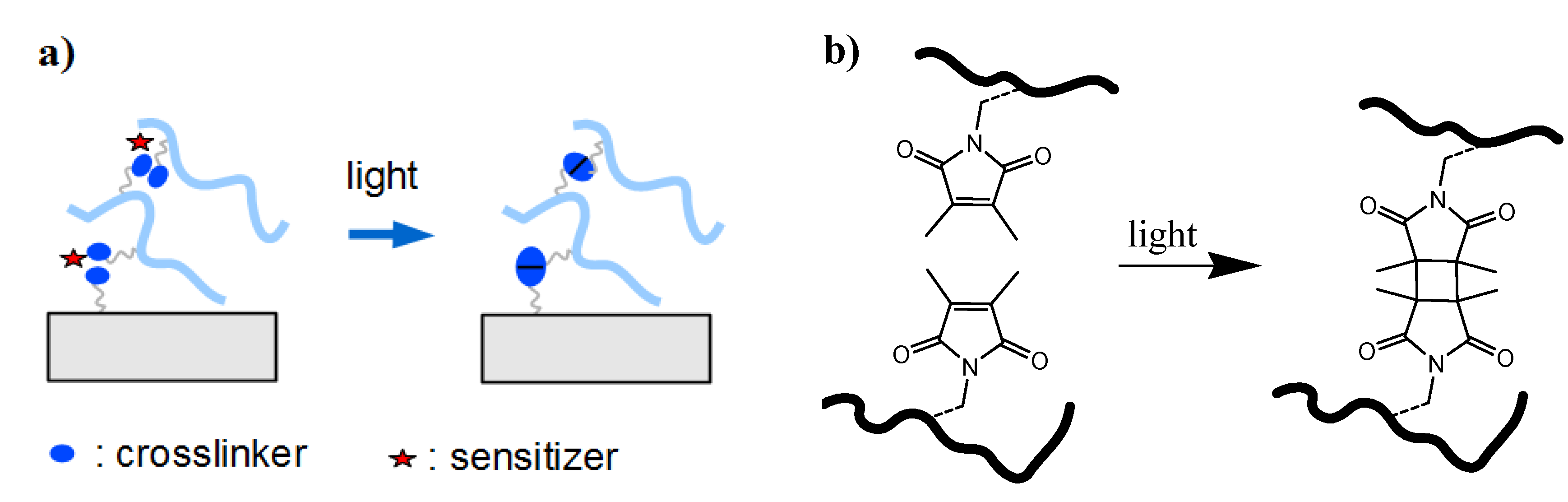
2.2.1.2. Photochemical “Grafting to”
- (1)
- Bond formation between photoactivated functional groups of the polymer chain with the substrate material, which usually also leads to crosslinking of the polymer film.
- (2)
- Reaction of photoreactive groups found at the substrate surface (e.g., from adhesion promotor layers) with the polymer chains of the hydrogel network.
2.2.2. “Grafting from” Method
2.3. Coating Methods
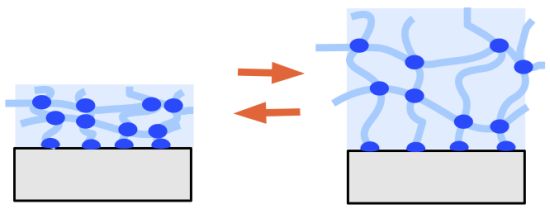
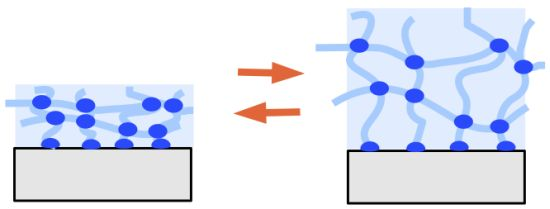
3. Properties of Hydrogel Layers
- spatial inhomogeneities of non-uniform distribution of crosslinks in space,
- topological inhomogeneities of the network such as loops, trapped entanglements, and dangling chain ends,
- connectivity inhomogeneities of the polymer cluster distribution in size and space, which is also related to variations of the branching architecture of the network.
4. Biosensor Implementations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Recognition element | Material | Detection method | Thickness(dh) | Analyte | LOD(sample) | Ref. |
|---|---|---|---|---|---|---|
| MIP | poly(2-vinylpyridine)-AuNPs | LSPR | 31 nm | cholesterol | n.a. (chloroform) | [90] |
| acrylamidophenylboronic acid- acrylamide | SPR | 22 nm | NAD(P)+ NAD(P)H | 1 µM (buffer) | [91] | |
| acrylic acid- | SPR | ~6 µm (dry) | dopamine | 1 nM (water) | [92] | |
| N-isopropylacrylamide- | ||||||
| N,N’-methylenbisacrylamide-AuNPs | ||||||
| poly(N-(N-propyl)acrylamide) | SPR | ~300 nm | Theophylline | 10 µM (buffer) | [93] | |
| methacylic acid-ethylene | SPR | 251 nm | atrazine | 5 pM (acetonitrile) | [94] | |
| glycol dimethacrylate-AuNPs | ||||||
| Enzyme | agaros-guar gum | OWS | 12 µm | sucrose | 25 pM (buffer) | [95] |
| polysaccharide | ||||||
| agarose co-polymer | OWS | 1 µm | paraoxon | 6 nM (buffer) | [96] | |
| agarose co-polymer | OWS-Fluorescence | 1 µm | glucose | 3 µM (buffer) | [96] | |
| alginate-gelatin-AgNPs | LSPR | 20 nm (dry) | glucose | 0.1 m (buffer) | [97]] | |
| acrylamide-bisacrylamide-AgNPs | LSPR | ~1 mm | glucose | 10 pM (buffer) | [98] | |
| Nucleic acid | POWT | SPR | 8 nm(dry) | DNA | n.a. (buffer) | [99] |
| aptamer-polyacrylamide | LSPR | n.a. | adenosine | n.a. (buffer) | [100] | |
| aptamer-polyacrylamide | LSPR | n.a. | cocanie | n.a. (buffer) | [101] | |
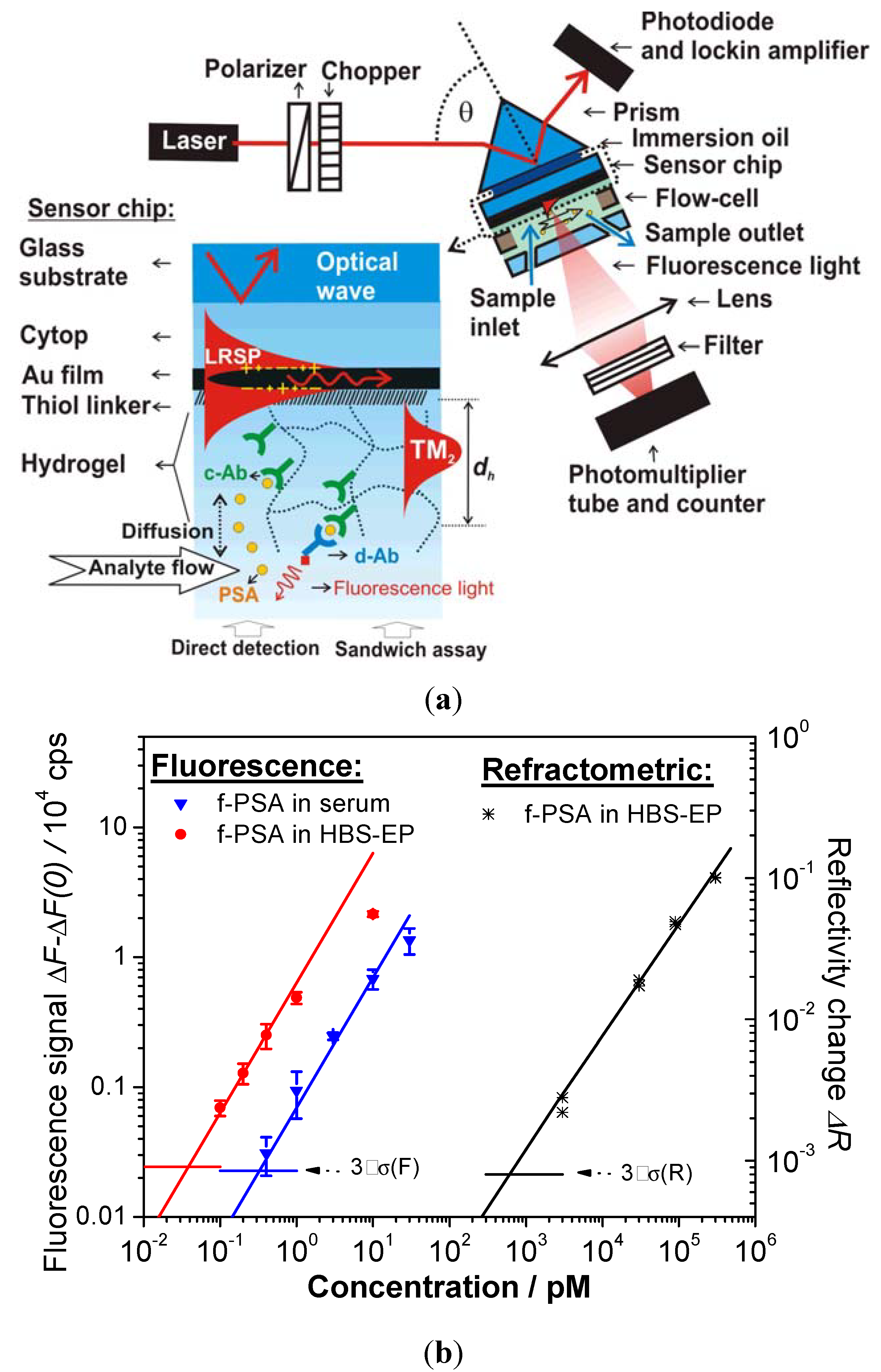
| Immuno-assay | carboxymethyl dextran | LR-SPR | ~1 µm | f-PSA | 0.68 n (buffer) | [23] |
| carboxymethyl dextran | LRSP-FS | ~1 µm | f-PSA | 34 fM (buffer) | [23] | |
| 330 fM (human serum) | ||||||
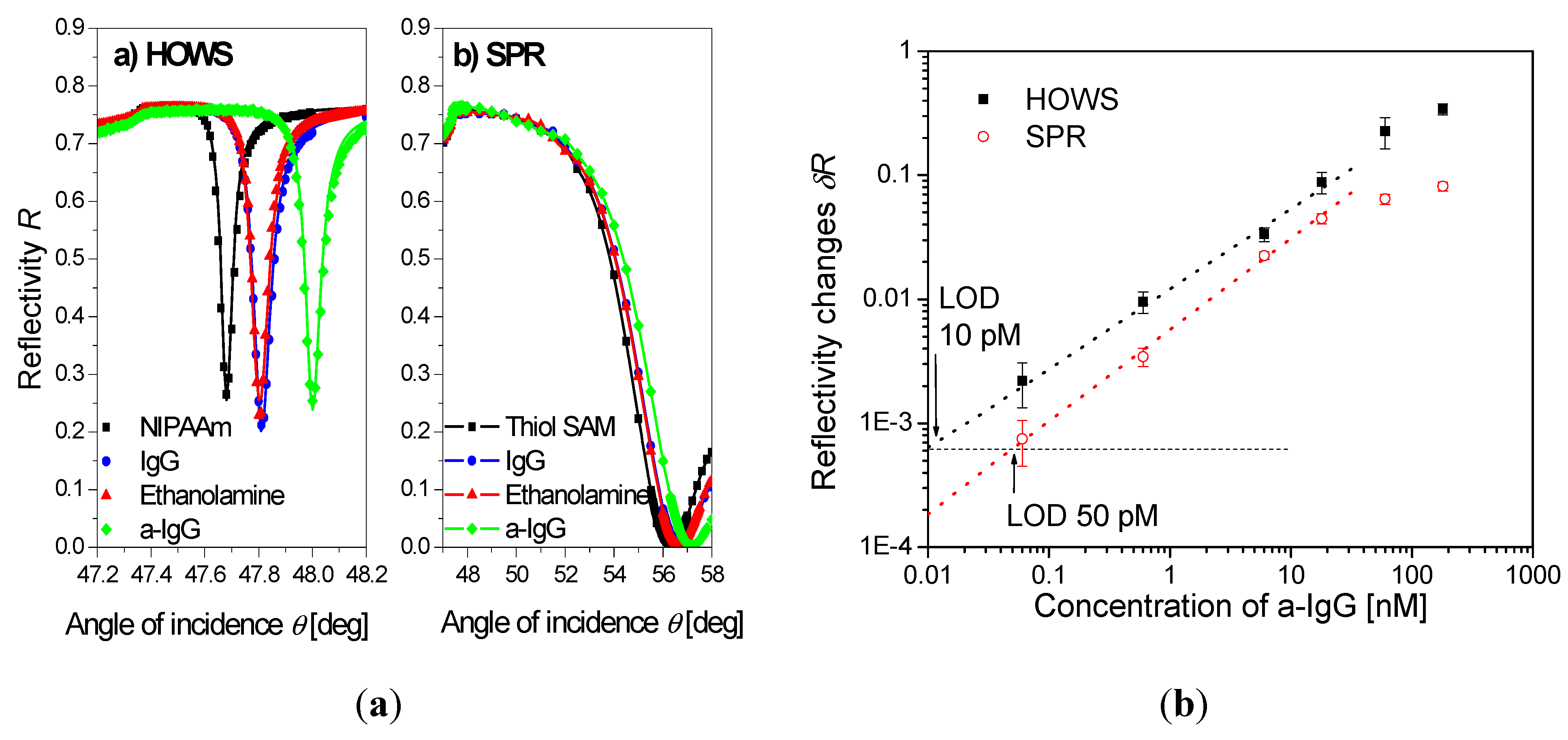
| poly(N-isopropylacrylamide) | HOWS | ~2 µm | IgG | 10 pM(buffer) | [102] | |
| poly(ethyleneglycol) methacrylate-2-hydroxyethyl methacrylate | SPRi | 5–45 nm | HSA and calmodulin | n.a.(buffer) | [25] |
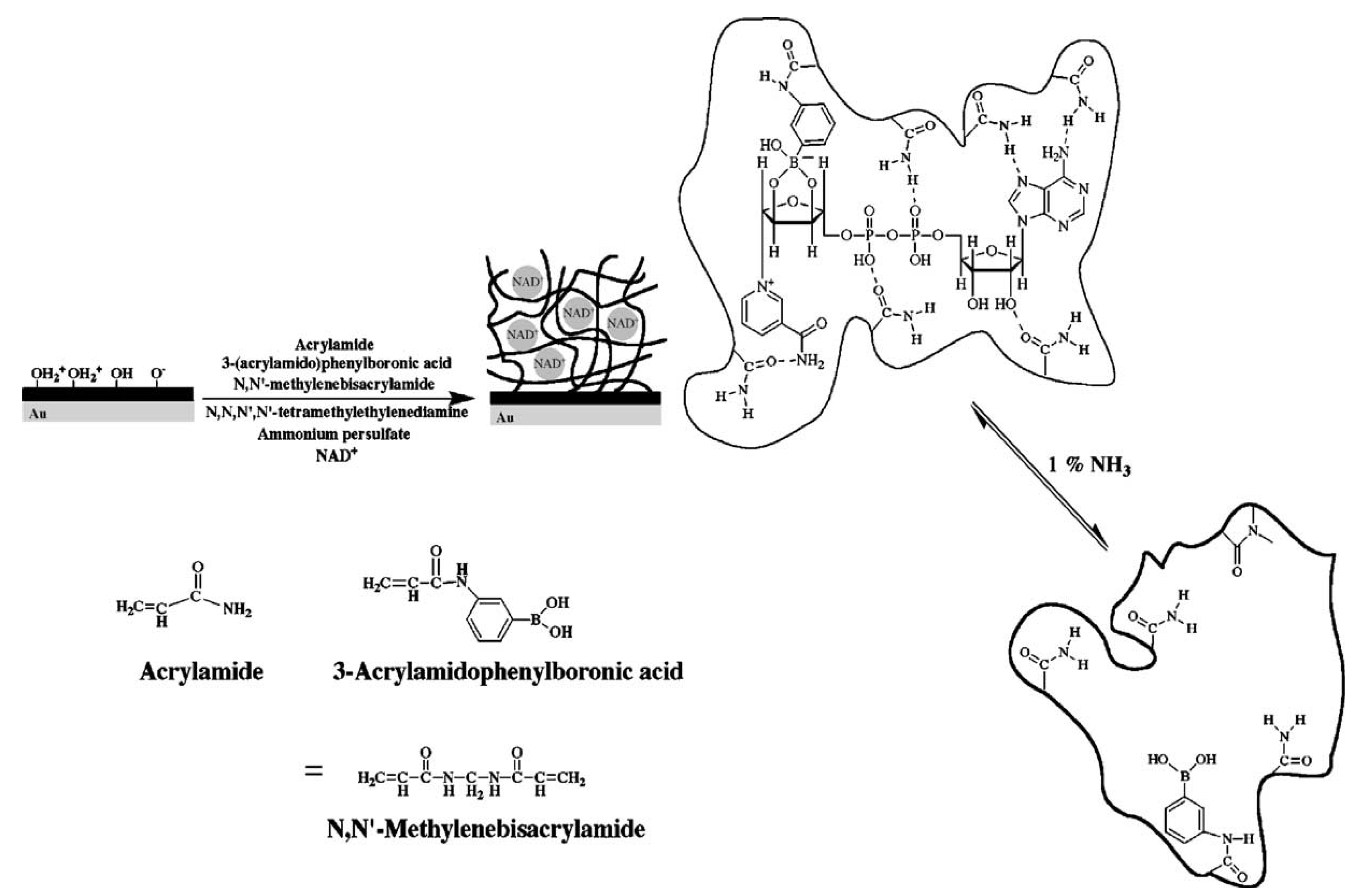
4.1. Molecular Imprinted Hydrogel-Based Biosensors
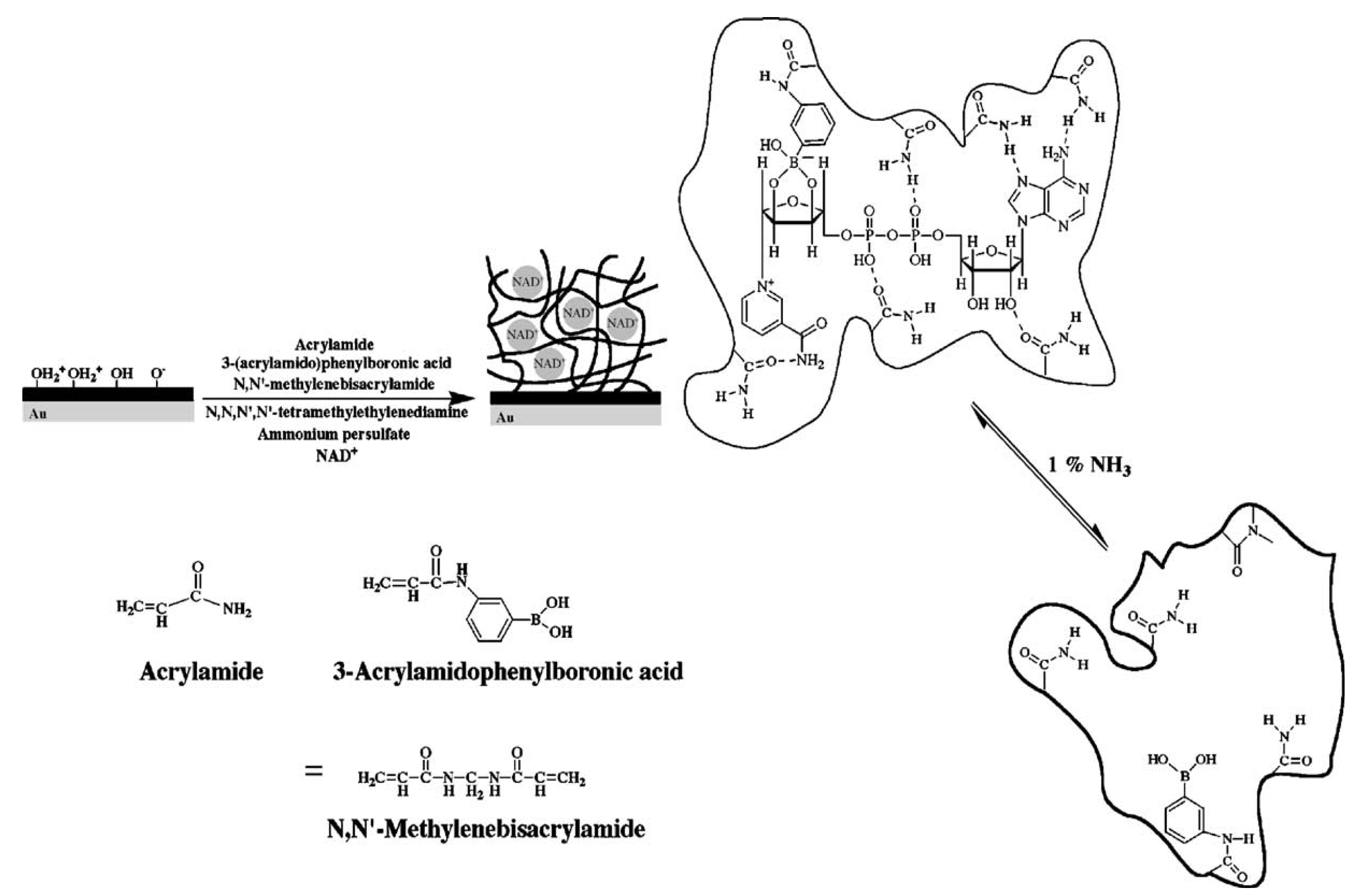
4.2. Enzyme-Based Biosensors
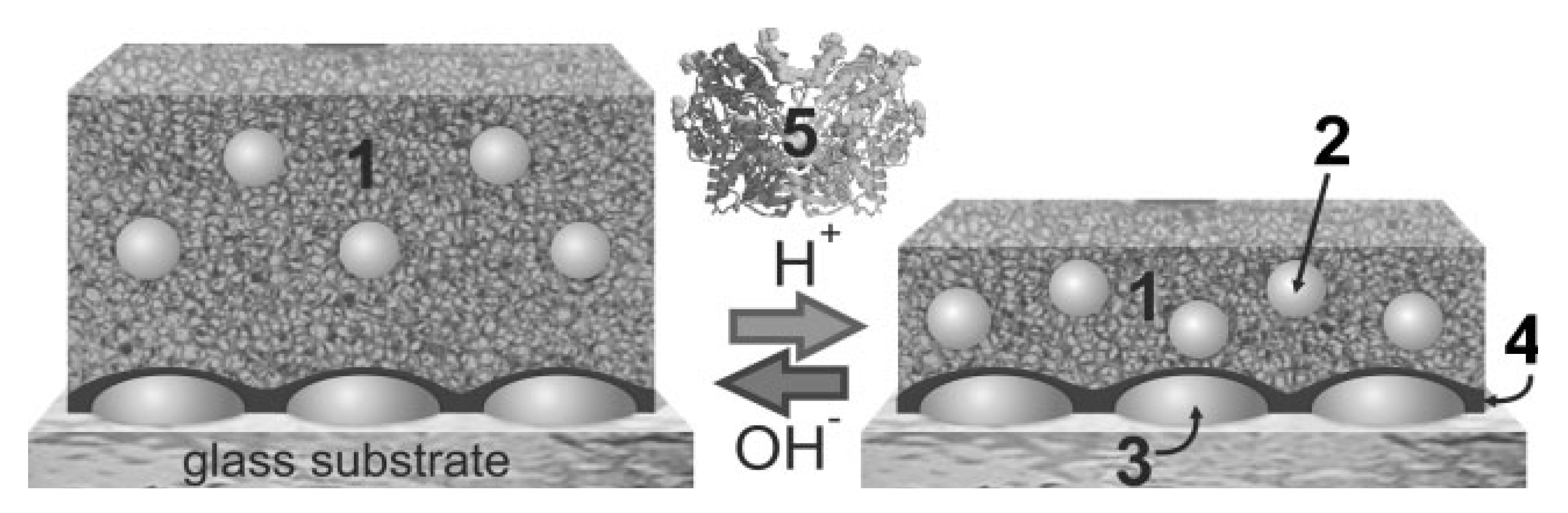
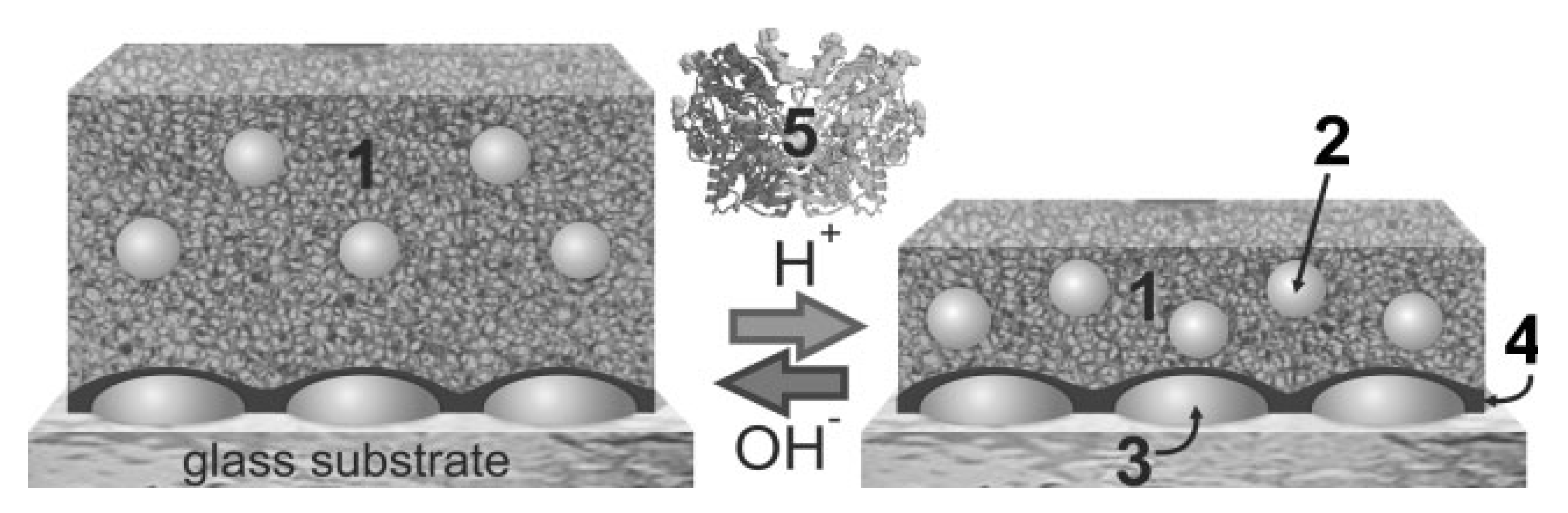
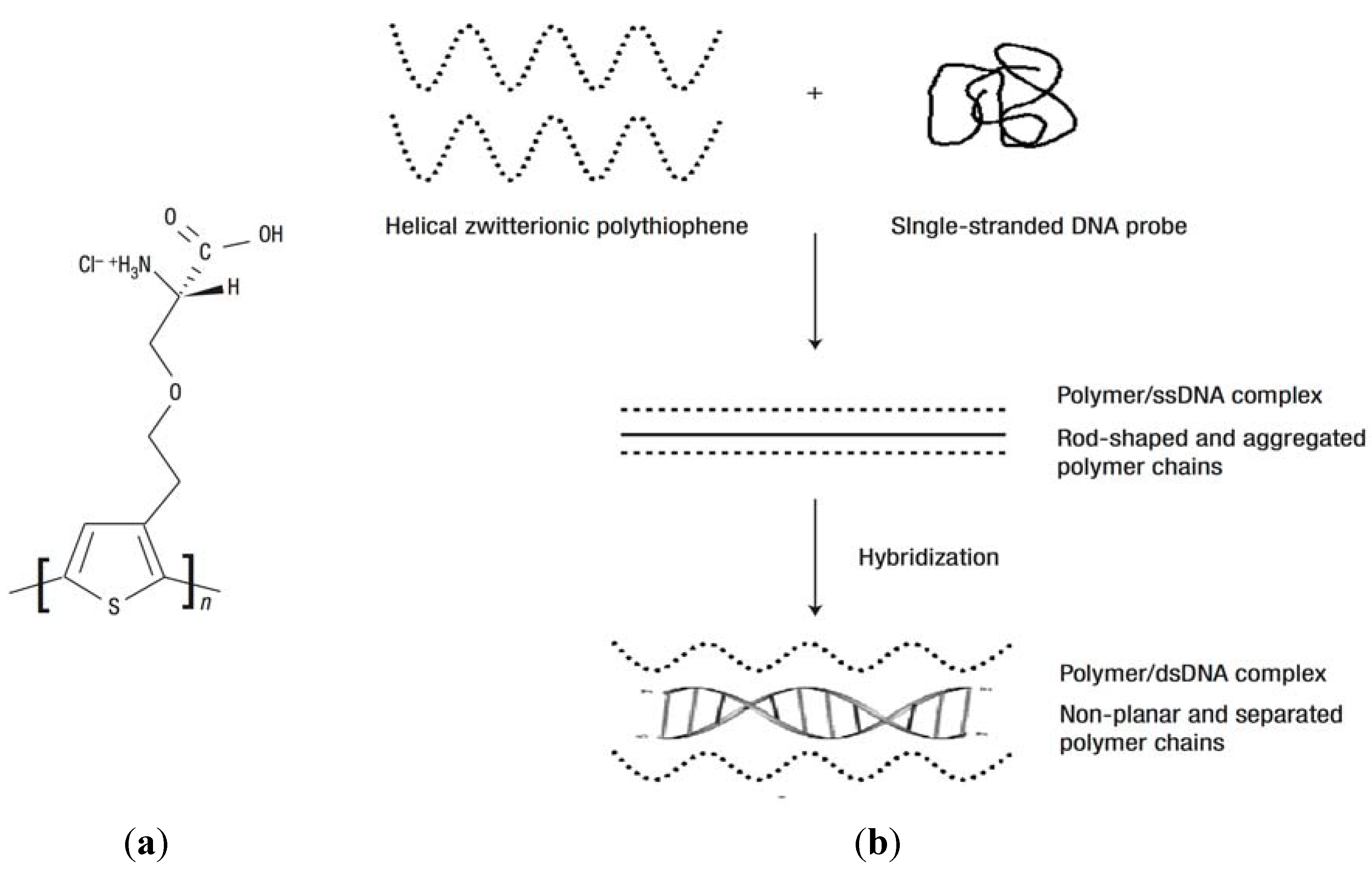
4.3. Nucleic Acids-Based Biosensors
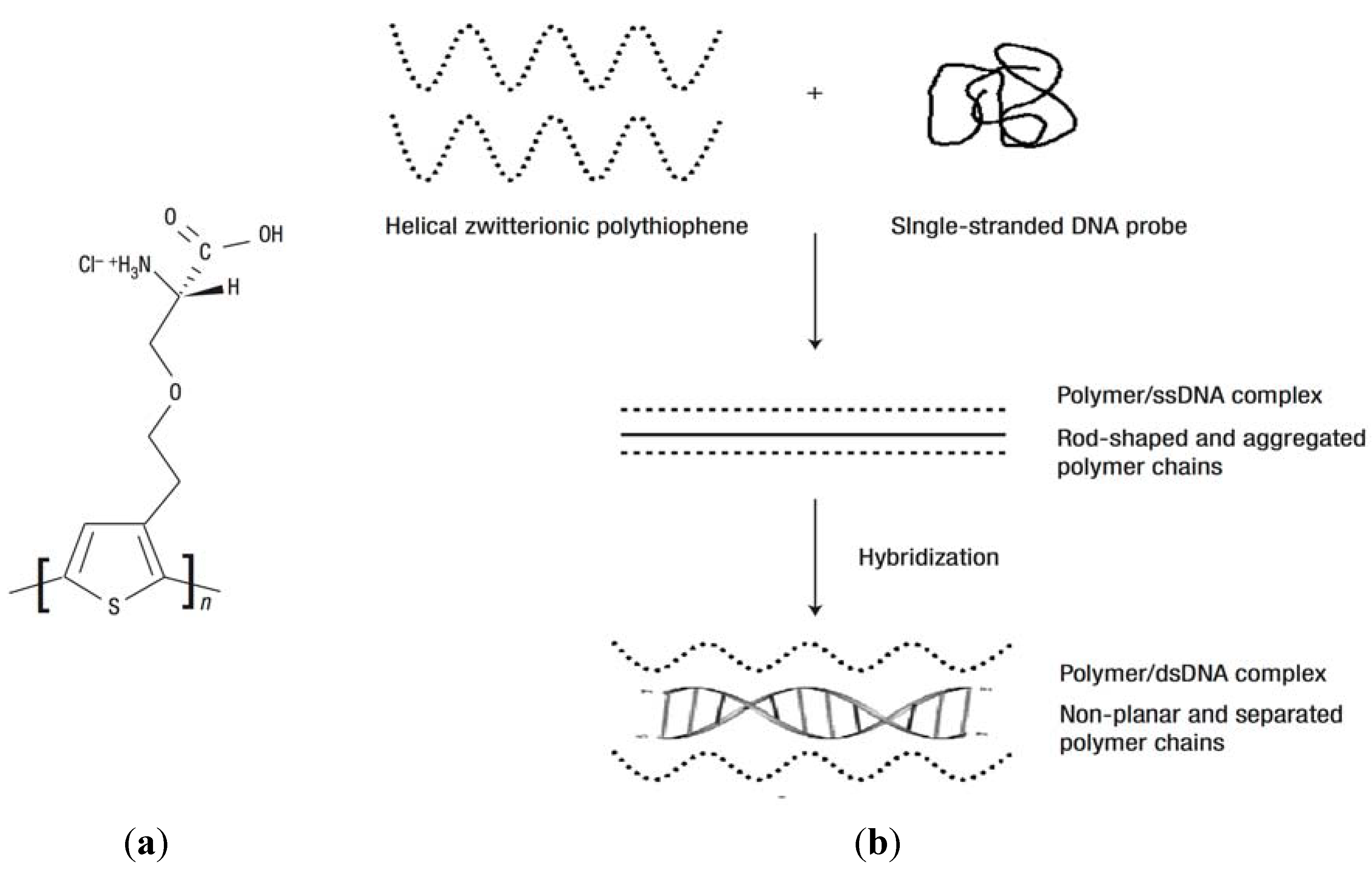
4.4. Immunoassay-Based Biosensors
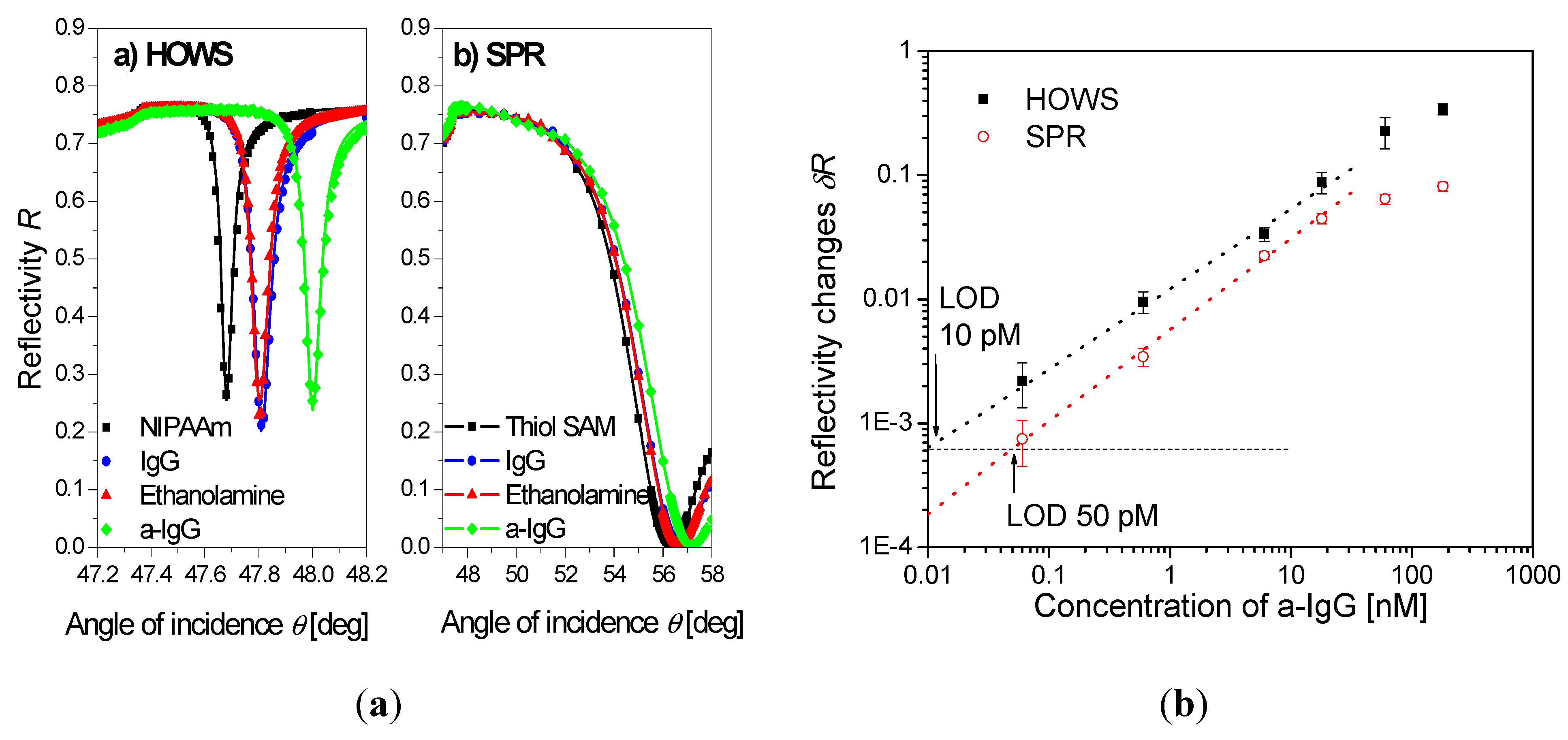
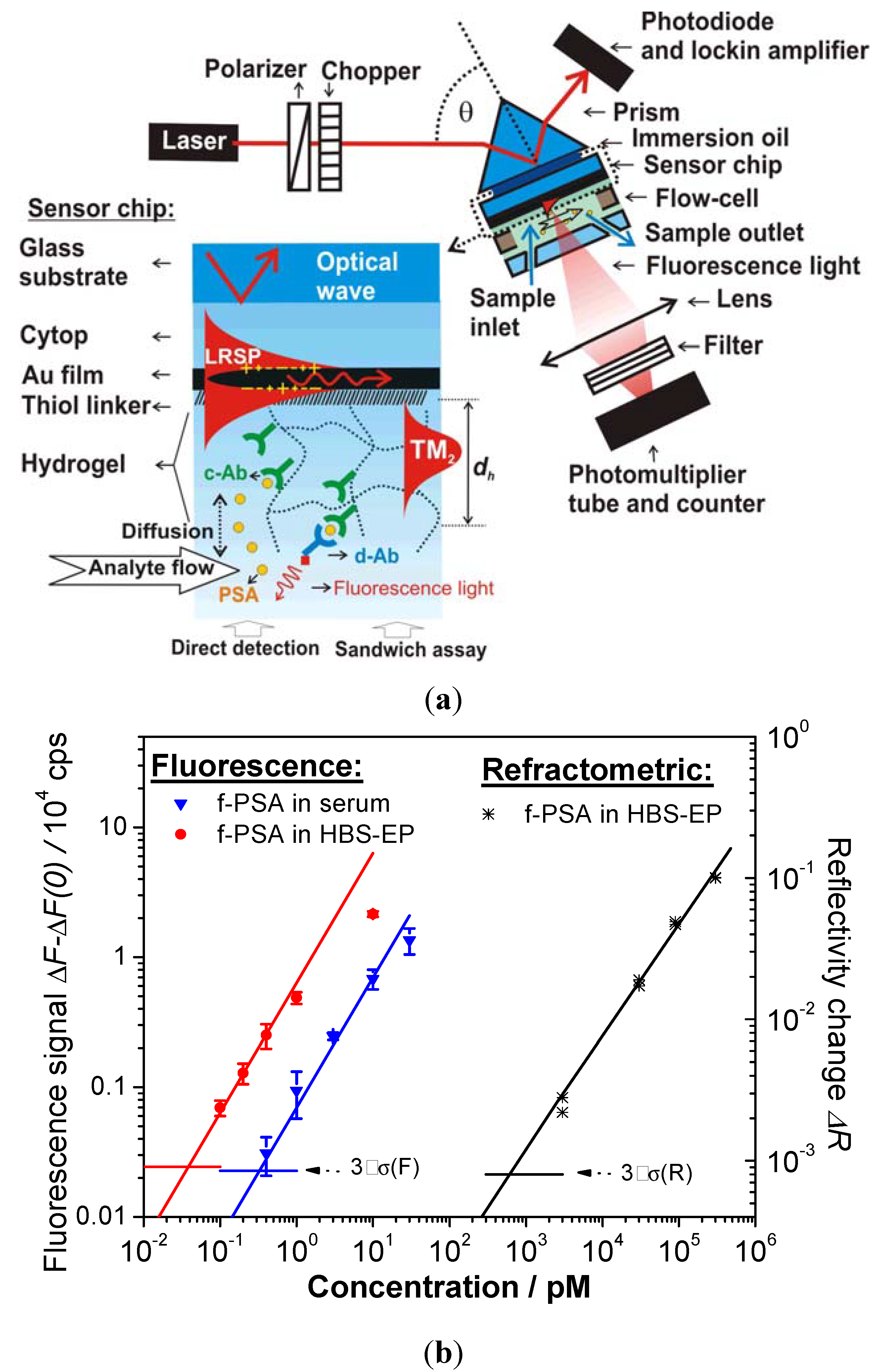
5. Conclusions
Acknowledgments
References
- Kuenzler, J.F. Hydrogels. In Encyclopedia of Polymer Science and Technology,3rd ed.; Mark, H.F., Ed.; Wiley-Interscience: New York, NY, USA, 2004; Volume 2, pp. 691–722. [Google Scholar]
- Estroff, L.A.; Hamilton, A.D. Water gelation by small organic molecules. Chem. Rev. 2004, 104, 1201–1217. [Google Scholar]
- Nicodemus, G.D.; Bryant, S.J. Cell encapsulation in biodegradable hydrogels for tissue engineering applications. Tissue Eng. Part B-Rev. 2008, 14, 149–165. [Google Scholar]
- Shoichet, M.S. Polymer scaffolds for biomaterials applications. Macromolecules 2010, 43, 581–591. [Google Scholar] [CrossRef]
- Slaughter, B.V.; Khurshid, S.S.; Fisher, O.Z.; Khademhosseini, A.; Peppas, N.A. Hydrogels in regenerative medicine. Adv. Mater. 2009, 21, 3307–3329. [Google Scholar]
- Hamidi, M.; Azadi, A.; Rafiei, P. Hydrogel nanoparticles in drug delivery. Adv. Drug Deliver. Rev. 2008, 60, 1638–1649. [Google Scholar] [CrossRef]
- Hoare, T.R.; Kohane, D.S. Hydrogels in drug delivery: Progress and challenges. Polymer 2008, 49, 1993–2007. [Google Scholar] [CrossRef]
- Bajpai, A.K.; Shukla, S.K.; Bhanu, S.; Kankane, S. Responsive polymers in controlled drug delivery. Prog. Polym. Sci. 2008, 33, 1088–1118. [Google Scholar] [CrossRef]
- Okano, T.; Yamada, N.; Sakai, H.; Sakurai, Y. A novel recovery-system for cultured-cells using plasma-treated polystyrene dishes grafted with poly(N-isopropylacrylamide). J. Biomed. Mater. Res. 1993, 27, 1243–1251. [Google Scholar] [CrossRef]
- Kwon, O.H.; Kikuchi, A.; Yamato, M.; Sakurai, Y.; Okano, T. Rapid cell sheet detachment from poly(N-isopropylacrylamide)-grafted porous cell culture membranes. J. Biomed. Mater. Res. 2000, 50, 82–89. [Google Scholar] [CrossRef]
- Nishida, K.; Yamato, M.; Hayashida, Y.; Watanabe, K.; Yamamoto, K.; Adachi, E.; Nagai, S.; Kikuchi, A.; Maeda, N.; Watanabe, H.; Okano, T.; Tano, Y. Corneal reconstruction with tissue-engineered cell sheets composed of autologous oral mucosal epithelium. N. Engl. J. Med. 2004, 351, 1187–1196. [Google Scholar] [CrossRef]
- Hillebrandt, H.; Wiegand, G.; Tanaka, M.; Sackmann, E. High electric resistance polymer/lipid composite films on indium-tin-oxide electrodes. Langmuir 1999, 15, 8451–8459. [Google Scholar] [CrossRef]
- Kuhner, M.; Tampe, R.; Sackmann, E. Lipid monolayer and bilayer supported on polymer-films- composite polymer-lipid films on solid substrates. Biophys. J. 1994, 67, 217–226. [Google Scholar] [CrossRef]
- Sackmann, E. Supported membranes: Scientific and practical applications. Science 1996, 271, 43–48. [Google Scholar]
- Simon, J.; Kuhner, M.; Ringsdorf, H.; Sackmann, E. Polymer-induced shape changes and capping in giant liposomes. Chem. Phys. Lipids 1995, 76, 241–258. [Google Scholar] [CrossRef]
- Tanaka, M.; Sackmann, E. Polymer-supported membranes as models of the cell surface. Nature 2005, 437, 656–663. [Google Scholar] [CrossRef]
- Urban, G.A.; Weiss, T. Hydrogels in biosensors. In Hydrogel Sensors and Actuators, 1st; Urban, G.A., Ed.; Springer: Berlin, Germany, 2009; Volume 6, pp. 197–220. [Google Scholar]
- Richter, A.; Paschew, G.; Klatt, S.; Lienig, J.; Arndt, K.F.; Adler, H.J.P. Review on hydrogel-based pH sensors and microsensors. Sensors 2008, 8, 561–581. [Google Scholar] [CrossRef]
- Tokarev, I.; Minko, S. Stimulli-responsive hydrogel thin films. Soft Matter 2009, 5, 511–524. [Google Scholar] [CrossRef]
- Davies, M.L.; Murphy, S.M.; Hamilton, C.J.; Tighe, B.J. Polymer membranes in clinical sensor applications. 3. Hydrogels as reactive matrix membranes in fiber optic sensors. Biomaterials 1992, 13, 991–999. [Google Scholar] [CrossRef]
- Murphy, S.M.; Hamilton, C.J.; Davies, M.L.; Tighe, B.J. Polymer membranes in clinical sensor applications. 2. The design and fabrication of permselective hydrogels for electrochemical devices. Biomaterials 1992, 13, 979–990. [Google Scholar] [CrossRef]
- Guenther, M.; Gerlach, G. Hydrogels for chemical sensors. In Hydrogel Sensors and Actuators, 1st; Urban, G.A., Ed.; Springer: Berlin, Germany, 2009; Volume 6, pp. 165–196. [Google Scholar]
- Wang, Y.; Brunsen, A.; Jonas, U.; Dostalek, J.; Knoll, W. Prostate specific antigen biosensor based on long range surface plasmon-enhanced fluorescence spectroscopy and dextran hydrogel binding matrix. Anal. Chem. 2009, 81, 9625–9632. [Google Scholar]
- Gerhke, S.H.; Uhden, L.H.; McBride, J.F. Enhanced loading and activity retention of bioactive proteins in hydrogel delivery systems. J. Control. Release 1998, 55, 21–33. [Google Scholar] [CrossRef]
- Andersson, O.; Larsson, A.; Ekbald, T.; Liedberg, B. Gradient hydrogel matrix for microarray and biosensor applications: An imaging SPR study. Biomacromolecules 2009, 10, 142–148. [Google Scholar] [CrossRef]
- Yang, X.P.; Pan, X.H.; Blyth, J.; Lowe, C.R. Towards the real-time monitoring of glucose in tear fluid: Holographic glucose sensors with reduced interference from lactate and pH. Biosens. Bioelectron. 2008, 23, 899–905. [Google Scholar] [CrossRef]
- Bhat, V.T.; James, N.R.; Jayakrishnan, A. A photochemical method for immobilization of azidated dextran onto aminated poly(ethylene terephthalate) surfaces. Polym. Int. 2008, 57, 124–132. [Google Scholar] [CrossRef]
- Glampedaki, P.; Jocic, D.; Warmoeskerken, M.M.C.G. Moisture absorption capacity of polyamide 6,6 fabrics surface functionalised by chitosan-based hydrogel finishes. Prog. Org. Coat. 2011, 72, 562–571. [Google Scholar]
- Tang, Y.; Lu, J.R.; Lewis, A.L.; Vick, T.A.; Stratford, P.W. Swelling of zwitterionic polymer films characterized by spectroscopic ellipsometry. Macromolecules 2001, 34, 8768–8776. [Google Scholar] [CrossRef]
- Toomey, R.; Freidank, D.; Ruhe, J. Swelling behavior of thin, surface-attached polymer networks. Macromolecules 2004, 37, 882–887. [Google Scholar] [CrossRef]
- Zhang, Y.F.; Ji, H.F.; Brown, G.M.; Thundat, T. Detection of CrO42− using a hydrogel swelling microcantilever sensor. Anal. Chem. 2003, 75, 4773–4777. [Google Scholar] [CrossRef]
- Kibrom, A.; Roskamp, R.F.; Jonas, U.; Menges, B.; Knoll, W.; Paulsene, H.; Naumann, R.L.C. Hydrogel-supported protein-tethered bilayer lipid membranes: A new approach toward polymer-supported lipid membranes. Soft Matter 2011, 7, 237–246. [Google Scholar]
- Tokarev, I.; Minko, S. Stimuli-responsive hydrogel thin films. Soft Matter 2009, 5, 511–524. [Google Scholar] [CrossRef]
- Anac, I.; Aulasevich, A.; Junk, M.J.N.; Jakubowicz, P.; Roskamp, R.F.; Menges, B.; Jonas, U.; Knoll, W. Optical characterization of co-nonsolvency effects in thin responsive PNIPAAm-based gel layers exposed to ethanol/water mixtures. Macromol. Chem. Phys. 2010, 211, 1018–1025. [Google Scholar] [CrossRef]
- Beines, P.W.; Klosterkamp, I.; Menges, B.; Jonas, U.; Knoll, W. Responsive thin hydrogel layers from photo-cross-linkable poly(N-isopropylacrylamide) terpolymers. Langmuir 2007, 23, 2231–2238. [Google Scholar] [CrossRef]
- Duval, J.F.L.; Zimmermann, R.; Cordeiro, A.L.; Rein, N.; Werner, C. Electrokinetics of diffuse soft interfaces. IV. Analysis of streaming current measurements at thermoresponsive thin films. Langmuir 2009, 25, 10691–10703. [Google Scholar] [CrossRef]
- Guenther, M.; Kuckling, D.; Corten, C.; Gerlach, G.; Sorber, J.; Suchaneck, G.; Arndt, K.F. Chemical sensors based on multiresponsive block copolymer hydrogels. Sensor. Actuator. B-Chem. 2007, 126, 97–106. [Google Scholar]
- Harmon, M.E.; Jakob, T.A.M.; Knoll, W.; Frank, C.W. A surface plasmon resonance study of volume phase transitions in N-isopropylacrylamide gel films. Macromolecules 2002, 35, 5999–6004. [Google Scholar] [CrossRef]
- Harmon, M.E.; Kuckling, D.; Frank, C.W. Photo-cross-linkable PNIPAAm copolymers. 5. Mechanical properties of hydrogel layers. Langmuir 2003, 19, 10660–10665. [Google Scholar] [CrossRef]
- Harmon, M.E.; Kuckling, D.; Frank, C.W. Photo-cross-linkable PNIPAAm copolymers. 2. Effects of constraint on temperature and pH-responsive hydrogel layers. Macromolecules 2003, 36, 162–172. [Google Scholar]
- Harmon, M.E.; Kuckling, D.; Pareek, P.; Frank, C.W. Photo-cross-linkable PNIPAAm copolymers. 4. Effects of copolymerization and cross-linking on the volume-phase transition in constrained hydrogel layers. Langmuir 2003, 19, 10947–10956. [Google Scholar]
- Junk, M.J.N.; Berger, R.; Jonas, U. Atomic force spectroscopy of thermoresponsive photo-cross-linked hydrogel films. Langmuir 2010, 26, 7262–7269. [Google Scholar] [CrossRef]
- Junk, M.J.N.; Jonas, U.; Hinderberger, D. EPR spectroscopy reveals nanoinhomogeneities in the structure and reactivity of thermoresponsive hydrogels. Small 2008, 4, 1485–1493. [Google Scholar] [CrossRef]
- Kuckling, D.; Harmon, M.E.; Frank, C.W. Photo-cross-linkable PNIPAAm copolymers. 1. Synthesis and characterization of constrained temperature-responsive hydrogel layers. Macromolecules 2002, 35, 6377–6383. [Google Scholar]
- Kuckling, D.; Hoffmann, J.; Plotner, M.; Ferse, D.; Kretschmer, K.; Adler, H.J.P.; Arndt, K.F.; Reicheltd, R. Photo cross-linkable poly (N-isopropylacrylamide) copolymers III: Micro-fabricated temperature responsive hydrogels. Polymer 2003, 44, 4455–4462. [Google Scholar] [CrossRef]
- Liao, K.S.; Fu, H.; Wan, A.; Batteas, J.D.; Bergbreiter, D.E. Designing surfaces with wettability that varies in response to solute identity and concentration. Langmuir 2009, 25, 26–28. [Google Scholar] [CrossRef]
- Schmaljohann, D.; Beyerlein, D.; Nitschke, M.; Werner, G. Thermo-reversible swelling of thin hydrogel films immobilized by low-pressure plasma. Langmuir 2004, 20, 10107–10114. [Google Scholar] [CrossRef]
- Vidyasagar, A.; Majewski, J.; Toomey, R. Temperature induced volume-phase transitions in surface-tethered poly(N-isopropylacrylamide) networks. Macromolecules 2008, 41, 919–924. [Google Scholar] [CrossRef]
- Lequieu, W.; Shtanko, N.I.; Du Prez, F.E. Track etched membranes with thermo-adjustable porosity and separation properties by surface immobilization of poly(N-vinylcaprolactam). J. Membr. Sci. 2005, 256, 64–71. [Google Scholar]
- Bashir, R.; Hilt, J.Z.; Elibol, O.; Gupta, A.; Peppas, N.A. Micromechanical cantilever as an ultrasensitive pH microsensor. Appl. Phys. Lett. 2002, 81, 3091–3093. [Google Scholar] [CrossRef]
- Richter, A.; Bund, A.; Keller, M.; Arndt, K.F. Characterization of a microgravimetric sensor based on pH sensitive hydrogels. Sensor. Actuator. B-Chem. 2004, 99, 579–585. [Google Scholar]
- Sorber, J.; Steiner, G.; Schulz, V.; Guenther, M.; Gerlach, G.; Salzer, R.; Arndt, K.F. Hydrogel-based piezoresistive pH sensors: Investigations using FT-IR attenuated total reflection spectroscopic imaging. Anal. Chem. 2008, 80, 2957–2962. [Google Scholar] [CrossRef]
- Xu, F.; Persson, B.; Lofas, S.; Knoll, W. Surface plasmon optical studies of carboxymethyl dextran brushes versus networks. Langmuir 2006, 22, 3352–3357. [Google Scholar] [CrossRef]
- Aulasevich, A.; Roskamp, R.F.; Jonas, U.; Menges, B.; Dostalek, J.; Knoll, W. Optical waveguide spectroscopy for the investigation of protein-functionalized hydrogel films. Macromol. Rapid Commun. 2009, 30, 872–877. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, C.J.; Jonas, U.; Wei, T.; Dostalek, J.; Knoll, W. Biosensor based on hydrogel optical waveguide spectroscopy. Biosens. Bioelectron. 2010, 25, 1663–1668. [Google Scholar] [CrossRef]
- Sanford, M.S.; Charles, P.T.; Commisso, S.M.; Roberts, J.C.; Conrad, D.W. Photoactivatable cross-linked polyacrylamide for the site-selective immobilization of antigens and antibodies. Chem. Mater. 1998, 10, 1510–1520. [Google Scholar] [CrossRef]
- Sidorenko, A.; Krupenkin, T.; Taylor, A.; Fratzl, P.; Aizenberg, J. Reversible switching of hydrogel-actuated nanostructures into complex micropatterns. Science 2007, 315, 487–490. [Google Scholar] [CrossRef]
- Revzin, A.; Russell, R.J.; Yadavalli, V.K.; Koh, W.G.; Deister, C.; Hile, D.D.; Mellott, M.B.; Pishko, M.V. Fabrication of poly(ethylene glycol) hydrogel microstructures using photolithography. Langmuir 2001, 17, 5440–5447. [Google Scholar] [CrossRef]
- Forch, R.; Zhang, Z.; Knoll, W. Soft plasma treated surfaces: Tailoring of structure and properties for biomaterial applications. Plasma Process. Polym. 2005, 2, 351–372. [Google Scholar] [CrossRef]
- Vickie Pan, Y.; Wesley, R.A.; Luginbuhl, R.; Denton, D.D.; Ratner, B.D. Plasma polymerized N-isopropylacrylamide: Synthesis and characterization of a smart thermally responsive coating. Biomacromolecules 2001, 2, 32–36. [Google Scholar] [CrossRef]
- Ito, Y.; Chen, G.P.; Guan, Y.Q.; Imanishi, Y. Patterned immobilization of thermoresponsive polymer. Langmuir 1997, 13, 2756–2759. [Google Scholar] [CrossRef]
- Schuh, K.; Prucker, O.; Ruhe, J. Surface attached polymer networks through thermally induced cross-linking of sulfonyl azide group containing polymers. Macromolecules 2008, 41, 9284–9289. [Google Scholar] [CrossRef]
- Liu, S.F.; Niu, J.R.; Gu, Z.Y. Temperature-sensitive poly(N-tert-butylacrylamide-co-acrylamide) hydrogels bonded on cotton fabrics by coating technique. J. Appl. Polym. Sci. 2009, 112, 2656–2662. [Google Scholar] [CrossRef]
- Bohanon, T.; Elender, G.; Knoll, W.; Koberle, P.; Lee, J.S.; Offenhausser, A.; Ringsdorf, H.; Sackmann, E.; Simon, J.; Tovar, G.; Winnik, F.M. Neural cell pattern formation on glass and oxidized silicon surfaces modified with poly(N-isopropylacrylamide). J. Biomat. Sci.-Polym. E 1996, 8, 19–39. [Google Scholar]
- Amos, R.A.; Anderson, A.B.; Clapper, D.L.; Duquette, P.H.; Duran, L.W.; Hohle, S.G.; Sogard, D.J.; Swanson, M.J.; Guire, P.E. Biomaterial surface modification using photochemical coupling technology. In Encyclopedic Handbook of Biomaterials and Bioengineering, Part A: Materials, 1st; Wise, D.L., Trantolo, D.J., Altobelli, D.E., Yaszemski, M.J., Gresser, J.D., Schwartz, E.R., Eds.; Marcel Dekker: New York, NY, USA, 1995; Volume 1, pp. 895–926. [Google Scholar]
- Prucker, O.; Naumann, C.A.; Ruhe, J.; Knoll, W.; Frank, C.W. Photochemical attachment of polymer films to solid surfaces via monolayers of benzophenone derivatives. J. Am. Chem. Soc. 1999, 121, 8766–8770. [Google Scholar] [CrossRef]
- Granville, A.M.; Brittain, W.J. Recent advances in polymer brush synthesis. In Polymer Brushes —Synthesis, Characterization, Applications, 1st; Advincula, R.C., Brittain, W.J., Caster, K.C., Ruhe, J., Eds.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2004; pp. 35–50. [Google Scholar]
- Minko, S. Grafting on solid surfaces: “Grafting to” and “grafting from” methods. In Polymer Surfaces and Interfaces—Characterization, Modification and Applications,, 1st; Stamm, M., Ed.; Springer: Berlin, Germany, 2008; pp. 215–234. [Google Scholar]
- Kobayashi, J.; Kikuchi, A.; Sakai, K.; Okano, T. Aqueous chromatography utilizing pH-/temperature responsive polymer stationary phases to separate ionic bioactive compounds. Anal. Chem. 2001, 73, 2027–2033. [Google Scholar] [CrossRef]
- Varvarenko, S.; Voronov, A.; Samaryk, V.; Tarnavchyk, I.; Nosova, N.; Kohut, A.; Voronov, S. Covalent grafting of polyacrylamide-based hydrogels to a polypropylene surface activated with functional polyperoxide. React. Funct. Polym. 2010, 70, 647–655. [Google Scholar] [CrossRef]
- Liang, L.; Feng, X.; Liu, J.; Rieke, P.C.; Fryxell, G.E. Reversible surface properties of glass plate and capillary tube grafted by photopolymerization of N-isopropylacrylamide. Macromolecules 1998, 31, 7845–7850. [Google Scholar] [CrossRef]
- Cole, M.A.; Voelcker, N.H.; Thissen, H.; Horn, R.G.; Griesser, H.J. Colloid probe AFM study of thermal collapse and protein interactions of poly(N-isopropylacrylamide) coatings. Soft Matter 2010, 6, 2657–2667. [Google Scholar] [CrossRef]
- Chen, J.; Yang, Z.Q.; Zhang, Q.F.; Wang, L.; Lu, Y. Grafting copolymerization of dimethylaminoethyl methacrylate and acrylic acid onto preirradiated polypropylene film by two-step reactions. J. Radioanal. Nucl. Chem. 2008, 275, 81–88. [Google Scholar] [CrossRef]
- Yamada, N.; Okano, T.; Sakai, H.; Karikusa, F.; Sawasaki, Y.; Sakurai, Y. Thermo-responsive polymeric surfaces; control of attachment and detachment of cultured cells. Makromol. Chem. Rapid Commun. 1990, 11, 571–576. [Google Scholar] [CrossRef]
- Kuckling, D. Responsive hydrogel layers—From synthesis to applications. Colloid Polym. Sci. 2009, 287, 881–891. [Google Scholar] [CrossRef]
- Hsu, T.P.; Ma, D.S.; Cohen, C. Effects of inhomogeneities in polyacrylamide gels on thermodynamic and transport properties. Polymer 1983, 24, 1273–1278. [Google Scholar] [CrossRef]
- Ikkai, F.; Shibayama, M. Inhomogeneity control in polymer gels. J. Polym. Sci. Pol. Phys. 2005, 43, 617–628. [Google Scholar] [CrossRef]
- Gianneli, M.; Beines, P.W.; Roskamp, R.F.; Koynov, K.; Fytas, G.; Knoll, W. Local and global dynamics of transient polymer networks and swollen gels anchored on solid surfaces. J. Phys. Chem. C 2007, 111, 13205–13211. [Google Scholar] [CrossRef]
- Gianneli, M.; Roskamp, R.F.; Jonas, U.; Loppinet, B.; Fytas, G.; Knoll, W. Dynamics of swollen gel layers anchored to solid surfaces. Soft Matter 2008, 4, 1443–1447. [Google Scholar] [CrossRef]
- Junk, M.J.N.; Ilke, A.; Menges, B.; Jonas, U. Analysis of optical gradient profiles during temperature- and salt-dependent swelling of thin responsive hydrogel films. Langmuir 2010, 26, 12253–12259. [Google Scholar]
- Matzelle, T.R.; Ivanov, D.A.; Landwehr, D.; Heinrich, L.A.; Herkt-Bruns, C.; Reichelt, R.; Kruse, N. Micromechanical properties of “smart” gels: Studies by scanning force and scanning electron microscopy of PNIPAAm. J. Phys. Chem. B 2002, 106, 2861–2866. [Google Scholar]
- Arndt, K.-F.; Krahl, F.; Richter, S.; Steiner, G. Swelling-related processes in hydrogels. In Hydrogel Sensors and Actuators, 1st; Gerlach G.;, Arndt, K.-F., Eds., Eds.; Springer: Berlin, Germany, 2009; pp. 69–136. [Google Scholar]
- Okay, O. General properties of hydrogels. In Hydrogel Sensors and Actuators, 1st; Gerlach, G., Arndt, K.-F., Eds.; Springer: Berlin, Germany, 2009; pp. 1–14. [Google Scholar]
- Yu, H.; Grainger, D.W. Thermosensitive swelling behaviour in cross-linked N-isopropylacrylamide networks -cationic, anionic, and ampholytic hydrogels. J. Appl. Polym. Sci. 1993, 49, 1553–1563. [Google Scholar] [CrossRef]
- Suzuki, A.; Wu, X.R.; Kuroda, M.; Ishiyama, E.; Kanama, D. Swelling properties of thin-plate hydrogels under mechanical constraint. Jpn J. Appl. Phys. Part 1 2003, 42, 564–569. [Google Scholar]
- Chen, J.; Park, K. Synthesis and characterization of superporous hydrogel composites. J. Control. Release 2000, 65, 73–82. [Google Scholar] [CrossRef]
- Irwin, E.F.; Ho, J.E.; Kane, S.R.; Healy, K.E. Analysis of interpenetrating polymer networks via quartz crystal microbalance with dissipation monitoring. Langmuir 2005, 21, 5529–5536. [Google Scholar] [CrossRef]
- Tamirisa, P.A.; Hess, D.W. Water and moisture uptake by plasma polymerized thermoresponsive hydrogel films. Macromolecules 2006, 39, 7092–7097. [Google Scholar] [CrossRef]
- Wang, Z.H.; Kuckling, D.; Johannsmann, D. Temperature-induced swelling and de-swelling of thin poly(N-isopropylacrylamide) gels in water: Combined acoustic and optical measurements. Soft Mater. 2003, 1, 353–364. [Google Scholar] [CrossRef]
- Tokareva, I.; Tokarev, I.; Minko, S.; Hutter, E.; Fendler, J.H. Ultrathin molecularly imprinted polymer sensors employing enhanced transmission surface plasmon resonance spectroscopy. Chem. Commun. 2006, 3343–3345. [Google Scholar]
- Raitman, O.A.; Chegel, V.I.; Kharitonov, A.B.; Zayats, M.; Katz, E.; Willner, I. Analysis of NAD(P)(+) and NAD(P)H cofactors by means of imprinted polymers associated with Au surfaces: A surface plasmon resonance study. Anal. Chim. Acta 2004, 504, 101–111. [Google Scholar] [CrossRef]
- Matsui, J.; Akamatsu, K.; Hara, N.; Miyoshi, D.; Nawafune, H.; Tamaki, K.; Sugimoto, N. SPR sensor chip for detection of small molecules using molecularly imprinted polymer with embedded gold nanoparticles. Anal. Chem. 2005, 77, 4282–4285. [Google Scholar]
- Lavine, B.K.; Westover, D.J.; Kaval, N.; Mirjankar, N.; Oxenford, L.; Mwangi, G.K. Swellable molecularly imprinted polyN-(N-propyl)acrylamide particles for detection of emerging organic contaminants using surface plasmon resonance spectroscopy. Talanta 2007, 72, 1042–1048. [Google Scholar] [CrossRef]
- Matsui, J.; Takayose, M.; Akamatsu, K.; Nawafune, H.; Tamaki, K.; Sugimoto, N. Molecularly imprinted nanocomposites for highly sensitive SPR detection of a non-aqueous atrazine sample. Analyst 2009, 134, 80–86. [Google Scholar] [CrossRef]
- Bagal, D.S.; Vijayan, A.; Aiyer, R.C.; Karekar, R.N.; Karve, M.S. Fabrication of sucrose biosensor based on single mode planar optical waveguide using co-immobilized plant invertase and GOD. Biosens. Bioelectron. 2007, 22, 3072–3079. [Google Scholar] [CrossRef]
- Zourob, M.; Goddard, N.J. Metal clad leaky waveguides for chemical and biosensing applications. Biosens. Bioelectron. 2005, 20, 1718–1727. [Google Scholar] [CrossRef]
- Tokarev, I.; Tokareva, I.; Gopishetty, V.; Katz, E.; Minko, S. Specific biochemical-to-optical signal transduction by responsive thin hydrogel films loaded with noble metal nanoparticles. Adv. Mater. 2010, 22, 1412–1416. [Google Scholar] [CrossRef]
- Endo, T.; Ikeda, R.; Yanagida, Y.; Hatsuzawa, T. Stimuli-responsive hydrogel-silver nanoparticles composite for development of localized surface plasmon resonance-based optical biosensor. Anal. Chim. Acta 2008, 611, 205–211. [Google Scholar] [CrossRef]
- Bjork, P.; Persson, N.K.; Peter, K.; Nilsson, R.; Asberg, P.; Inganas, O. Dynamics of complex formation between biological and luminescent conjugated polyelectrolytes—A surface plasmon resonance study. Biosens. Bioelectron. 2005, 20, 1764–1771. [Google Scholar] [CrossRef]
- Yang, H.H.; Liu, H.P.; Kang, H.Z.; Tan, W.H. Engineering target-responsive hydrogels based on aptamer - Target interactions. J. Am. Chem. Soc. 2008, 130, 6320–6321. [Google Scholar]
- Zhu, Z.; Wu, C.C.; Liu, H.P.; Zou, Y.; Zhang, X.L.; Kang, H.Z.; Yang, C.J.; Tan, W.H. An aptamer cross-linked hydrogel as a colorimetric platform for visual detection. Angew. Chem. Int. Edit. 2010, 49, 1052–1056. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, C.J.; Jonas, U.; Wei, T.X.; Dostalek, J.; Knoll, W. Biosensor based on hydrogel optical waveguide spectroscopy. Biosens. Bioelectron. 2010, 25, 1663–1668. [Google Scholar] [CrossRef]
- Byrne, M.E.; Park, K.; Peppas, N.A. Molecular imprinting within hydrogels. Adv. Drug Deliver. Rev. 2002, 54, 149–161. [Google Scholar]
- Byrne, M.E.; Salian, V. Molecular imprinting within hydrogels II: Progress and analysis of the field. Int. J. Pharm. 2008, 364, 188–212. [Google Scholar] [CrossRef]
- Matsui, J.; Akamatsu, K.; Nishiguchi, S.; Miyoshi, D.; Nawafune, H.; Tamaki, K.; Sugimoto, N. Composite of Au nanoparticles and molecularly imprinted polymer as a sensing material. Anal. Chem. 2004, 76, 1310–1315. [Google Scholar] [CrossRef]
- Stuart, M.A.C.; Huck, W.T.S.; Genzer, J.; Muller, M.; Ober, C.; Stamm, M.; Sukhorukov, G.B.; Szleifer, I.; Tsukruk, V.V.; Urban, M.; Winnik, F.; Zauscher, S.; Luzinov, I.; Minko, S. Emerging applications of stimuli-responsive polymer materials. Nat. Mater. 2010, 9, 101–113. [Google Scholar]
- Nilsson, K.P.R.; Inganas, O. Chip and solution detection of DNA hybridization using a luminescent zwitterionic polythiophene derivative. Nat. Mater. 2003, 2, 419–424. [Google Scholar] [CrossRef]
- Nilsson, K.P.R.; Andersson, M.R.; Inganas, O. Conformational transitions of a free amino-acid-functionalized polythiophene induced by different buffer systems. J. Phys.Condens. Mat. 2002, 14, 10011–10020. [Google Scholar] [CrossRef]
- Esmi European Soft Matter Infrastructure Website . Available online: www.esmi-fp7.net accessed on 6 February 2012.
- NILaustria Nanoimprint Lithography Project Cluster Website. Available online: www.NILAustria.at accessed on 6 February 2012.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mateescu, A.; Wang, Y.; Dostalek, J.; Jonas, U. Thin Hydrogel Films for Optical Biosensor Applications. Membranes 2012, 2, 40-69. https://doi.org/10.3390/membranes2010040
Mateescu A, Wang Y, Dostalek J, Jonas U. Thin Hydrogel Films for Optical Biosensor Applications. Membranes. 2012; 2(1):40-69. https://doi.org/10.3390/membranes2010040
Chicago/Turabian StyleMateescu, Anca, Yi Wang, Jakub Dostalek, and Ulrich Jonas. 2012. "Thin Hydrogel Films for Optical Biosensor Applications" Membranes 2, no. 1: 40-69. https://doi.org/10.3390/membranes2010040
APA StyleMateescu, A., Wang, Y., Dostalek, J., & Jonas, U. (2012). Thin Hydrogel Films for Optical Biosensor Applications. Membranes, 2(1), 40-69. https://doi.org/10.3390/membranes2010040