
Mitochondrial Membranes and Mitochondrial Genome: Interactions and Clinical Syndromes


Abstract
:1. Introduction
2. Mitochondrial DNA Synthesis
3. Mitochondrial Membranes and the Organization of the Mitochondrial Genome
3.1. Molecular Basis of Mitochondrial Genome Organization
3.2. Clinical Syndromes due to Defects in Mitochondrial Genome Organization
4. Mitochondrial Fission and Fusion and Maintaining Mitochondrial Genome
4.1. Molecular Basis of Mitochondrial Fission and Fusion
4.2. Clinical Syndromes due to Defects in Mitochondrial Fission
4.3. Clinical Syndromes due to Defects in Mitochondrial Fusion
5. Cardiolipin and Mitochondrial-Genome Stability
5.1. Role of Cardiolipin in mtDNA Maintenance
5.2. Clinical Syndromes due to Defects in Cardiolipin Metabolism
6. Mitochondrial-Membrane Transporters and mtDNA Maintenance
6.1. Role of Mitochondrial Membrane Transporters in mtDNA Maintenance
6.2. Clinical Syndromes due to Defects in Mitochondrial-Membrane Transporters
7. Therapeutic Strategies
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Anderson, A.J.; Jackson, T.D.; Stroud, D.A.; Stojanovski, D. Mitochondria—Hubs for regulating cellular biochemistry: Emerging concepts and networks. Open Biol. 2019, 9, 190126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Hattab, A.W.; Craigen, W.J.; Scaglia, F. Mitochondrial DNA maintenance defects. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1539–1555. [Google Scholar] [CrossRef] [PubMed]
- Kühlbrandt, W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 2015, 13, 89. [Google Scholar] [CrossRef] [Green Version]
- Wohlrab, H. Transport proteins (carriers) of mitochondria. IUBMB Life 2009, 61, 40–46. [Google Scholar] [CrossRef]
- Mannella, C.A. Consequences of Folding the Mitochondrial Inner Membrane. Front Physiol. 2020, 11, 536. [Google Scholar] [CrossRef]
- Rius, R.; Cowley, M.J.; Riley, L.; Puttick, C.; Thorburn, D.R.; Christodoulou, J. Biparental inheritance of mitochondrial DNA in humans is not a common phenomenon. Genet. Med. 2019, 21, 2823–2826. [Google Scholar] [CrossRef] [PubMed]
- Castellani, C.A.; Longchamps, R.J.; Sun, J.; Guallar, E.; Arking, D.E. Thinking outside the nucleus: Mitochondrial DNA copy number in health and disease. Mitochondrion 2020, 53, 214–223. [Google Scholar] [CrossRef]
- Almannai, M.; El-Hattab, A.W.; Scaglia, F. Mitochondrial DNA replication: Clinical syndromes. Essays Biochem. 2018, 62, 297–308. [Google Scholar] [CrossRef]
- Chen, H.; Vermulst, M.; Wang, Y.E.; Chomyn, A.; Prolla, T.A.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 2010, 141, 280–289. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.R.; Han, J. Mitochondrial Nucleoid: Shield and Switch of the Mitochondrial Genome. Oxidative Med. Cell. Longev. 2017, 2017, 8060949. [Google Scholar] [CrossRef]
- Chapman, J.; Ng, Y.S.; Nicholls, T.J. The Maintenance of Mitochondrial DNA Integrity and Dynamics by Mitochondrial Membranes. Life 2020, 10, 164. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F.; Hudson, G. Mitochondrial genetics. Br. Med Bull. 2013, 106, 135–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taanman, J.-W. The mitochondrial genome: Structure, transcription, translation and replication. Biochim. Biophys. Acta BBA-Bioenergies 1999, 1410, 103–123. [Google Scholar] [CrossRef] [Green Version]
- Nicholls, T.J.; Minczuk, M. In D-loop: 40 years of mitochondrial 7S DNA. Exp. Gerontol. 2014, 56, 175–181. [Google Scholar] [CrossRef]
- Fusté, J.M.; Shi, Y.; Wanrooij, S.; Zhu, X.; Jemt, E.; Persson, Ö.; Sabouri, N.; Gustafsson, C.M.; Falkenberg, M. In Vivo Occupancy of Mitochondrial Single-Stranded DNA Binding Protein Supports the Strand Displacement Mode of DNA Replication. PLoS Genet. 2014, 10, e1004832. [Google Scholar] [CrossRef]
- Graziewicz, M.A.; Longley, M.J.; Copeland, W.C. DNA Polymerase γ in Mitochondrial DNA Replication and Repair. Chem. Rev. 2005, 106, 383–405. [Google Scholar] [CrossRef]
- Milenkovic, D.; Matic, S.; Kühl, I.; Ruzzenente, B.; Freyer, C.; Jemt, E.; Park, C.B.; Falkenberg, M.; Larsson, N.-G. TWINKLE is an essential mitochondrial helicase required for synthesis of nascent D-loop strands and complete mtDNA replication. Hum. Mol. Genet. 2013, 22, 1983–1993. [Google Scholar] [CrossRef]
- Oliveira, M.T.; Pontes, C.D.B.; Ciesielski, G.L. Roles of the mitochondrial replisome in mitochondrial DNA deletion formation. Genet. Mol. Biol. 2020, 43 (Suppl. S1), e20190069. [Google Scholar] [CrossRef] [Green Version]
- Fontana, G.A.; Gahlon, H.L. Mechanisms of replication and repair in mitochondrial DNA deletion formation. Nucleic Acids Res. 2020, 48, 11244–11258. [Google Scholar] [CrossRef]
- Gredilla, R. DNA Damage and Base Excision Repair in Mitochondria and Their Role in Aging. J. Aging Res. 2011, 2011, e257093. [Google Scholar] [CrossRef] [Green Version]
- Kukat, C.; Wurm, C.A.; Spåhr, H.; Falkenberg, M.; Larsson, N.-G.; Jakobs, S. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc. Natl. Acad. Sci. USA 2011, 108, 13534–13539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilkerson, R.; Bravo, L.; Garcia, I.; Gaytan, N.; Herrera, A.; Maldonado, A.; Quintanilla, B. The Mitochondrial Nucleoid: Integrating Mitochondrial DNA into Cellular Homeostasis. Cold Spring Harb. Perspect. Biol. 2013, 5, a011080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonekamp, N.A.; Larsson, N.-G. SnapShot: Mitochondrial Nucleoid. Cell 2018, 172, 388–388.e1. [Google Scholar] [CrossRef]
- Kopek, B.G.; Shtengel, G.; Xu, C.S.; Clayton, D.A.; Hess, H.F. Correlative 3D superresolution fluorescence and electron microscopy reveal the relationship of mitochondrial nucleoids to membranes. Proc. Natl. Acad. Sci. USA 2012, 109, 6136–6141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Zhou, R.; Zhang, C.; He, S.; Su, Z. Mitochondria-Associated Endoplasmic Reticulum Membranes in the Pathogenesis of Type 2 Diabetes Mellitus. Front. Cell Dev. Biol. 2020, 8, 571554. [Google Scholar] [CrossRef]
- Lewis, S.C.; Uchiyama, L.F.; Nunnari, J. ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 2016, 353, aaf5549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, J.; Guo, Y.; Xue, B.; Shi, P.; Chen, Y.; Su, Q.P.; Hao, H.; Zhao, S.; Wu, C.; Yu, L.; et al. ER-mitochondria contacts promote mtDNA nucleoids active transportation via mitochondrial dynamic tubulation. Nat. Commun. 2020, 11, 4471. [Google Scholar] [CrossRef]
- Ngo, H.B.; Lovely, G.A.; Phillips, R.; Chan, D.C. Distinct structural features of TFAM drive mitochondrial DNA packaging versus transcriptional activation. Nat. Commun. 2014, 5, 3077. [Google Scholar] [CrossRef] [Green Version]
- Aasumets, K.; Basikhina, Y.; Pohjoismäki, J.L.; Goffart, S.; Gerhold, J. TFAM knockdown-triggered mtDNA-nucleoid aggregation and a decrease in mtDNA copy number induce the reorganization of nucleoid populations and mitochondria-associated ER-membrane contacts. Biochem. Biophys. Rep. 2021, 28, 101142. [Google Scholar] [CrossRef]
- Rajala, N.; Gerhold, J.M.; Martinsson, P.; Klymov, A.; Spelbrink, J.N. Replication factors transiently associate with mtDNA at the mitochondrial inner membrane to facilitate replication. Nucleic Acids Res. 2013, 42, 952–967. [Google Scholar] [CrossRef] [Green Version]
- So, M.; Stiban, J.; Ciesielski, G.L.; Hovde, S.L.; Kaguni, L.S. Implications of Membrane Binding by the Fe-S Cluster-Containing N-Terminal Domain in the Drosophila Mitochondrial Replicative DNA Helicase. Front. Genet. 2021, 12, 790521. [Google Scholar] [CrossRef] [PubMed]
- Gerhold, J.M.; Cansiz-Arda, Ş.; Lõhmus, M.; Engberg, O.; Reyes, A.; Van Rennes, H.; Sanz, A.; Holt, I.J.; Cooper, H.M.; Spelbrink, J.N. Human Mitochondrial DNA-Protein Complexes Attach to a Cholesterol-Rich Membrane Structure. Sci. Rep. 2015, 5, 15292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Mao, C.-C.; Reyes, A.; Sembongi, H.; Di Re, M.; Granycome, C.; Clippingdale, A.B.; Fearnley, I.M.; Harbour, M.; Robinson, A.J.; et al. The AAA+ protein ATAD3 has displacement loop binding properties and is involved in mitochondrial nucleoid organization. J. Cell Biol. 2007, 176, 141–146. [Google Scholar] [CrossRef]
- Arguello, T.; Peralta, S.; Antonicka, H.; Gaidosh, G.; Diaz, F.; Tu, Y.-T.; Garcia, S.; Shiekhattar, R.; Barrientos, A.; Moraes, C.T. ATAD3A has a scaffolding role regulating mitochondria inner membrane structure and protein assembly. Cell Rep. 2021, 37, 110139. [Google Scholar] [CrossRef]
- Stiles, A.R.; Simon, M.; Stover, A.; Eftekharian, S.; Khanlou, N.; Wang, H.L.; Magaki, S.; Lee, H.; Partynski, K.; Dorrani, N.; et al. Mutations in TFAM, encoding mitochondrial transcription factor A, cause neonatal liver failure associated with mtDNA depletion. Mol. Genet. Metab. 2016, 119, 91–99. [Google Scholar] [CrossRef]
- Ullah, F.; Rauf, W.; Khan, K.; Khan, S.; Bell, K.M.; de Oliveira, V.C.; Tariq, M.; Bakhshalizadeh, S.; Touraine, P.; Katsanis, N.; et al. A recessive variant in TFAM causes mtDNA depletion associated with primary ovarian insufficiency, seizures, intellectual disability and hearing loss. Qual. Life Res. 2021, 140, 1733–1751. [Google Scholar] [CrossRef]
- Van Hove, J.L.; Cunningham, V.; Rice, C.; Ringel, S.P.; Zhang, Q.; Chou, P.-C.; Truong, C.K.; Wong, L.-J.C. Finding twinkle in the eyes of a 71-year-old lady: A case report and review of the genotypic and phenotypic spectrum of TWINKLE-related dominant disease. Am. J. Med Genet. Part A 2009, 149, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Pierce, S.B.; Gulsuner, S.; Stapleton, G.A.; Walsh, T.; Lee, M.K.; Mandell, J.B.; Morales, A.; Klevit, R.E.; King, M.-C.; Rogers, R.C. Infantile onset spinocerebellar ataxia caused by compound heterozygosity for Twinkle mutations and modeling of Twinkle mutations causing recessive disease. Mol. Case Stud. 2016, 2, a001107. [Google Scholar] [CrossRef] [Green Version]
- Hakonen, A.H.; Goffart, S.; Marjavaara, S.; Paetau, A.; Cooper, H.; Mattila, K.; Lampinen, M.; Sajantila, A.; Lönnqvist, T.; Spelbrink, J.N.; et al. Infantile-onset spinocerebellar ataxia and mitochondrial recessive ataxia syndrome are associated with neuronal complex I defect and mtDNA depletion. Hum. Mol. Genet. 2008, 17, 3822–3835. [Google Scholar] [CrossRef] [Green Version]
- Fekete, B.; Pentelényi, K.; Rudas, G.; Gál, A.; Grosz, Z.; Illés, A.; Idris, J.; Csukly, G.; Domonkos, A.; Molnar, M.J. Broadening the phenotype of the TWNK gene associated Perrault syndrome. BMC Med. Genet. 2019, 20, 198. [Google Scholar] [CrossRef] [Green Version]
- Harel, T.; Yoon, W.H.; Garone, C.; Gu, S.; Coban-Akdemir, Z.; Eldomery, M.K.; Posey, J.; Jhangiani, S.N.; Rosenfeld, J.A.; Cho, M.T.; et al. Recurrent De Novo and Biallelic Variation of ATAD3A, Encoding a Mitochondrial Membrane Protein, Results in Distinct Neurological Syndromes. Am. J. Hum. Genet. 2016, 99, 831–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peralta, S.; Goffart, S.; Williams, S.L.; Diaz, F.; Garcia, S.; Nissanka, N.; Area-Gomez, E.; Pohjoismäki, J.; Moraes, C.T. ATAD3 controls mitochondrial cristae structure in mouse muscle, influencing mtDNA replication and cholesterol levels. J. Cell Sci. 2018, 131, jcs217075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, R.; Frazier, A.E.; Durigon, R.; Patel, H.; Jones, A.W.; Dalla Rosa, I.; Lake, N.J.; Compton, A.G.; Mountford, H.S.; Tucker, E.J.; et al. ATAD3 gene cluster deletions cause cerebellar dysfunction associated with altered mitochondrial DNA and cholesterol metabolism. Brain 2017, 140, 1595–1610. [Google Scholar] [CrossRef] [PubMed]
- Gunning, A.C.; Strucinska, K.; Oreja, M.M.; Parrish, A.; Caswell, R.; Stals, K.L.; Durigon, R.; Durlacher-Betzer, K.; Cunningham, M.H.; Grochowski, C.M.; et al. Recurrent De Novo NAHR Reciprocal Duplications in the ATAD3 Gene Cluster Cause a Neurogenetic Trait with Perturbed Cholesterol and Mitochondrial Metabolism. Am. J. Hum. Genet. 2020, 106, 272–279. [Google Scholar] [CrossRef] [Green Version]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef] [Green Version]
- Ranieri, M.; Brajkovic, S.; Riboldi, G.; Ronchi, D.; Rizzo, F.; Bresolin, N.; Corti, S.; Comi, G.P. Mitochondrial Fusion Proteins and Human Diseases. Neurol. Res. Int. 2013, 2013, 293893. [Google Scholar] [CrossRef] [Green Version]
- Silva Ramos, E.; Motori, E.; Brüser, C.; Kühl, I.; Yeroslaviz, A.; Ruzzenente, B.; Kauppila, J.H.K.; Busch, J.D.; Hultenby, K.; Habermann, B.H.; et al. Mitochondrial fusion is required for regulation of mitochondrial DNA replication. PLoS Genet. 2019, 15, e1008085. [Google Scholar] [CrossRef] [Green Version]
- Yoon, G.; Malam, Z.; Paton, T.; Marshall, C.R.; Hyatt, E.; Ivakine, Z.; Scherer, S.W.; Lee, K.-S.; Hawkins, C.; Cohn, R.D.; et al. Lethal Disorder of Mitochondrial Fission Caused by Mutations in DNM1L. J. Pediatrics 2016, 171, 313–316.e2. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Z.; Li, D.; Lei, M.; Li, Q.; Liu, X.; Zhang, P. DNM1L-Related Mitochondrial Fission Defects Presenting as Encephalopathy: A Case Report and Literature Review. Front. Pediatr. 2021, 9, 626657. [Google Scholar] [CrossRef]
- Gerber, S.; Charif, M.; Chevrollier, A.; Chaumette, T.; Angebault, C.; Kane, M.S.; Paris, A.; Alban, J.; Quiles, M.; Delettre, C.; et al. Mutations in DNM1L, as in OPA1, result in dominant optic atrophy despite opposite effects on mitochondrial fusion and fission. Brain 2017, 140, 2586–2596. [Google Scholar] [CrossRef] [PubMed]
- Nasca, A.; Nardecchia, F.; Commone, A.; Semeraro, M.; Legati, A.; Garavaglia, B.; Ghezzi, D.; Leuzzi, V. Clinical and Biochemical Features in a Patient With Mitochondrial Fission Factor Gene Alteration. Front. Genet. 2018, 9, 625. Available online: https://www.frontiersin.org/article/10.3389/fgene.2018.00625 (accessed on 8 May 2022). [CrossRef] [PubMed] [Green Version]
- Koch, J.; Feichtinger, R.G.; Freisinger, P.; Pies, M.; Schrödl, F.; Iuso, A.; Sperl, W.; Mayr, J.A.; Prokisch, H.; Haack, T.B. Disturbed mitochondrial and peroxisomal dynamics due to loss of MFF causes Leigh-like encephalopathy, optic atrophy and peripheral neuropathy. J. Med. Genet. 2016, 53, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Amati-Bonneau, P.; Valentino, M.L.; Reynier, P.; Gallardo, M.E.; Bornstein, B.; Boissière, A.; Campos, Y.; Rivera, H.; de la Aleja, J.G.; Carroccia, R.; et al. OPA1 mutations induce mitochondrial DNA instability and optic atrophy ’plus’ phenotypes. Brain 2007, 131, 338–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carelli, V.; Sabatelli, M.; Carrozzo, R.; Rizza, T.; Schimpf, S.; Wissinger, B.; Zanna, C.; Rugolo, M.; La Morgia, C.; Caporali, L.; et al. ‘Behr syndrome’ with OPA1 compound heterozygote mutations. Brain 2015, 138, e321. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, R.; Saada, A.; Flannery, P.J.; Burté, F.; Soiferman, D.; Khayat, M.; Eisner, V.; Vladovski, E.; Taylor, R.W.; Bindoff, L.A.; et al. Fatal infantile mitochondrial encephalomyopathy, hypertrophic cardiomyopathy and optic atrophy associated with a homozygous OPA1 mutation. J. Med. Genet. 2015, 53, 127–131. [Google Scholar] [CrossRef] [Green Version]
- Rouzier, C.; Bannwarth, S.; Chaussenot, A.; Chevrollier, A.; Verschueren, A.; Bonello-Palot, N.; Fragaki, K.; Cano, A.; Pouget, J.; Pellissier, J.-F.; et al. The MFN2 gene is responsible for mitochondrial DNA instability and optic atrophy ’plus’ phenotype. Brain 2012, 135, 23–34. [Google Scholar] [CrossRef] [Green Version]
- Züchner, S.; Mersiyanova, I.V.; Muglia, M.; Bissar-Tadmouri, N.; Rochelle, J.; Dadali, E.L.; Zappia, M.; Nelis, E.; Patitucci, A.; Senderek, J.; et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004, 36, 449–451. [Google Scholar] [CrossRef]
- Pipis, M.; Feely, S.M.E.; Polke, J.M.; Skorupinska, M.; Perez, L.; Shy, R.R.; Laura, M.; Morrow, J.M.; Moroni, I.; Pisciotta, C.; et al. Natural history of Charcot-Marie-Tooth disease type 2A: A large international multicentre study. Brain 2020, 143, 3589–3602. [Google Scholar] [CrossRef]
- Schenkel, L.C.; Bakovic, M. Formation and Regulation of Mitochondrial Membranes. Int. J. Cell Biol. 2014, 2014, e709828. [Google Scholar] [CrossRef] [Green Version]
- Dudek, J. Role of Cardiolipin in Mitochondrial Signaling Pathways. Front. Cell Dev. Biol. 2017, 5, 90. Available online: https://www.frontiersin.org/article/10.3389/fcell.2017.00090 (accessed on 1 April 2022). [CrossRef] [PubMed] [Green Version]
- Ren, M.; Phoon, C.K.; Schlame, M. Metabolism and function of mitochondrial cardiolipin. Prog. Lipid Res. 2014, 55, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ikon, N.; Ryan, R.O. Cardiolipin and mitochondrial cristae organization. Biochim. Biophys. Acta 2017, 1859, 1156–1163. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; De Benedictis, V.; Ruggiero, F.M.; Petrosillo, G. Functional role of cardiolipin in mitochondrial bioenergetics. Biochim. Biophys. Acta 2014, 1837, 408–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Role of Cardiolipin in Mitochondrial Function and Dynamics in Health and Disease: Molecular and Pharmacological Aspects. Cells 2019, 8, 728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kameoka, S.; Adachi, Y.; Okamoto, K.; Iijima, M.; Sesaki, H. Phosphatidic Acid and Cardiolipin Coordinate Mitochondrial Dynamics. Trends Cell Biol. 2017, 28, 67–76. [Google Scholar] [CrossRef]
- Ban, T.; Ishihara, T.; Kohno, H.; Saita, S.; Ichimura, A.; Maenaka, K.; Oka, T.; Mihara, K.; Ishihara, N. Molecular basis of selective mitochondrial fusion by heterotypic action between OPA1 and cardiolipin. Nat. Cell Biol. 2017, 19, 856–863. [Google Scholar] [CrossRef]
- Stepanyants, N.; Macdonald, P.J.; Francy, C.A.; Mears, J.A.; Qi, X.; Ramachandran, R. Cardiolipin’s propensity for phase transition and its reorganization by dynamin-related protein 1 form a basis for mitochondrial membrane fission. Mol. Biol. Cell 2015, 26, 3104–3116. [Google Scholar] [CrossRef]
- Luévano-Martínez, L.A.; Forni, M.F.; dos Santos, V.T.; Souza-Pinto, N.C.; Kowaltowski, A.J. Cardiolipin is a key determinant for mtDNA stability and segregation during mitochondrial stress. Biochim. Biophys. Acta 2015, 1847, 587–598. [Google Scholar] [CrossRef] [Green Version]
- Houtkooper, R.H.; Vaz, F.M. Cardiolipin, the heart of mitochondrial metabolism. Cell Mol. Life Sci. CMLS 2008, 65, 2493–2506. [Google Scholar] [CrossRef]
- Ye, C.; Shen, Z.; Greenberg, M.L. Cardiolipin remodeling: A regulatory hub for modulating cardiolipin metabolism and function. J. Bioenerg. Biomembr. 2014, 48, 113–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houtkooper, R.H.; Turkenburg, M.; Poll-The, B.T.; Karall, D.; Pérez-Cerdá, C.; Morrone, A.; Malvagia, S.; Wanders, R.J.; Kulik, W.; Vaz, F.M. The enigmatic role of tafazzin in cardiolipin metabolism. Biochim. Biophys. Acta (BBA) Biomembr. 2009, 1788, 2003–2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finsterer, J. Barth syndrome: Mechanisms and management. Appl. Clin. Genet. 2019, 12, 95–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, C.; Rao, E.S.; Pierre, G.; Chronopoulou, E.; Hornby, B.; Heyman, A.; Vernon, H.J. Clinical presentation and natural history of Barth Syndrome: An overview. J. Inherit. Metab. Dis. 2021, 45, 7–16. [Google Scholar] [CrossRef]
- Suzuki-Hatano, S.; Sriramvenugopal, M.; Ramanathan, M.; Soustek, M.; Byrne, B.J.; Cade, W.T.; Kang, P.B.; Pacak, C.A. Increased mtDNA Abundance and Improved Function in Human Barth Syndrome Patient Fibroblasts Following AAV-TAZ Gene Delivery. Int. J. Mol. Sci. 2019, 20, 3416. [Google Scholar] [CrossRef] [Green Version]
- Wang, L. Mitochondrial purine and pyrimidine metabolism and beyond. Nucleosides Nucleotides Nucleic Acids 2016, 35, 578–594. [Google Scholar] [CrossRef]
- Palmieri, F. Mitochondrial transporters of the SLC25 family and associated diseases: A review. J. Inherit. Metab. Dis. 2014, 37, 565–575. [Google Scholar] [CrossRef]
- Baldwin, S.A.; Beal, P.R.; Yao, S.Y.; King, A.E.; Cass, C.E.; Young, J.D. The equilibrative nucleoside transporter family, SLC29. Pflugers Arch. 2004, 447, 735–743. [Google Scholar] [CrossRef]
- Di Noia, M.A.; Todisco, S.; Cirigliano, A.; Rinaldi, T.; Agrimi, G.; Iacobazzi, V.; Palmieri, F. The Human SLC25A33 and SLC25A36 Genes of Solute Carrier Family 25 Encode Two Mitochondrial Pyrimidine Nucleotide Transporters. J. Biol. Chem. 2014, 289, 33137–33148. [Google Scholar] [CrossRef] [Green Version]
- Antonenkov, V.D.; Isomursu, A.; Mennerich, D.; Vapola, M.H.; Weiher, H.; Kietzmann, T.; Hiltunen, J.K. The Human Mitochondrial DNA Depletion Syndrome Gene MPV17 Encodes a Non-selective Channel That Modulates Membrane Potential. J. Biol. Chem. 2015, 290, 13840–13861. [Google Scholar] [CrossRef] [Green Version]
- Sperl, L.E.; Hagn, F. NMR Structural and Biophysical Analysis of the Disease-Linked Inner Mitochondrial Membrane Protein MPV17. J. Mol. Biol. 2021, 433, 167098. [Google Scholar] [CrossRef]
- Rosa, I.D.; Cámara, Y.; Durigon, R.; Moss, C.F.; Vidoni, S.; Akman, G.; Hunt, L.; Johnson, M.A.; Grocott, S.; Wang, L.; et al. MPV17 Loss Causes Deoxynucleotide Insufficiency and Slow DNA Replication in Mitochondria. PLoS Genet. 2016, 12, e1005779. [Google Scholar] [CrossRef]
- Martorano, L.; Peron, M.; Laquatra, C.; Lidron, E.; Facchinello, N.; Meneghetti, G.; Tiso, N.; Rasola, A.; Ghezzi, D.; Argenton, F. The zebrafish orthologue of the human hepatocerebral disease gene MPV17 plays pleiotropic roles in mitochondria. Dis. Model. Mech. 2019, 12, dmm037226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonzo, J.R.; Venkataraman, C.; Field, M.; Stover, P.J. The mitochondrial inner membrane protein MPV17 prevents uracil accumulation in mitochondrial DNA. J. Biol. Chem. 2018, 293, 20285–20294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Chen, X.J. Adenine Nucleotide Translocase, Mitochondrial Stress, and Degenerative Cell Death. Oxidative Med. Cell. Longev. 2013, 2013, 146860. [Google Scholar] [CrossRef] [Green Version]
- Brower, J.V.; Rodic, N.; Seki, T.; Jorgensen, M.; Fliess, N.; Yachnis, A.T.; McCarrey, J.R.; Oh, S.P.; Terada, N. Evolutionarily Conserved Mammalian Adenine Nucleotide Translocase 4 Is Essential for Spermatogenesis. J. Biol. Chem. 2007, 282, 29658–29666. [Google Scholar] [CrossRef] [Green Version]
- Epand, R.M.; Epand, R.F.; Berno, B.; Pelosi, L.; Brandolin, G. Association of Phosphatidic Acid with the Bovine Mitochondrial ADP/ATP Carrier. Biochemistry 2009, 48, 12358–12364. [Google Scholar] [CrossRef]
- Clémençon, B.; Babot, M.; Trézéguet, V. The mitochondrial ADP/ATP carrier (SLC25 family): Pathological implications of its dysfunction. Mol. Asp. Med. 2013, 34, 485–493. [Google Scholar] [CrossRef] [Green Version]
- Ruprecht, J.J.; King, M.S.; Zögg, T.; Aleksandrova, A.; Pardon, E.; Crichton, P.G.; Steyaert, J.; Kunji, E.R. The Molecular Mechanism of Transport by the Mitochondrial ADP/ATP Carrier. Cell 2019, 176, 435–447.e15. [Google Scholar] [CrossRef] [Green Version]
- Brustovetsky, N. The Role of Adenine Nucleotide Translocase in the Mitochondrial Permeability Transition. Cells 2020, 9, 2686. [Google Scholar] [CrossRef]
- Kaukonen, J.; Juselius, J.K.; Tiranti, V.; Kyttälä, A.; Zeviani, M.; Comi, G.P.; Keränen, S.; Peltonen, L.; Suomalainen, A. Role of Adenine Nucleotide Translocator 1 in mtDNA Maintenance. Science 2000, 289, 782–785. [Google Scholar] [CrossRef] [PubMed]
- Spinazzola, A.; Viscomi, C.; Fernandez-Vizarra, E.; Carrara, F.; D’Adamo, A.P.; Calvo, S.; Marsano, R.M.; Donnini, C.; Weiher, H.; Strisciuglio, P.; et al. MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat. Genet. 2006, 38, 570–575. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W.; Wang, J.; Dai, H.; Almannai, M.; Staufner, C.; Alfadhel, M.; Gambello, M.J.; Prasun, P.; Raza, S.; Lyons, H.J.; et al. MPV17-related mitochondrial DNA maintenance defect: New cases and review of clinical, biochemical, and molecular aspects. Hum. Mutat. 2018, 39, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Siciliano, G.; Tessa, A.; Petrini, S.; Mancuso, M.; Bruno, C.; Grieco, G.; Malandrini, A.; DeFlorio, L.; Martini, B.; Federico, A.; et al. Autosomal dominant external ophthalmoplegia and bipolar affective disorder associated with a mutation in the ANT1 gene. Neuromuscul. Disord. 2003, 13, 162–165. [Google Scholar] [CrossRef]
- Palmieri, L.; Alberio, S.; Pisano, I.; Lodi, T.; Meznaric-Petrusa, M.; Zidar, J.; Santoro, A.; Scarcia, P.; Fontanesi, F.; Lamantea, E.; et al. Complete loss-of-function of the heart/muscle-specific adenine nucleotide translocator is associated with mitochondrial myopathy and cardiomyopathy. Hum. Mol. Genet. 2005, 14, 3079–3088. [Google Scholar] [CrossRef] [Green Version]
- Mayr, J.A.; Haack, T.B.; Graf, E.; Zimmermann, F.A.; Wieland, T.; Haberberger, B.; Superti-Furga, A.; Kirschner, J.; Steinmann, B.; Baumgartner, M.R.; et al. Lack of the Mitochondrial Protein Acylglycerol Kinase Causes Sengers Syndrome. Am. J. Hum. Genet. 2012, 90, 314–320. [Google Scholar] [CrossRef] [Green Version]
- Haghighi, A.; Haack, T.B.; Atiq, M.; Mottaghi, H.; Haghighi-Kakhki, H.; Bashir, R.A.; Ahting, U.; Feichtinger, R.G.; Mayr, J.A.; Rötig, A.; et al. Sengers syndrome: Six novel AGK mutations in seven new families and review of the phenotypic and mutational spectrum of 29 patients. Orphanet J. Rare Dis. 2014, 9, 119. [Google Scholar] [CrossRef] [Green Version]
- Almannai, M.; El-Hattab, A.W.; Ali, M.; Soler-Alfonso, C.; Scaglia, F. Clinical trials in mitochondrial disorders, an update. Mol. Genet. Metab. 2020, 131, 1–13. [Google Scholar] [CrossRef]
- Jüschke, C.; Klopstock, T.; Catarino, C.B.; Owczarek-Lipska, M.; Wissinger, B.; Neidhardt, J. Autosomal dominant optic atrophy: A novel treatment for OPA1 splice defects using U1 snRNA adaption. Mol. Ther. Nucleic Acids 2021, 26, 1186–1197. [Google Scholar] [CrossRef]
- Weiss, J.N.; Levy, S. Stem Cell Ophthalmology Treatment Study (SCOTS): Bone marrow derived stem cells in the treatment of Dominant Optic Atrophy. Stem Cell Investig. 2019, 6, 41. [Google Scholar] [CrossRef]
- Sarzi, E.; Seveno, M.; Piro-Mégy, C.; Elzière, L.; Quilès, M.; Péquignot, M.; Müller, A.; Hamel, C.P.; Lenaers, G.; Delettre, C. OPA1 gene therapy prevents retinal ganglion cell loss in a Dominant Optic Atrophy mouse model. Sci. Rep. 2018, 8, 2468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Úbeda, M.; Semenzato, M.; Franco-Romero, A.; Junquera, E.; Aicart, E.; Scorrano, L.; López-Montero, I. Transgene expression in mice of the Opa1 mitochondrial transmembrane protein through bicontinuous cubic lipoplexes containing gemini imidazolium surfactants. J. Nanobiotechnology 2021, 19, 425. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.; Kitsis, R.N.; Fleischer, J.A.; Gavathiotis, E.; Kornfeld, O.S.; Gong, G.; Biris, N.; Benz, A.; Qvit, N.; Donnelly, S.; et al. Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature 2016, 540, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Rocha, A.G.; Franco, A.; Krezel, A.M.; Rumsey, J.M.; Alberti, J.M.; Knight, W.C.; Biris, N.; Zacharioudakis, E.; Janetka, J.W.; Baloh, R.H.; et al. MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot-Marie-Tooth disease type 2A. Science 2018, 360, 336–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, X.; Zhang, L.; Franco, A.; Li, J.; Rocha, A.G.; Devanathan, S.; Dolle, R.E.; Bernstein, P.R.; Dorn, I.G.W. Discovery of 6-Phenylhexanamide Derivatives as Potent Stereoselective Mitofusin Activators for the Treatment of Mitochondrial Diseases. J. Med. Chem. 2020, 63, 7033–7051. [Google Scholar] [CrossRef]
- Szeto, H.H.; Schiller, P.W. Novel Therapies Targeting Inner Mitochondrial Membrane—From Discovery to Clinical Development. Pharm. Res. 2011, 28, 2669–2679. [Google Scholar] [CrossRef]
- Szeto, H.H. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. J. Cereb. Blood Flow Metab. 2014, 171, 2029–2050. [Google Scholar] [CrossRef] [Green Version]
- Thompson, R.; Manuel, R.; Aiudi, A.; Jones, J.J.; Carr, J.; Hornby, B.; Vernon, H. Elamipretide in patients with barth syndrome: A randomized, double-blind, placebo-controlled clinical trial followed by 36-week open-label extension. J. Am. Coll. Cardiol. 2020, 75 (Suppl. S1), 957. [Google Scholar] [CrossRef]
- Bottani, E.; Giordano, C.; Civiletto, G.; Di Meo, I.; Auricchio, A.; Ciusani, E.; Marchet, S.; Lamperti, C.; D’Amati, G.; Viscomi, C.; et al. AAV-mediated Liver-specific MPV17 Expression Restores mtDNA Levels and Prevents Diet-induced Liver Failure. Mol. Ther. 2014, 22, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Di Punzio, G.; Gilberti, M.; Baruffini, E.; Lodi, T.; Donnini, C.; Dallabona, C. A Yeast-Based Repurposing Approach for the Treatment of Mitochondrial DNA Depletion Syndromes Led to the Identification of Molecules Able to Modulate the dNTP Pool. Int. J. Mol. Sci. 2021, 22, 12223. [Google Scholar] [CrossRef]
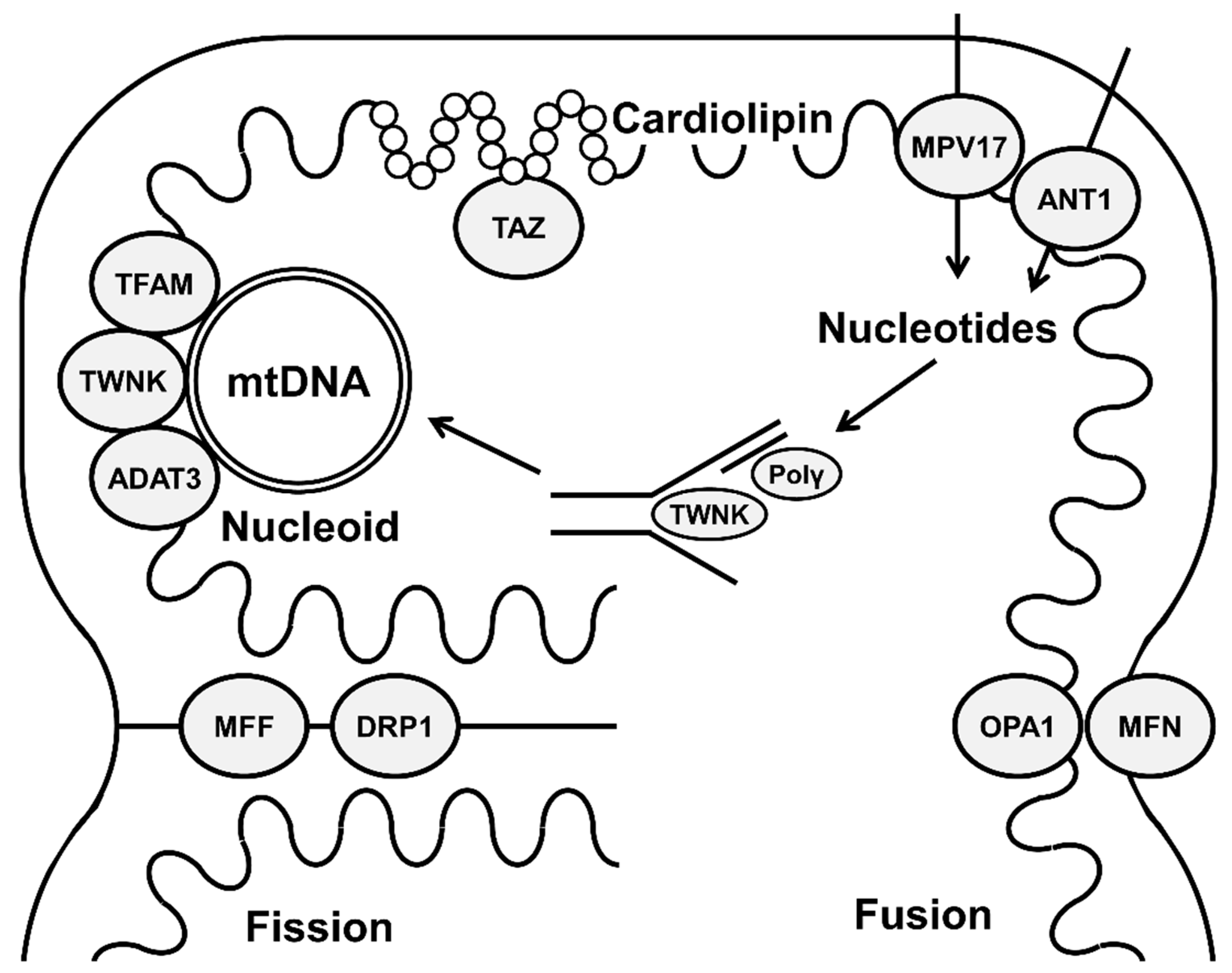

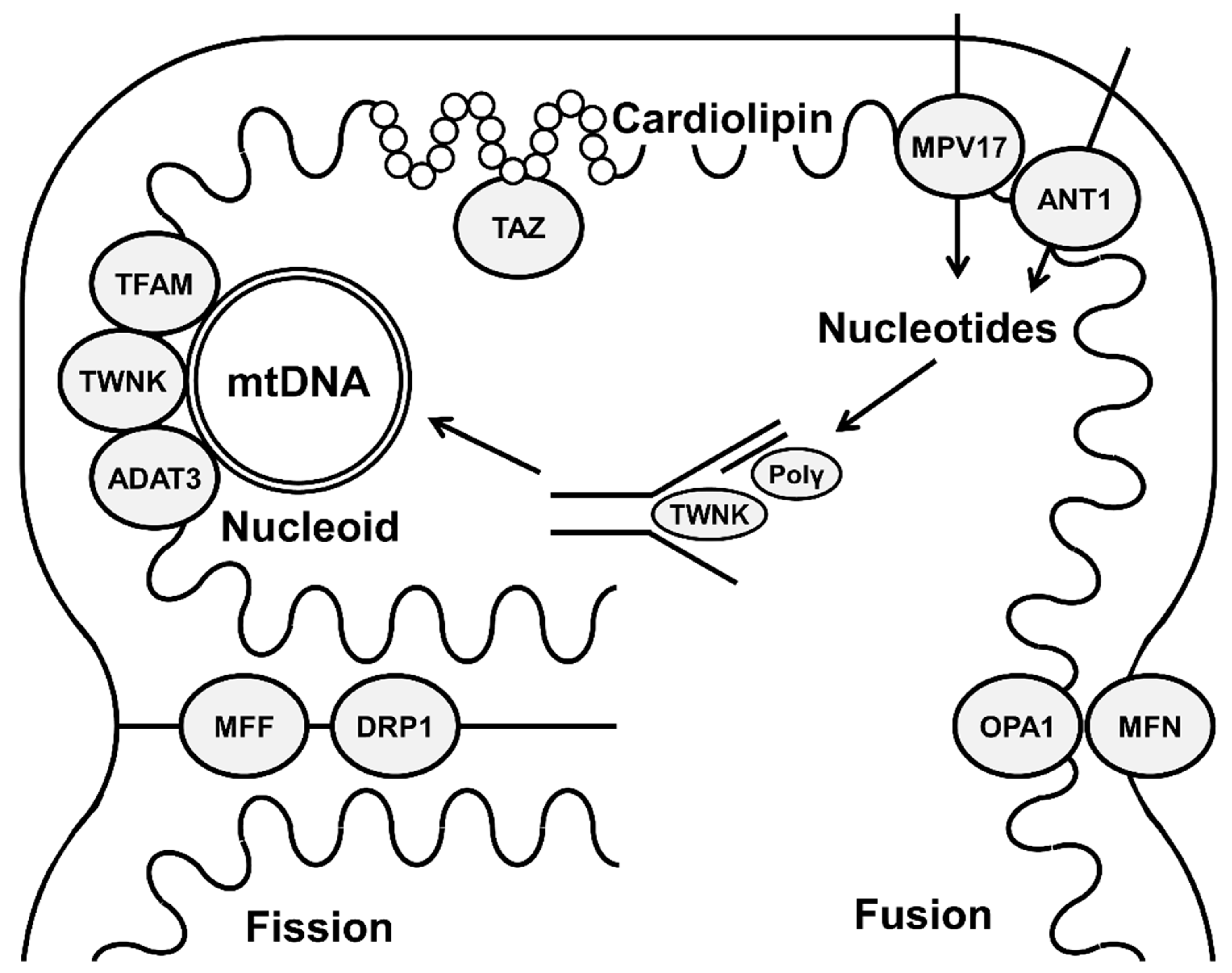{kind=link}
| Mechanism | Gene | Disease | Inheritance |
|---|---|---|---|
| Nucleoid Organization | TFAM | Mitochondrial DNA depletion syndrome 15 (hepatocerebral type) (MIM#617156) | AR |
| TWNK | autosomal recessive infantile-onset spinocerebellar ataxia (IOSCA) (MIM# 271245) | AR | |
| Perrault syndrome (MIM#:616138) | AR | ||
| ATAD3A | Harel–Yoon syndrome (MIM#617183) | AR/AD | |
| Cerebellar hypoplasia, hypotonia, and respiratory insufficiency syndrome, neonatal lethal (MIM#618810) | AR | ||
| Mitochondrial fission and fusion | DNM1L | Encephalopathy, lethal, due to defective mitochondrial peroxisomal fission 1 (MIM#614388) | AR/AD |
| Optic atrophy 5 (MIM#610708) | AD | ||
| MFF | Encephalopathy due to defective mitochondrial and peroxisomal fission 2 (MIM#617068) | AR | |
| OPA1 | Mitochondrial DNA depletion syndrome 14 (encephalocardiomyopathic type) (MIM#616896) | AR | |
| Behr syndrome (MIM#210000) | AR | ||
| Optic atrophy 1 (MIM#165500) | AD | ||
| Optic atrophy plus syndrome (MIM#125250) | AD | ||
| MFN2 | Charcot–Marie–Tooth disease, axonal, type 2A2 (MIM# 609260/617087) | AD/AR | |
| Optic atrophy plus syndrome | AD | ||
| Cardiolipin Remodeling | TAFFAZIN | Barth syndrome (MIM#302060) | XLR |
| Mitochondrial membrane transporters | MPV17 | Mitochondrial DNA depletion syndrome 6 (hepatocerebral type) (MIM#256810) | AR |
| Charcot–Marie–Tooth disease, axonal, type 2EE (MIM#618400) | AR | ||
| SLC25A4 | Progressive external ophthalmoplegia with mitochondrial DNA deletions, autosomal dominant 2 (MIM#609382) | AD | |
| Mitochondrial DNA depletion syndrome 12A (cardiomyopathic type) (MIM#617184/615418) | AR/AD |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almannai, M.; Salah, A.; El-Hattab, A.W. Mitochondrial Membranes and Mitochondrial Genome: Interactions and Clinical Syndromes. Membranes 2022, 12, 625. https://doi.org/10.3390/membranes12060625
Almannai M, Salah A, El-Hattab AW. Mitochondrial Membranes and Mitochondrial Genome: Interactions and Clinical Syndromes. Membranes. 2022; 12(6):625. https://doi.org/10.3390/membranes12060625
Chicago/Turabian StyleAlmannai, Mohammed, Azza Salah, and Ayman W. El-Hattab. 2022. "Mitochondrial Membranes and Mitochondrial Genome: Interactions and Clinical Syndromes" Membranes 12, no. 6: 625. https://doi.org/10.3390/membranes12060625
APA StyleAlmannai, M., Salah, A., & El-Hattab, A. W. (2022). Mitochondrial Membranes and Mitochondrial Genome: Interactions and Clinical Syndromes. Membranes, 12(6), 625. https://doi.org/10.3390/membranes12060625