1. Introduction
Click chemistry is versatile and has been lauded for bio-based applications due to high specificity, high fidelity (i.e., no side products), mild reaction conditions, and its ability to operate at low concentrations. The term “click” chemistry originally was coined to describe the Cu(I)-catalyzed alkyne–azide reaction chemistry developed by Sharpless and coworkers in 2002 [
1]. Since then, it has been utilized widely in the fields of chemical biology for many applications, including preparation of antibody-drug conjugates and vaccines; imaging and profiling of all classes of biomolecules including but not limited to proteins, glycans, lipids, metabolites; immobilization of biomolecules onto stationary supports; and the development of new classes of biorthogonal chemistry like tetrazine ligation chemistry [
2,
3]. While Cu(I) alkyne–azide chemistry has been used to prepare chromatography media [
4,
5,
6,
7], there have been no instances in the literature where it has been used specifically for preparation of Protein A chromatography media.
While Cu(I)-catalyzed alkyne–azide click chemistry remains popular for the reasons mentioned above, the requirement to use copper is a significant drawback because it is cytotoxic, and for development of chromatographic supports, it might bind to the surface of the chromatography medium during and after ligand immobilization [
5]. Strain-promoted click chemistry was developed to overcome these copper-related issues. Bertozzi and coworkers [
8] found that the same benefits of Cu(I) alkyne–azide click chemistry could be realized without using copper by replacing the terminal alkyne with an internal alkyne within a ring structure. This class of compounds, termed cyclooctynes, undergo cycloaddition with azides under physiological conditions, with the ring strain acting as the catalyst instead of Cu(I). One of the most applied uses of strain-promoted click chemistry has been for reactions in living systems [
8,
9]. In this study, the main interest was to use this chemistry for covalent protein immobilization onto a chromatography support.
In the field of protein immobilization onto solid supports, immobilization chemistry plays a major role in immobilization yield, activity of immobilized protein, and performance of the solid support. In a recent study by Zhu and Sun [
10], multiple chemistries were evaluated for Protein A/G immobilization onto poly(vinyl alcohol-
co-ethylene) nanofibers under similar conditions. These chemistries included: epibromohydrin (EP), 1,4-butanediol diglycidyl ether (BDGE), oxalyl chloride (OA), nitrophenyl chloroformate (NP), cyanuric chloride (CC), carbonyldiimidazole (CDI), gluteraldehyde (GA), and disuccinimidyl carbonate (DSC). Of these chemistries, GA and DSC resulted in the highest activities and immobilization yields.
Based on the strong affinity of Protein A to the Fc-region of antibodies, Protein A membrane adsorbers can be used for the rapid purification of antibodies and other Fc-fusion proteins [
11]. Previous attempts to develop Protein A membrane adsorbers have used other coupling chemistries such as EDC/NHS (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide/N-hydroxysuccinimide) [
12], EDC/Sulfo-NHS [
13], cyanogen bromide [
14], and GA [
10]. To our knowledge, there has been no attempt at preparing Protein A membrane adsorbers using strain-promoted click chemistry. We therefore present a new approach for their preparation via dibenzocyclooctyne (DBCO)-azide strain-promoted click immobilization of Protein A onto regenerated cellulose membrane supports. The synthetic approach includes three main steps: incorporation of DBCO groups into Protein A via NHS ester bioconjugation; preparation of azide functionalized membranes using the DSC functionalization of hydroxyl groups on the membrane supports, followed by reaction with an azido-PEG
3-amine molecule; and finally, a click reaction between DBCO-conjugated Protein A and the azide-functionalized membrane surface.
2. Materials and Methods
2.1. Materials
The following materials were purchased from MilliporeSigma: 3-azido-1-propanol (>96%), 4-dimethylaminopyridine (DMAP, >99%), acetonitrile (HPLC grade, >99.9%), alpha-cyano-4-hydroxycinnamic acid (α-CHCA, 99%), copper(II) sulfate pentahydrate (CuSO4-5H2O, >98.0%), dimethyl sulfoxide (anhydrous, >99.9%), dimethylformamide (anhydrous, >99.8%), Folin–Ciocalteu’s phenol reagent (F&C reagent, product no. 47641), formic acid (reagent grade, >95%), guanidine hydrochloride (>99%), hydrochloric acid (HCl, 37%), potassium sodium tartrate tetrahydrate (>99%), phosphate-buffered saline (PBS, powder, product number P3813), sodium carbonate (Na2CO3, >99.5%), sodium chloride (NaCl, 98%), sodium dodecyl sulfate (SDS, >98%), sodium phosphate dibasic (>99%), trisbase (>99.7%), N,N′-disuccinimidyl carbonate (DSC, >95%), Whatman regenerated cellulose membrane filters (RC-58, diameter 47 mm, pore size 0.2 µm).
Polyclonal Immunoglobulin G (IgG) from human plasma was purchased from Lee Biosolutions (Product number 340-21, >95%). Native recombinant Staphylococcal Protein A ligand was purchased from Repligen (rSPA, Product number 10-2001-0M). DBCO-PEG5-NHS ester was purchased from Click Chemistry Tools (Catalog number A102P-100, >95% by HPLC). Azido-PEG3-amine was purchased from BroadPharm (Catalog number BP20580, 98%).
2.2. Preparation of DBCO-Conjugated Protein A
2.2.1. Conjugation with DBCO-PEG5-NHS Ester
DBCO-PEG5-NHS ester reacted with Protein A at different molar ratios. The following procedure outlines the preparation using a 15 to 1 molar ratio with respect to Protein A. Protein A stock solution was supplied by Repligen at 50 mg/mL in a sodium chloride buffer solution. A 50 mM stock DBCO-PEG5-NHS ester solution was prepared in anhydrous DMSO. One milliliter of 50 mg/mL Protein A was diluted with 3.6 mL of 140 mM sodium phosphate buffer at pH 8.4. For a 15 to 1 molar ratio, 321 µL of the 50 mM DBCO-PEG5-NHS ester solution was added drop wise to the Protein A solution while gently mixing with a stir bar. The resulting Protein A was 10 mg/mL in 100 mM phosphate buffer at pH 8.3, and the reaction was performed for 12 h at room temperature.
After bioconjugation, excess DBCO reagent was removed using size exclusion chromatography. One milliliter of 10 mg/mL DBCO-Protein A was injected into Cytiva Superdex 75 10/300 GL at 0.4 mL/min. The mobile phase was 100 mM sodium phosphate at pH 8.3.
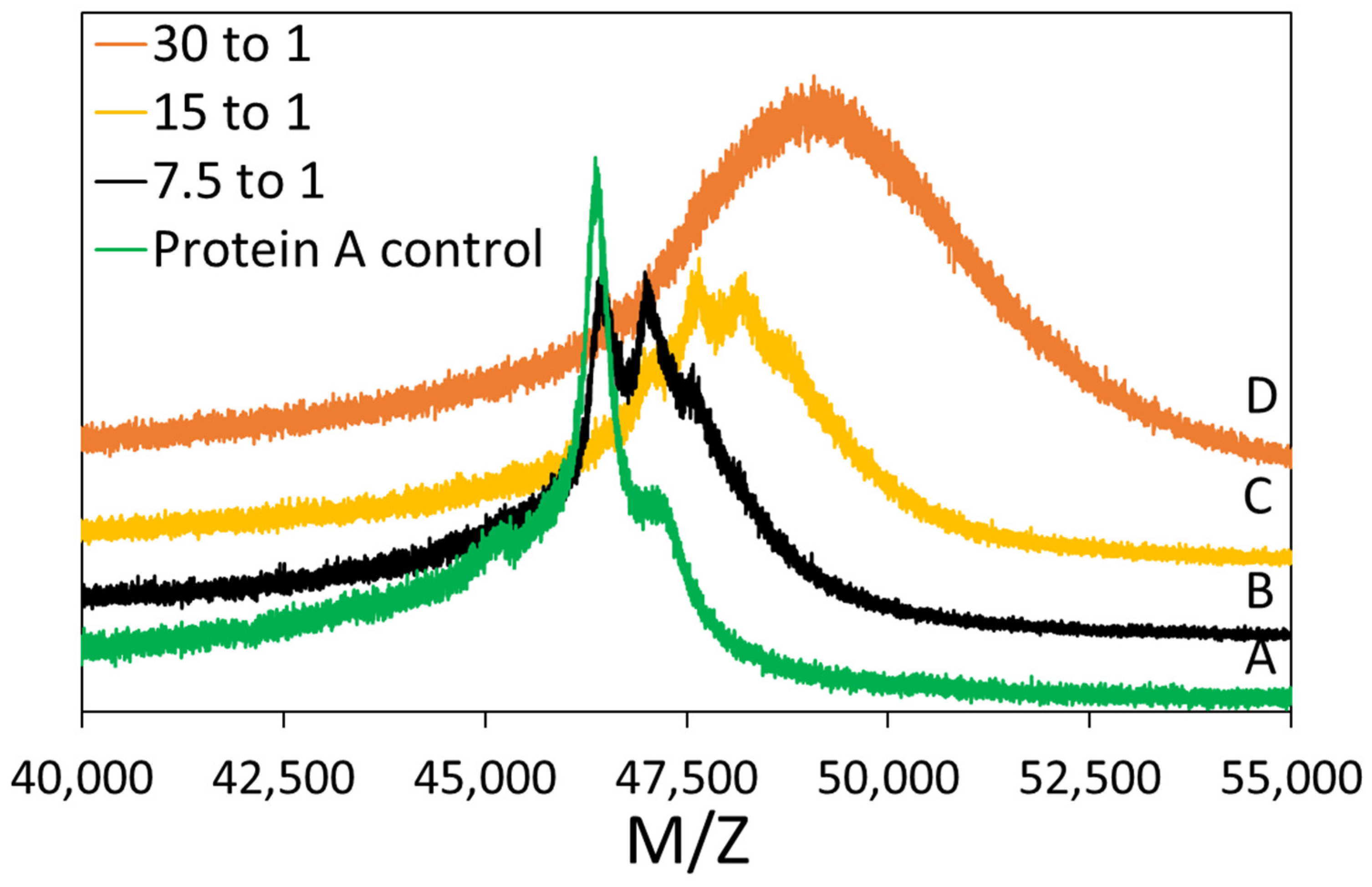
Figure S1, in Supporting Information, shows the resulting chromatograms. The first peak of the chromatogram, spanning 5 mL in elution volume, corresponds to DBCO-Protein A and was collected for further use and analysis.
2.2.2. Matrix Assisted Laser Desorption Ionization-Time of Flight (MALDI-TOF) Mass Spectrometry
MALDI-TOF mass spectrometry was performed to determine the success of the conjugation reaction. The matrix was prepared with 20 mg/mL α-CHCA in a 70/30 (v/v) ACN:5% formic acid (aq.) mixture. The plating procedure was as follows: 1 µL of matrix was plated and then allowed to dry. Then, 1 µL of 2 mg/mL Protein A solution was plated. Finally, 1.5 µL of matrix was added. All samples were analyzed using a Bruker MicroflexTM LRF MALDI-TOF in linear mode (105 cm flight path). Laser level was between 20 and 70%, detector gain was 2827 V, and the analysis used positive ion spectrum. The same procedure was carried out for DBCO-Protein A. Reported MW/charge (M/Z) values are the average of two peak measurements. The standard deviation of these measurement was reported as the error bars.
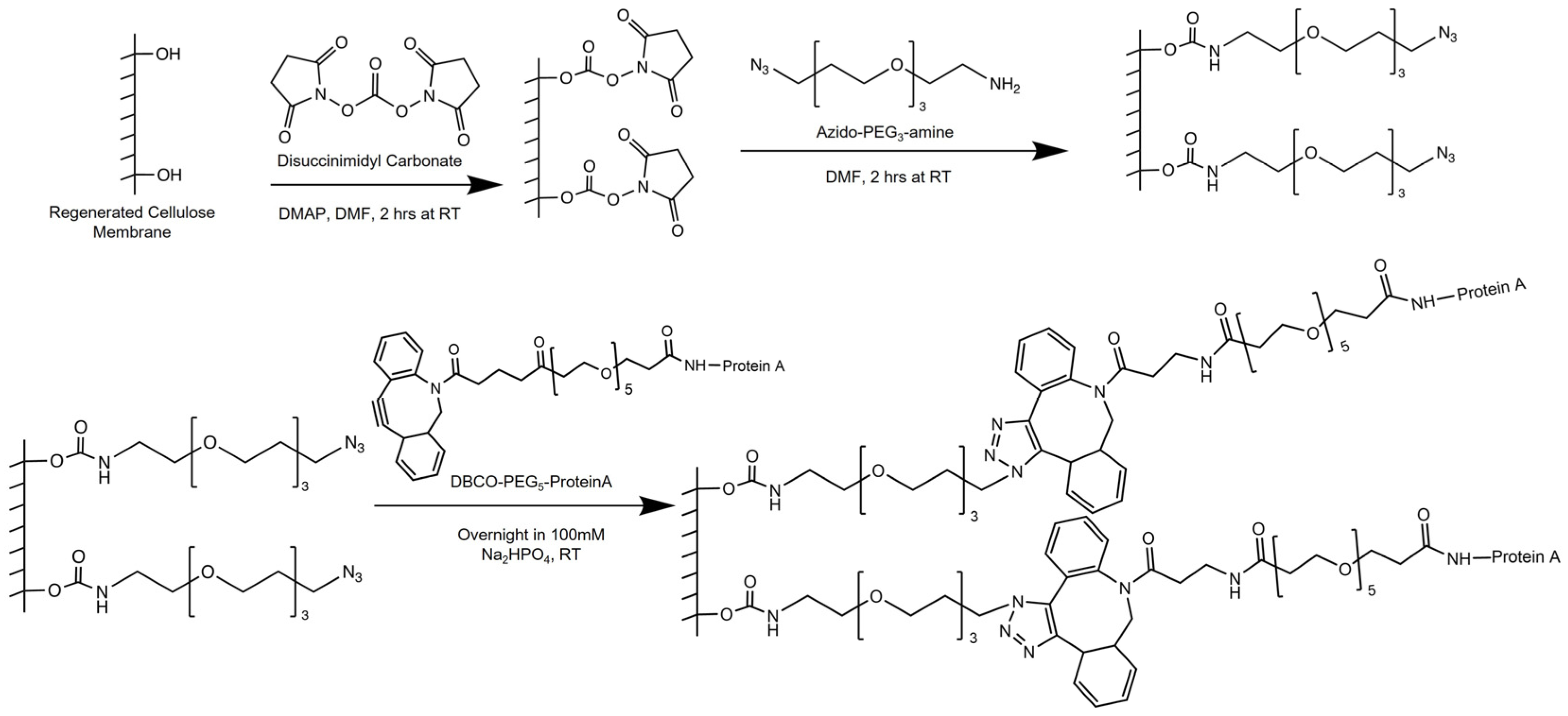
2.3. Membrane Surface Modification
Scheme 1 illustrates the functionalization procedure of the regenerated cellulose (RC-58) substrates. First, activated NHS ester molecules are created by reacting the hydroxyl groups with DSC. Activated NHS ester molecules undergo nucleophilic attack by primary amines of the azido-PEG
3-amine molecules. The resulting scaffold is a membrane surface modified by azide moieties. DBCO-PEG
5-conjugated Protein A is then clicked onto the azide groups of the membrane. A DBCO-PEG
5-conjugated Protein A, prepared using a 30 to 1 molar ratio of DBCO-PEG
5-NHS ester to Protein A, was used for the click reaction. The volumes used per membrane sample and final solution concentration of each substance are provided in the sections that follow.
2.3.1. DSC Functionalization of Regenerated Cellulose Membranes
DMAP (579 mM, 0.991 g) was added to dry DMF (14 mL) and the solution was sonicated until DMAP dissolved fully. The RC-58 membrane was rinsed by immersing in 10 mL of dry DMF for 10 min. After rinsing, the membrane was soaked in the DMAP/DMF solution for a few minutes. DSC (343 mM, 1.232 g) was added, and the solution was placed on a shaker at 120 rpm. The solution turned brown as the reaction proceeded at room temperature for 2 h. After the reaction, the membrane was rinsed with DMF, DMSO, and 2-propanol. Functionalized membranes were stored in 2-propanol until the next reaction step.
2.3.2. Azide Functionalization of Regenerated Cellulose Membranes
Azide-PEG3-amine (500 mM, 0.3 mL) was added into DMF (2.7 mL). A DSC-modified RC-58 membrane was immersed into the solution and reacted for at least 2 h at room temperature. Unreacted DSC groups were quenched using the following rinsing procedure at room temperature: (i) 1 M Trisbase, pH 9.1 (30 min); (ii) water (1 min, twice); (iii) 6 M guanidine, pH 7 (30 min); (iv) 1 M NaCl (10 min, three times). After the rinsing procedure, membranes were stored in water until the next step.
2.3.3. Click Reaction with Azide Functionalized Membrane
An azide-modified RC-58 membrane was immersed into 2.5 mL of 100 mM PBS solution containing 10 mg/mL DBCO-conjugated Protein A. The solution was placed on a shaker at 120 rpm. The reaction was performed at room temperature for 8 h.
2.4. Characterization of Modified Membranes
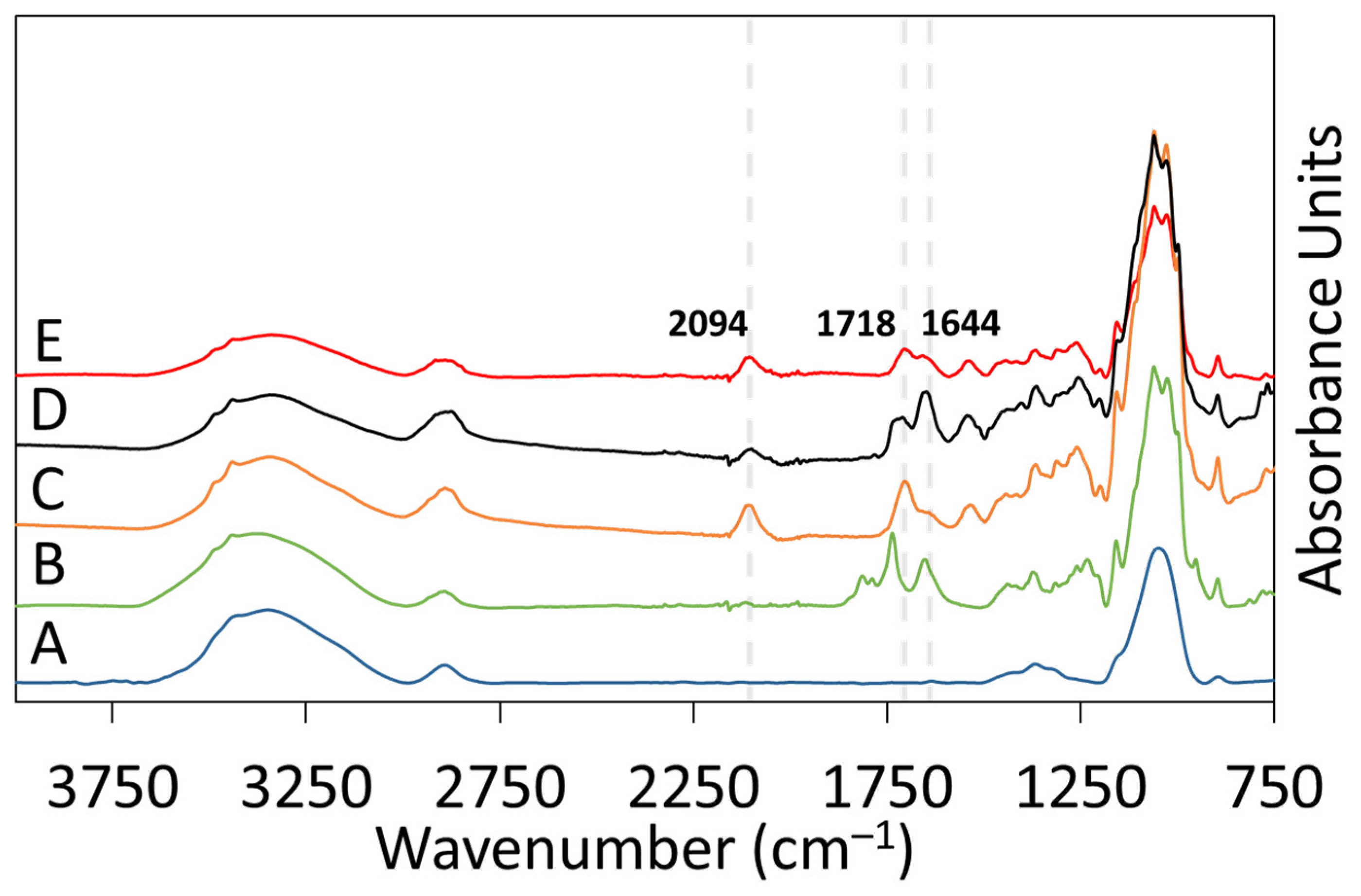
2.4.1. Attenuated Total Reflectance Fourier-Transform Infrared (ATR-FTIR) Spectroscopy
ATR-FTIR spectroscopy was used to analyze the surface chemistry of RC-58 membranes after each reaction step: DSC modification, azido-PEG3-amine attachment, and DBCO-Protein A click reaction. A Perkin Elmer Spectrum Two FTIR was used for analysis of all samples. The instrument was equipped with a Universal ATR accessory using a single reflection diamond crystal, and each spectrum was obtained using 32 scans at a resolution of 4 cm−1. Background correction, baseline correction, ATR-FTIR correction, and peak analysis were performed using PerkinElmer Spectrum 10 software.
2.4.2. X-ray Photoelectron Spectroscopy (XPS)
XPS measurements were performed using a Physical Electronics PHI 5000 Versa Probe III Scanning ESCA Microprobe (Chanhassen, MN, USA). The instrument featured a 180° hemispherical electron energy analyzer and a monochromatic Al Kα scanning microprobe X-ray source that used a 15 kV beam volage and 100 µm spot size. High resolution scans were performed for C(1s), N(1s), and O(1s) using a step size of 0.125 eV and pass energy of 69 eV. Measurements were made under vacuum at <1.3 × 10−6 Pa.
2.4.3. Ligand Density
Ligand density measurements were performed using a Lowry Assay. Stock solution A was prepared comprising 1/1/100 (v/v/v) aqueous mixture of 1 wt% copper(II) sulfate pentahydrate: 2.7 wt% potassium sodium tartrate: 2 wt% sodium carbonate. All solutions were prepared using DI water. Stock solution B was prepared comprising 1/1 (v/v) F&C reagent:DI water. One hundred microliter Protein A solutions were prepared at concentrations ranging from 0.05 to 1 mg/mL in small test tubes. The same was conducted for DBCO-Protein A samples. These samples were used as calibration standards for calculating the amount of covalently immobilized Protein A on the membranes. Protein A clicked membranes were cut into 5 mm diameter coupons and inserted into test tubes containing 100 µL of water.
To begin the assay, 1 mL of solution A was added to each tube, vortexed and incubated at room temperature for 10 min. One hundred microliters of solution B were added to each tube, vortexed and incubated at room temperature for 20 min. Three hundred microliters from each test tube were aliquoted into a clear 96 well plate and absorbance values were measured at a wavelength of 750 nm. The 750 nm absorbance readings of samples from test tubes containing DBCO-Protein A clicked membranes were converted to masses of DBCO-Protein A using the calibration standards. The mass values were divided by membrane volume to determine ligand densities in units of mg/mL membrane.
2.5. Performance of Protein A Membranes
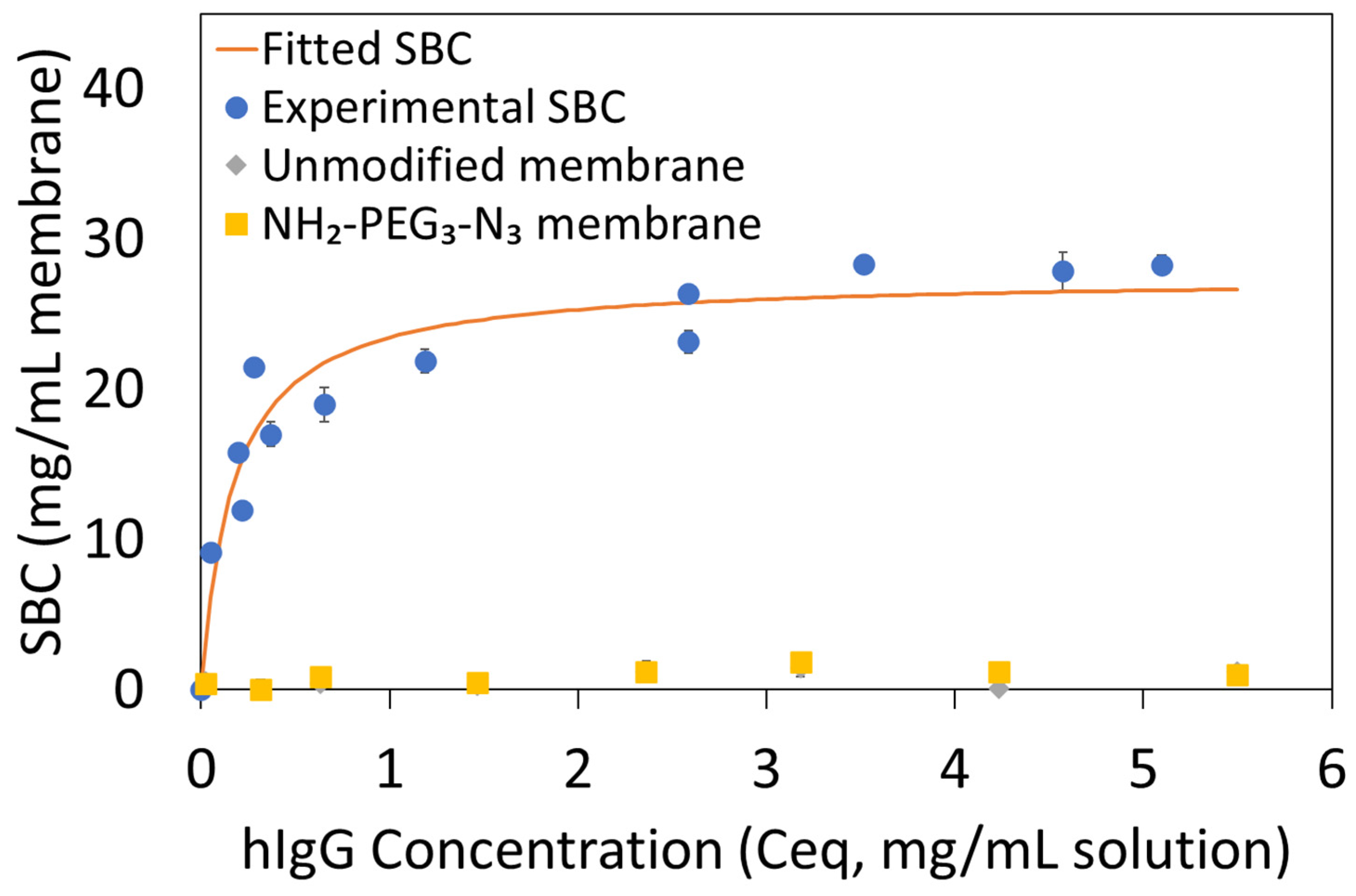
2.5.1. Static Binding Capacity (SBC)
SBCs of clicked Protein A membranes were measured using hIgG. A typical procedure for this experiment is as follows: using 1× PBS at pH 7.4 as the buffer solution, hIgG solutions were prepared at initial concentrations ranging from 0.1 to 5 mg/mL. One 2.5 cm diameter membrane was placed into a glass vial containing 2 mL of hIgG solution at a known concentration. The glass vial was put in a shaker bath at room temperature and 100 rpm for at least 12 h until equilibrium was reached. The equilibrium hIgG concentration was measured with a Nanodrop™ One Microvolume UV-Vis Spectrophotometer at 280 nm. A mass balance was performed using the initial (C
o) and equilibrium (C
eq) hIgG concentrations, the solution volume (V
sol) and the membrane volume (V
mem) to determine SBC, q (in mg hIgG adsorbed per mL of membrane) (Equation (1)). Each measurement was repeated in duplicate. Thermodynamic parameters, q
max and apparent K
d, were determined by fitting SBC values using the Langmuir adsorption isotherm model (Equation (2)).
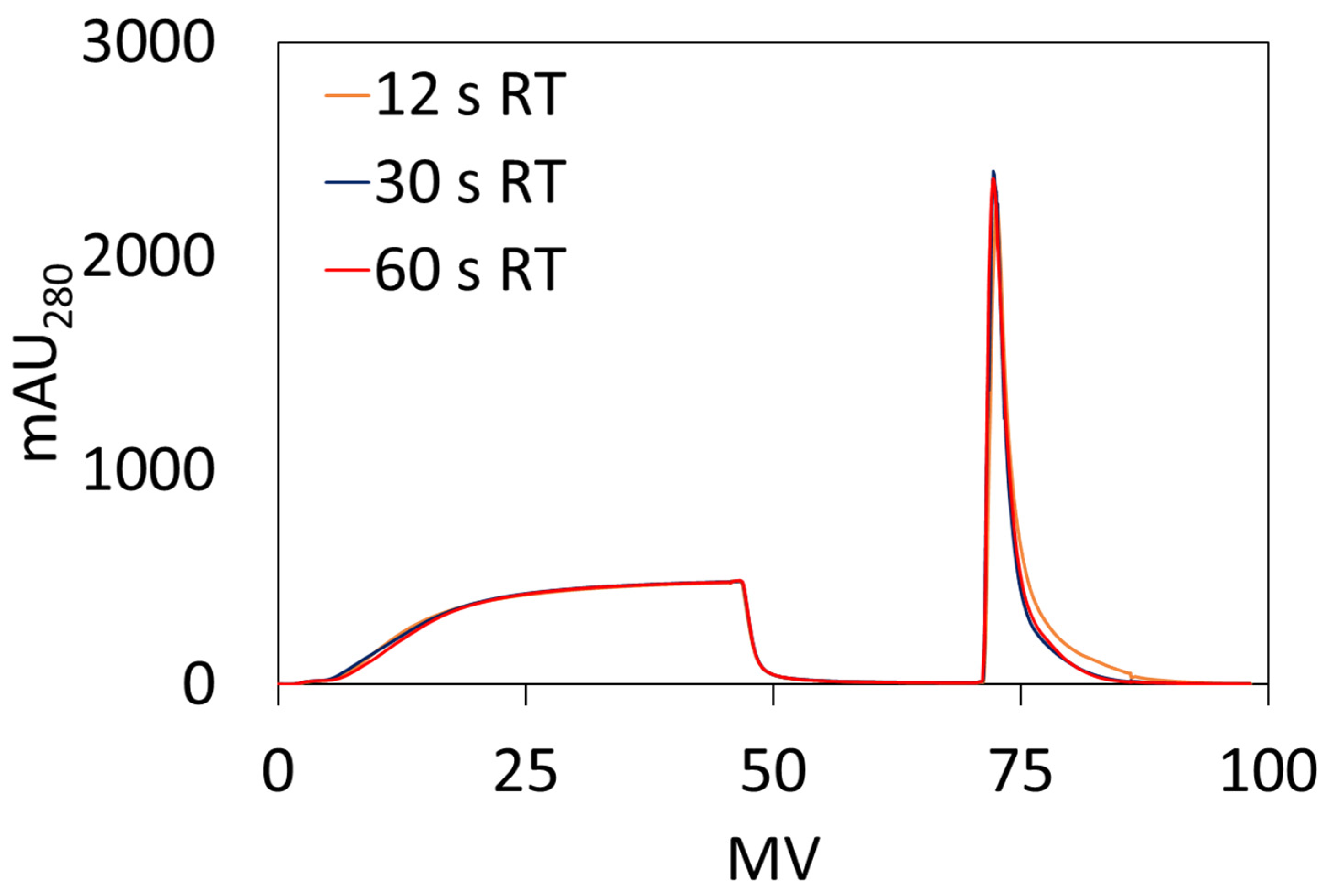
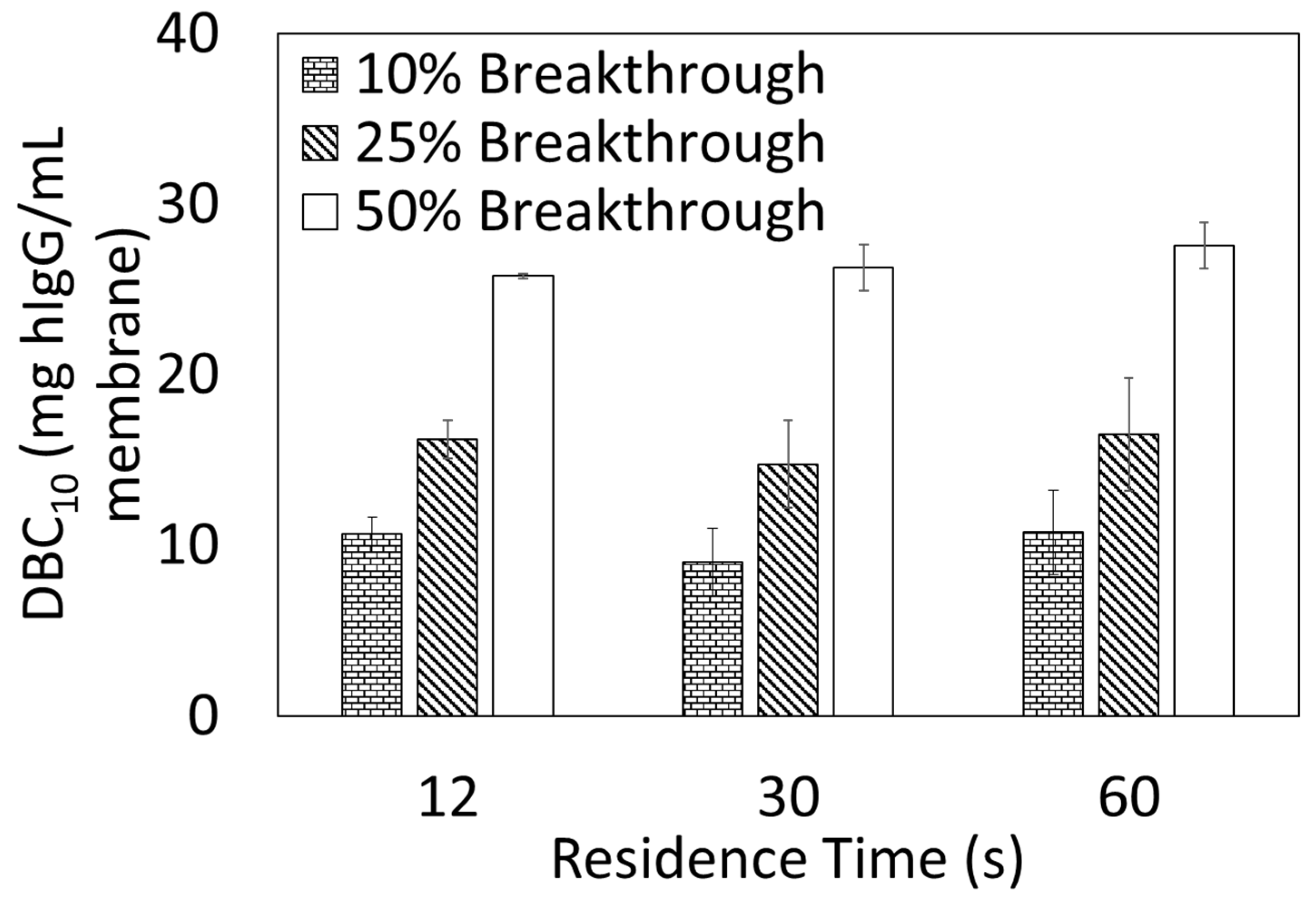
2.5.2. Dynamic Binding Capacity (DBC10)
DBC
10 measurements were performed using an AKTA Pure 25 from Cytiva. Ten DBCO-Protein A clicked membranes, 23 mm diameter each, were stacked into a membrane holder in an axial format. Bind and elute studies were performed using hIgG solutions prepared by dissolving hIgG in the loading buffer (1× PBS, pH 7.4) and subsequently filtering through a 0.2 µm cellulose acetate syringe filter prior to use. DBC
10 values were calculated at 10% breakthrough using Equation (3) where V
break is the effluent volume (mL) at 10% breakthrough, V
dead is the dead volume of the system (mL), C
o is the feed concentration (mg/mL), and V
mem is the volume of the membrane adsorber (mL).
4. Discussion
This study demonstrated the preparation of Protein A affinity membranes using copper-free DBCO-azide click chemistry. Performance of these Protein A membranes, in terms of binding capacity, can be compared to other Protein A membranes described in the literature and to those that are commercially available. In 2015, Akashi et al. prepared Protein A membranes by immobilizing Protein A onto acrylic acid-grafted PVDF and PES membranes using EDC/Sulfo-NHS chemistry [
13]. The resulting membranes had static binding capacities ranging from 0.55 to 0.75 mg IgG/mL membrane [
13]. In 2009, Ma et al. prepared Protein A membrane adsorbers by immobilizing Protein A onto electrospun PES supports using EDC/NHS ester chemistry [
12]. The resulting membranes showed a SBC of 4.5 mg IgG/mL membrane [
12]. In 2008, Boi et al. reported the performance of a new Protein A membrane adsorber with SBC of 11.67 mg IgG/mL membrane and DBC
10 of 5.76 mg IgG/mL membrane [
17]. The preparation procedure for the membrane adsorber was not reported [
17].
Commercially, up until 2017, the only available Protein A membrane was the Sartorius Sartobind
® Protein A, which reported a DBC
10 of 5.9 mg IgG/mL membrane [
18]. Newer commercial Protein A membranes, Purilogics’ Purexa™ PrA and Cytiva’s Fibro™ PrismA, were evaluated by Osuofa and Husson and shown to have ~70 mg/mL DBC
10 at 5 s residence time and ~90 mg/mL SBC for hIgG [
19]. Sartorius’ new convecdiff Protein A membranes have reported DBC
10 values of ~43 mg/mL at 12 s residence time [
20]. For these newer membrane devices, the preparation procedures are proprietary. In this study, the Protein A membranes prepared using the new click chemistry approach have shown binding capacity like other Protein A membranes in the literature.
The apparent K
d value also was benchmarked against other Protein A membranes. Previously, we measured K
d values of 7.07 × 10
−2 ± 1.8 × 10
−2 mg/mL for Purilogics Purexa™ PrA and 7.63 × 10
−2 mg/mL for Sartorius Sartobind
® Protein A membranes [
19]. K
d values from Protein A resins have been reported to be between 4.5 × 10
−2 and 1.20 × 10
−1 mg/mL by Hahn et al. [
21]. Pabst et al. [
22] reported K
d for Protein A resins to be between 7.99 × 10
−3 and 1.05 × 10
−2 mg/mL, depending on the mAb used for SBC experiments. By comparison, the K
d value of 1.72 × 10
−1 ± 4.03 × 10
−2 mg/mL for membranes prepared in this study is relatively high. Since 1/K
d is a mathematical representation of the affinity between immobilized Protein A and hIgG in solution, this relatively high K
d value (indicating weaker interaction between hIgG and this membrane compared to other Protein A media) may be a result of conjugating Protein A with DBCO-PEG
5 molecules, which may interfere with the Fc binding domain.
The measured ligand density using Lowry assay was 3.47 ± 0.53 mg DBCO-Protein A/mL membrane, which results in a molar ratio of 2.60 + 0.18 hIgG molecules per Protein A molecule. Although the K
d value may be affected by the conjugation of Protein A, the binding efficiency remains within the expected range from approximately 2.54 to 3.3 antibody molecules to one Protein A molecule [
23]. Additionally, other factors that may contribute to the ratio of hIgG to Protein A include intra- and inter- ligand steric hindrances [
23], spatial distribution of the ligand on the membrane surface and pores [
24], and multipoint attachment of the ligand during covalent immobilization [
10]. The support itself can play a role on the spatial distribution of the ligand on the membrane. We selected regenerated cellulose over such supports as polyvinylidene fluoride and polyethersulfone, as the density of native surface functional groups is significantly higher for regenerated cellulose. Indeed, In the case of PVDF and PES membranes, surface treatments are normally performed to increase the number of reactive groups prior to ligand immobilization.
EV values for membrane adsorbers typically are not reported in the literature, but the measured EV of 8.78 ± 0.55 MVs is similar to values that we have measured for research scale Protein A membrane adsorbers [
19], which ranged from 0.8 to 8.96 MVs. For Protein A resins, Pabst et al. [
22] found that EVs range from 1.8 to 3.8 CVs. Higher EVs for membrane adsorbers can be attributed to the larger dead volume in membrane devices.














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}