
HIV-Induced Hyperactivity of Striatal Neurons Is Associated with Dysfunction of Voltage-Gated Calcium and Potassium Channels at Middle Age

Abstract
1. Introduction
2. Materials and Methods
3. Results
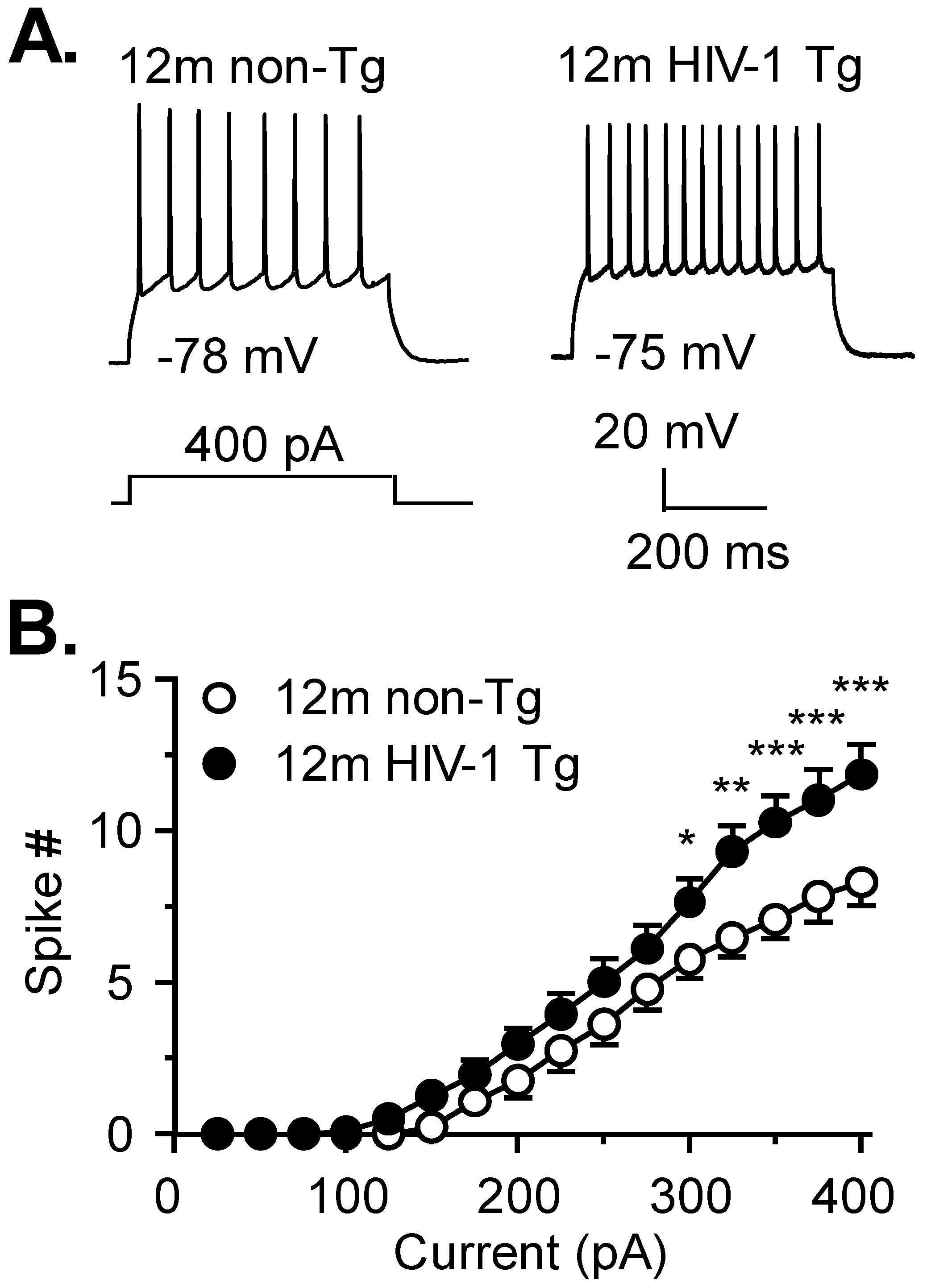
3.1. Firing of Striatal MSNs Is Abnormally Increased in 12mo HIV-1 Tg Rats
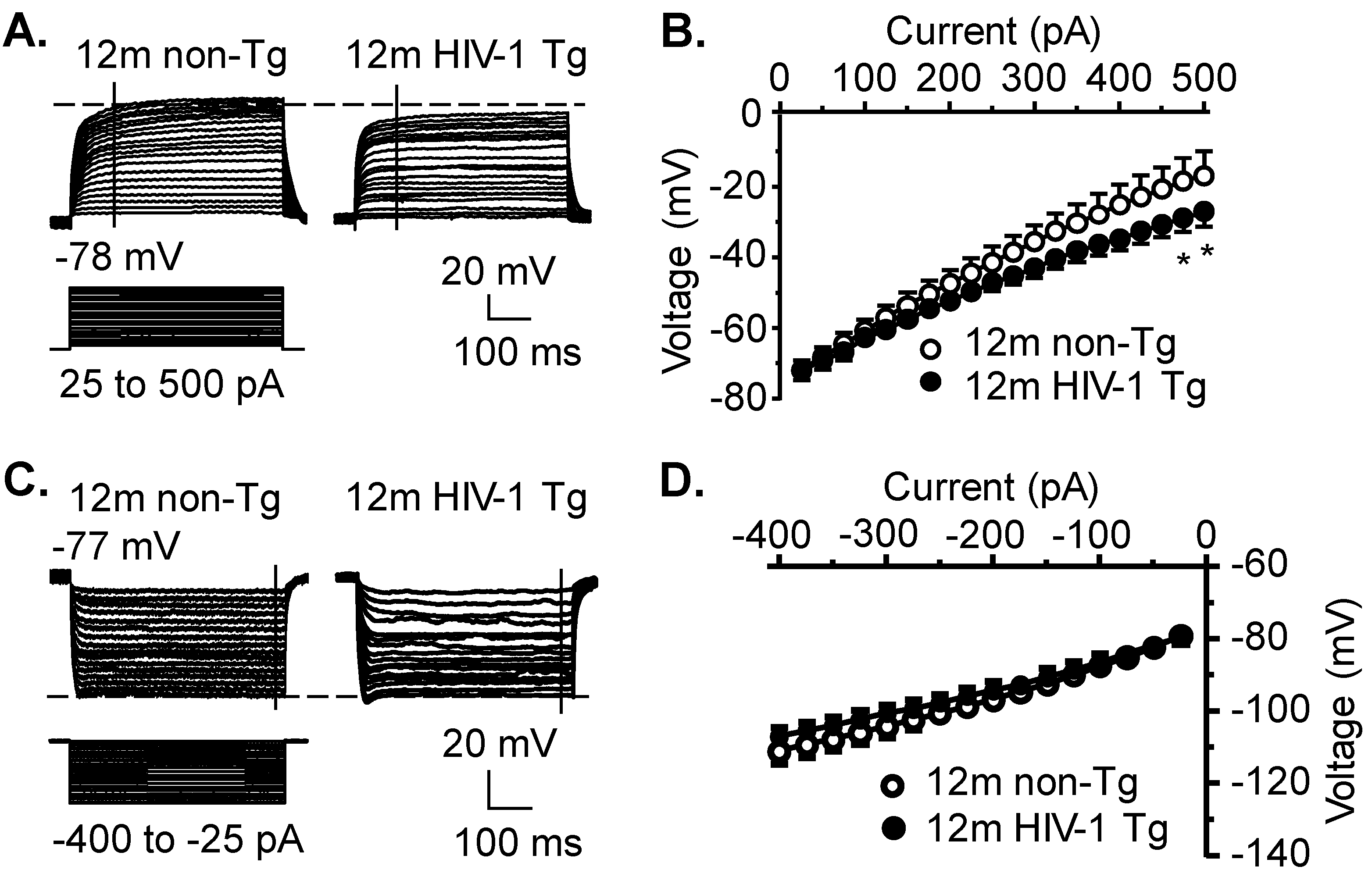
3.2. Activity of VGCCs Is Reduced in Striatal MSNs of 12mo HIV-1 Tg Rats
3.3. Expression of a Shorter and Less Functional 150 kDa Cav1.2 L-Channel Form Is Significantly Increased in the Striatum of 12mo Rats in the Context of neuroHIV
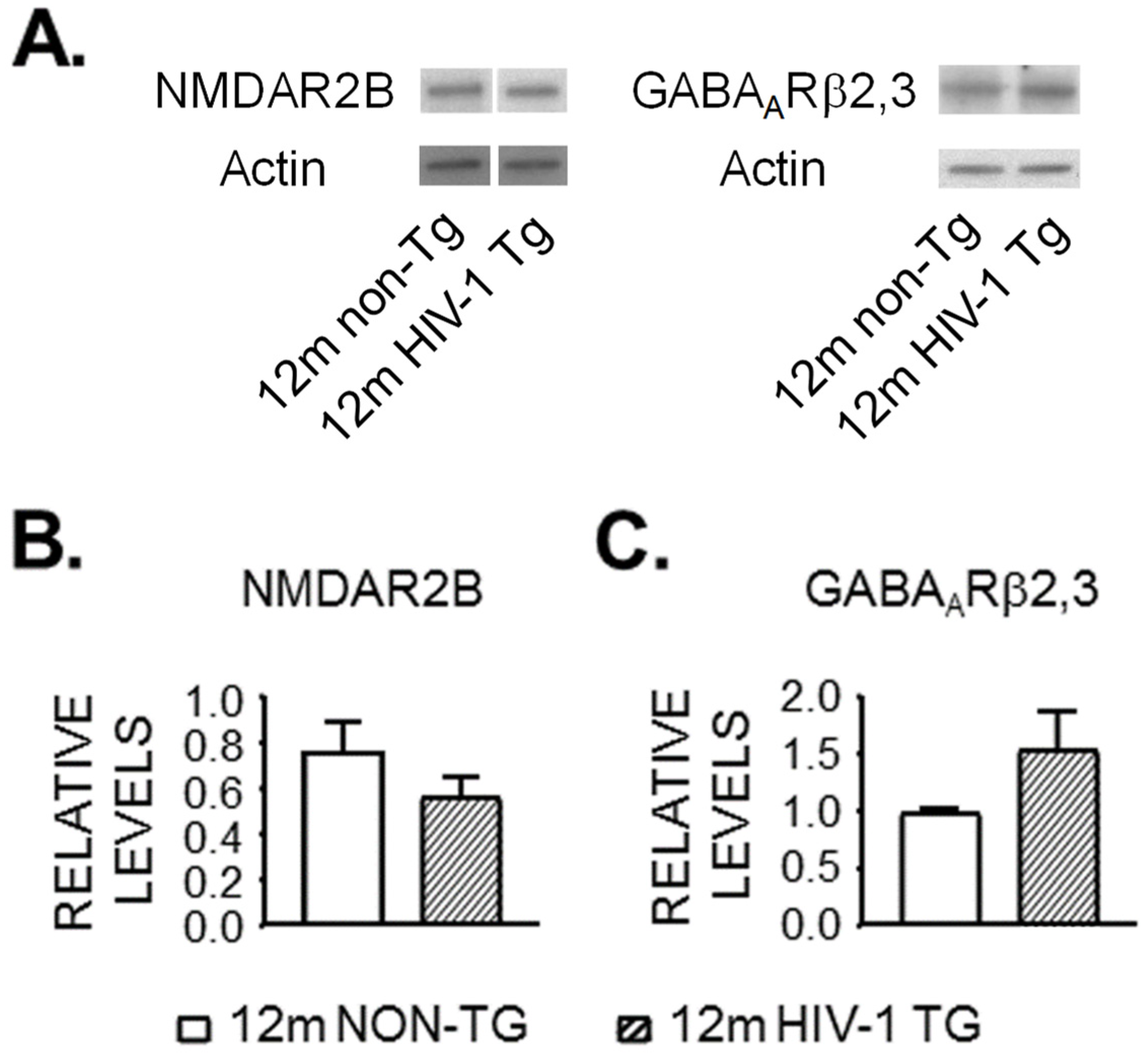
3.4. Expression of NMDAR and GABAAR Is Unaltered in the Striatum of 12mo Rats by neuroHIV
3.5. Kv Channel Activity Is Significantly Increased, While Activity of Kir and K2P Channels Is Not Significantly Altered, in Striatal MSNs in the Context of neuroHIV
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Antinori, A.; Arendt, G.; Becker, J.T.; Brew, B.J.; Byrd, D.A.; Cherner, M.; Clifford, D.B.; Cinque, P.; Epstein, L.G.; Goodkin, K.; et al. Updated research nosology for HIV-associated neurocognitive disorders. Neurology 2007, 69, 1789–1799. [Google Scholar] [CrossRef]
- McArthur, J.C.; Steiner, J.; Sacktor, N.; Nath, A. Human immunodeficiency virus-associated neurocognitive disorders: Mind the gap. Ann. Neurol. 2010, 67, 699–714. [Google Scholar] [PubMed]
- Heaton, R.K.; Clifford, D.B.; Franklin, D.R.; Woods, S.P.; Ake, C.; Vaida, F.; Ellis, R.J.; Letendre, S.L.; Marcotte, T.D.; Atkinson, J.H.; et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology 2010, 75, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Simioni, S.; Cavassini, M.; Annoni, J.-M.; Abraham, A.R.; Bourquin, I.; Schiffer, V.; Calmy, A.; Chave, J.-P.; Giacobini, E.; Hirschel, B.; et al. Cognitive dysfunction in HIV patients despite long-standing suppression of viremia. AIDS 2010, 24, 1243–1250. [Google Scholar] [CrossRef]
- Ferris, M.J.; Mactutus, C.F.; Booze, R.M. Neurotoxic profiles of HIV, psychostimulant drugs of abuse, and their concerted effect on the brain: Current status of dopamine system vulnerability in NeuroAIDS. Neurosci. Biobehav. Rev. 2008, 32, 883–909. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.-T. HIV-1 Tat-induced calcium dysregulation and neuronal dysfunction in vulnerable brain regions. Curr. Drug Targets 2016, 17, 4–14. [Google Scholar] [CrossRef] [PubMed]
- SSchier, C.J.; Marks, W.; Paris, J.J.; Barbour, A.J.; McLane, V.D.; Maragos, W.F.; McQuiston, A.R.; Knapp, P.E.; Hauser, K.F. Selective Vulnerability of Striatal D2 versus D1 Dopamine Receptor-Expressing Medium Spiny Neurons in HIV-1 Tat Transgenic Male Mice. J. Neurosci. 2017, 37, 5758–5769. [Google Scholar] [CrossRef]
- Stauch, K.L.; Emanuel, K.; Lamberty, B.G.; Morsey, B.; Fox, H.S. Central nervous system-penetrating antiretrovirals impair energetic reserve in striatal nerve terminals. J. Neurovirol. 2017, 23, 795–807. [Google Scholar] [CrossRef] [PubMed]
- Packard, M.G.; Knowlton, B.J. Learning and memory functions of the Basal Ganglia. Annu. Rev. Neurosci. 2002, 25, 563–593. [Google Scholar] [CrossRef]
- Thompson, P.M.; Dutton, R.A.; Hayashi, K.M.; Toga, A.W.; Lopez, O.L.; Aizenstein, H.J.; Becker, J.T. Thinning of the cerebral cortex visualized in HIV/AIDS reflects CD4+ T lymphocyte decline. Proc. Natl. Acad. Sci. USA 2005, 102, 15647–15652. [Google Scholar] [CrossRef]
- Tepper, J.M.; Abercrombie, E.D.; Bolam, J.P. Basal ganglia macrocircuits. Prog. Brain Res. 2007, 160, 3–7. [Google Scholar] [PubMed]
- Sesack, S.R.; Deutch, A.Y.; Roth, R.H.; Bunney, B.S. Topographical organization of the efferent projections of the medial prefrontal cortex in the rat: An anterograde tract-tracing study with phaseolus vulgaris leucoagglutinin. J. Comp. Neurol. 1989, 290, 213–242. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Haughey, N.J.; Nath, A. Cell death in HIV dementia. Cell Death Differ. 2005, 12 (Suppl. S1), 893–904. [Google Scholar] [CrossRef]
- Yu, C.; Narasipura, S.D.; Richards, M.H.; Hu, X.T.; Yamamoto, B.; Al-Harthi, L. HIV and drug abuse mediate astrocyte senescence in a beta-catenin-dependent manner leading to neuronal toxicity. Aging Cell, 2017; in press. [Google Scholar]
- Lutgen, V.; Narasipura, S.D.; Sharma, A.; Min, S.; Al-Harthi, L. β-Catenin signaling positively regulates glutamate uptake and metabolism in astrocytes. J. Neuroinflamm. 2016, 13, 242. [Google Scholar] [CrossRef]
- Haughey, N.J.; Mattson, M.P. Calcium dysregulation and neuronal apoptosis by the HIV-1 proteins Tat and gp120. J. Acquir. Immune Defic. Syndr. 2002, 31 (Suppl. S2), S55–S61. [Google Scholar] [CrossRef]
- Schifitto, G.; Navia, B.A.; Yiannoutsos, C.T.; Marra, C.M.; Chang, L.; Ernst, T.; Jarvik, J.G.; Miller, E.; Singer, E.J.; Ellis, R.; et al. Memantine and HIV-associated cognitive impairment: A neuropsychological and proton magnetic resonance spectroscopy study. AIDS 2007, 21, 1877–1886. [Google Scholar] [CrossRef]
- Reid, W.; Sadowska, M.; Denaro, F.; Rao, S.; Foulke, J.; Hayes, N.; Jones, O.; Doodnauth, D.; Davis, H.; Sill, A.; et al. An HIV-1 transgenic rat that develops HIV-related pathology and immunologic dysfunction. Proc. Natl. Acad. Sci. USA 2001, 98, 9271–9276. [Google Scholar] [CrossRef]
- Khodr, C.E.; Chen, L.; Dave, S.; Al-Harthi, L.; Hu, X.T. Combined chronic blockade of hyper-active L-type calcium channels and NMDA receptors ameliorates HIV-1 associated hyper-excitability of mPFC pyramidal neurons. Neurobiol. Dis. 2016, 94, 85–94. [Google Scholar] [CrossRef][Green Version]
- Khodr, C.E.; Chen, L.; Al-Harthi, L.; Hu, X.T. Aging alters voltage-gated calcium channels in prefrontal cortex pyramidal neurons in the HIV brain. J. Neurovirol. 2018, 24, 113–118. [Google Scholar] [CrossRef]
- Wayman, W.N.; Chen, L.; Napier, T.C.; Hu, X.T. Cocaine self-administration enhances excitatory responses of pyramidal neurons in the rat medial prefrontal cortex to human immunodeficiency virus-1 Tat. Eur. J. Neurosci. 2015, 41, 1195–1206. [Google Scholar] [CrossRef] [PubMed]
- Wayman, W.N.; Chen, L.; Hu, X.T.; Napier, T.C. HIV-1 transgenic rat prefrontal cortex hyper-excitability is enhanced by cocaine self-administration. Neuropsychopharmacology 2016, 41, 1965–1973. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Khodr, C.E.; Al-Harthi, L.; Hu, X.T. Aging and HIV-1 alter the function of specific K(+) channels in prefrontal cortex pyramidal neurons. Neurosci. Lett. 2019, 708, 134341. [Google Scholar] [CrossRef] [PubMed]
- Vigorito, M.; Connaghan, K.P.; Chang, S.L. The HIV-1 transgenic rat model of neuroHIV. Brain Behav. Immun. 2015, 48, 336–349. [Google Scholar] [CrossRef]
- Peng, J.; Vigorito, M.; Liu, X.; Zhou, D.; Wu, X.; Chang, S.L. The HIV-1 transgenic rat as a model for HIV-1 infected individuals on HAART. J. Neuroimmunol. 2010, 218, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, P. The Laboratory Rat: Relating Its Age with Human’s. Int. J. Prev. Med. 2013, 4, 624–630. [Google Scholar] [PubMed]
- Hu, X.-T.; Basu, S.; White, F.J. Repeated cocaine administration suppresses HVA-Ca2+ potentials and enhances activity of K+ channels in rat nucleus accumbens neurons. J. Neurophysiol. 2004, 92, 1597–1607. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Brini, M.; Cali, T.; Ottolini, D.; Carafoli, E. Neuronal calcium signaling: Function and dysfunction. Cell Mol. Life Sci. 2014, 71, 2787–2814. [Google Scholar] [CrossRef] [PubMed]
- Lipscombe, D. L-type calcium channels—Highs and new lows. Circ. Res. 2002, 90, 933–935. [Google Scholar] [CrossRef]
- Michailidis, I.E.; Abele-Henckels, K.; Zhang, W.K.; Lin, B.; Yu, Y.; Geyman, L.S.; Ehlers, M.D.; Pnevmatikakis, E.A.; Yang, J. Age-related homeostatic midchannel proteolysis of neuronal L-type voltage-gated Ca2+ channels. Neuron 2014, 82, 1045–1057. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Baudry, M. Developmental changes in NMDA neurotoxicity reflect developmental changes in subunit composition of NMDA receptors. J. Neurosci. 2006, 26, 2956–2963. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wong, T.P.; Aarts, M.; Rooyakkers, A.; Liu, L.; Lai, T.W.; Wu, D.C.; Lu, J.; Tymianski, M.; Craig, A.M.; et al. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J. Neurosci. 2007, 27, 2846–2857. [Google Scholar] [CrossRef] [PubMed]
- Goetz, T.; Arslan, A.; Wisden, W.; Wulff, P. GABA(A) receptors: Structure and function in the basal ganglia. Prog. Brain Res. 2007, 160, 21–41. [Google Scholar]
- Napier, T.C.; Chen, L.; Kashanchi, F.; Hu, X.-T. Repeated cocaine treatment enhances HIV-1 Tat-induced cortical excitability via over-activation of L-type calcium channels. J. Neuroimmune Pharmacol. 2014, 9, 354–368. [Google Scholar] [CrossRef] [PubMed]
- Wayman, W.N.; Dodiya, H.B.; Persons, A.L.; Kashanchi, F.; Kordower, J.H.; Hu, X.-T.; Napier, T.C. Enduring cortical alterations after a single in vivo treatment of HIV-1 Tat. Neuroreport 2012, 23, 825–829. [Google Scholar] [CrossRef] [PubMed]
- Groth, R.D.; Tirko, N.N.; Tsien, R.W. CaV1.2 calcium channels: Just cut out to be regulated? Neuron 2014, 82, 939–940. [Google Scholar] [CrossRef] [PubMed]
- Gleichmann, M.; Mattson, M.P. Neuronal calcium homeostasis and dysregulation. Antioxid. Redox. Signal. 2011, 14, 1261–1273. [Google Scholar] [CrossRef] [PubMed]
- Chami, M.; Oules, B.; Paterlini-Brechot, P. Cytobiological consequences of calcium-signaling alterations induced by human viral proteins. Biochim. Biophys. Acta 2006, 1763, 1344–1362. [Google Scholar] [CrossRef]
- Hulme, J.T.; Yarov-Yarovoy, V.; Lin, T.W.; Scheuer, T.; Catterall, W.A. Autoinhibitory control of the CaV1.2 channel by its proteolytically processed distal C-terminal domain. J. Physiol. 2006, 576, 87–102. [Google Scholar] [CrossRef]
- Green, E.M.; Barrett, C.F.; Bultynck, G.; Shamah, S.M.; Dolmetsch, R.E. The tumor suppressor eIF3e mediates calcium-dependent internalization of the L-type calcium channel CaV1.2. Neuron 2007, 55, 615–632. [Google Scholar] [CrossRef] [PubMed]
- Zamponi, G.W.; Currie, K.P. Regulation of Ca(V)2 calcium channels by G protein coupled receptors. Biochim. Biophys. Acta 2013, 1828, 1629–1643. [Google Scholar] [CrossRef] [PubMed]
- Arikkath, J.; Campbell, K.P. Auxiliary subunits: Essential components of the voltage-gated calcium channel complex. Curr. Opin. Neurobiol. 2003, 13, 298–307. [Google Scholar] [CrossRef]
- Brailoiu, G.C.; Deliu, E.; Barr, J.L.; Console-Bram, L.M.; Ciuciu, A.M.; Abood, M.E.; Unterwald, E.M.; Brailoiu, E. HIV Tat excites D1 receptor-like expressing neurons from rat nucleus accumbens. Drug Alcohol Depend. 2017, 178, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.P.; Sampair, C.S.; Kofuji, P.; Nath, A.; Ding, J.M. HIV protein, transactivator of transcription, alters circadian rhythms through the light entrainment pathway. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R656–R662. [Google Scholar] [CrossRef] [PubMed]
- Brittain, J.M.; Duarte, D.B.; Wilson, S.M.; Zhu, W.; Ballard, C.; Johnson, P.; Liu, N.; Xiong, W.; Ripsch, M.S.; Wang, Y.; et al. Suppression of inflammatory and neuropathic pain by uncoupling CRMP-2 from the presynaptic Ca2+ channel complex. Nat. Med. 2011, 17, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Ton, H.; Xiong, H. Astrocyte Dysfunctions and HIV-1 Neurotoxicity. J. AIDS Clin. Res. 2013, 4, 255. [Google Scholar] [PubMed]
- Melendez, R.I.; Roman, C.; Capo-Velez, C.M.; Lasalde-Dominicci, J.A. Decreased glial and synaptic glutamate uptake in the striatum of HIV-1 gp120 transgenic mice. J. Neurovirol. 2016, 22, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Gelman, B.B.; Chen, T.; Lisinicchia, J.G.; Soukup, V.M.; Carmical, J.R.; Starkey, J.; Masliah, E.; Commins, D.L.; Brandt, D.; Grant, I.; et al. The National NeuroAIDS Tissue Consortium brain gene array: Two types of HIV-associated neurocognitive impairment. PLoS ONE 2012, 7, e46178. [Google Scholar] [CrossRef] [PubMed]
- Buzhdygan, T.; Lisinicchia, J.; Patel, V.; Johnson, K.; Neugebauer, V.; Paessler, S.; Jennings, K.; Gelman, B. Neuropsychological, Neurovirological and Neuroimmune Aspects of Abnormal GABAergic Transmission in HIV Infection. J. Neuroimmune Pharmacol. 2016, 11, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Tsuruta, F.; Green, E.M.; Rousset, M.; Dolmetsch, R.E. PIKfyve regulates CaV1.2 degradation and prevents excitotoxic cell death. J. Cell Biol. 2009, 187, 279–294. [Google Scholar] [CrossRef]
- Liu, X.; Shi, S.; Yang, H.; Qu, C.; Chen, Y.; Liang, J.; Yang, B. The activation of N-methyl-d-aspartate receptors downregulates transient outward potassium and L-type calcium currents in rat models of depression. Am. J. Physiol.-Cell Physiol. 2017, 313, C187–C196. [Google Scholar] [CrossRef] [PubMed]
- Mangiavacchi, S.; Wolf, M.E. Stimulation of N-methyl-D-aspartate receptors, AMPA receptors or metabotropic glutamate receptors leads to rapid internalization of AMPA receptors in cultured nucleus accumbens neurons. Eur. J. Neurosci. 2004, 20, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Derkach, V.A.; Oh, M.C.; Guire, E.S.; Soderling, T.R. Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat. Rev. Neurosci. 2007, 8, 101–113. [Google Scholar] [PubMed]
- Lau, C.G.; Zukin, R.S. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat. Rev. Neurosci. 2007, 8, 413–426. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A. Membrane Property | 12mo Non-Tg | Cell # | 12mo HIV-1 Tg | Cell # | p Value |
|---|---|---|---|---|---|
| Rat Number | 7 | 14 | |||
| RMP (mV) | −78.5 ± 1.3 | 13 | −75.1 ± 1.2 | 22 | 0.074 |
| Rin (MΩ) | 97.1 ± 6.7 | 13 | 108.3 ± 9.6 | 21 | 0.410 |
| Rheobase (pA) | 203.8 ± 12.0 | 13 | 191.7 ± 13.0 | 21 | 0.526 |
| Threshold (mV) | −39.3 ± 0.8 | 13 | −38.5 ± 1.3 | 22 | 0.671 |
| Spike Amplitude (mV) | 79.4 ± 2.5 | 13 | 73.0 ± 2.8 | 22 | 0.133 |
| 1/2 Peak Duration (ms) | 2.1 ± 0.1 | 13 | 1.9 ± 0.1 | 22 | 0.371 |
| Time Constant (ms) | 11.5 ± 1.0 | 13 | 11.1 ± 1.1 | 21 | 0.816 |
| AHP (mV) | −14.0 ± 0.6 | 13 | −13.6 ± 0.6 | 21 | 0.659 |
| B. Ca2+ Spike | 12mo Non-Tg | 12mo HIV-1 Tg | p Value | ||
| Mean ± SEM | Cell # | Mean ± SEM | Cell # | ||
| Rheobase (pA) | 555.8 ± 43.5 | 12 | 743.1 ± 75.8 * | 8 | 0.0335 |
| Threshold (mV) | −8.3 ± 2.0 | 8 | −16.4 ± 3.4 | 7 | 0.8465 |
| Amplitude (mV) | 43.5 ± 2.1 | 8 | 48.2 ± 5.3 | 7 | 0.3992 |
| 1/2 amplitude duration (ms) | 473.8 ± 91.7 | 8 | 282.3 ± 29.6 | 7 | 0.0838 |
| Stage 1 duration (s) | 1.01 ± 0.12 | 12 | 0.56 ± 0.07 ** | 8 | 0.0092 |
| Stage 1 area (KmV × ms) | 89.9 ± 11.7 | 12 | 49.2 ± 6.2 * | 8 | 0.0158 |
| Ion Channels/Ionotropic Receptors | Location | Participation in Unique Findings |
|---|---|---|
| 1. L-type VGCC (high/low voltage-activated) | the dorsal striatum | Increased expression and ratio of dysfunctional (less active) HVA-Cav1.2 L-channel protein |
| 2. non-L-type VGCC (high/low voltage-activated) | the dorsal striatum | Not assessed |
| 3. Potassium channels | the dorsal striatum | Increased Kv channel activity; but no significant change in the activity of K2P or inwardly rectifying K+ channels |
| 4. NMDAR-2B and GABAAR | the dorsal striatum | No significant change in the protein levels of either one |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khodr, C.E.; Chen, L.; Al-Harthi, L.; Hu, X.-T. HIV-Induced Hyperactivity of Striatal Neurons Is Associated with Dysfunction of Voltage-Gated Calcium and Potassium Channels at Middle Age. Membranes 2022, 12, 737. https://doi.org/10.3390/membranes12080737
Khodr CE, Chen L, Al-Harthi L, Hu X-T. HIV-Induced Hyperactivity of Striatal Neurons Is Associated with Dysfunction of Voltage-Gated Calcium and Potassium Channels at Middle Age. Membranes. 2022; 12(8):737. https://doi.org/10.3390/membranes12080737
Chicago/Turabian StyleKhodr, Christina E., Lihua Chen, Lena Al-Harthi, and Xiu-Ti Hu. 2022. "HIV-Induced Hyperactivity of Striatal Neurons Is Associated with Dysfunction of Voltage-Gated Calcium and Potassium Channels at Middle Age" Membranes 12, no. 8: 737. https://doi.org/10.3390/membranes12080737
APA StyleKhodr, C. E., Chen, L., Al-Harthi, L., & Hu, X.-T. (2022). HIV-Induced Hyperactivity of Striatal Neurons Is Associated with Dysfunction of Voltage-Gated Calcium and Potassium Channels at Middle Age. Membranes, 12(8), 737. https://doi.org/10.3390/membranes12080737