
Influence of Degassing Treatment on the Ink Properties and Performance of Proton Exchange Membrane Fuel Cells


Abstract
:1. Introduction
2. Experimental Procedures
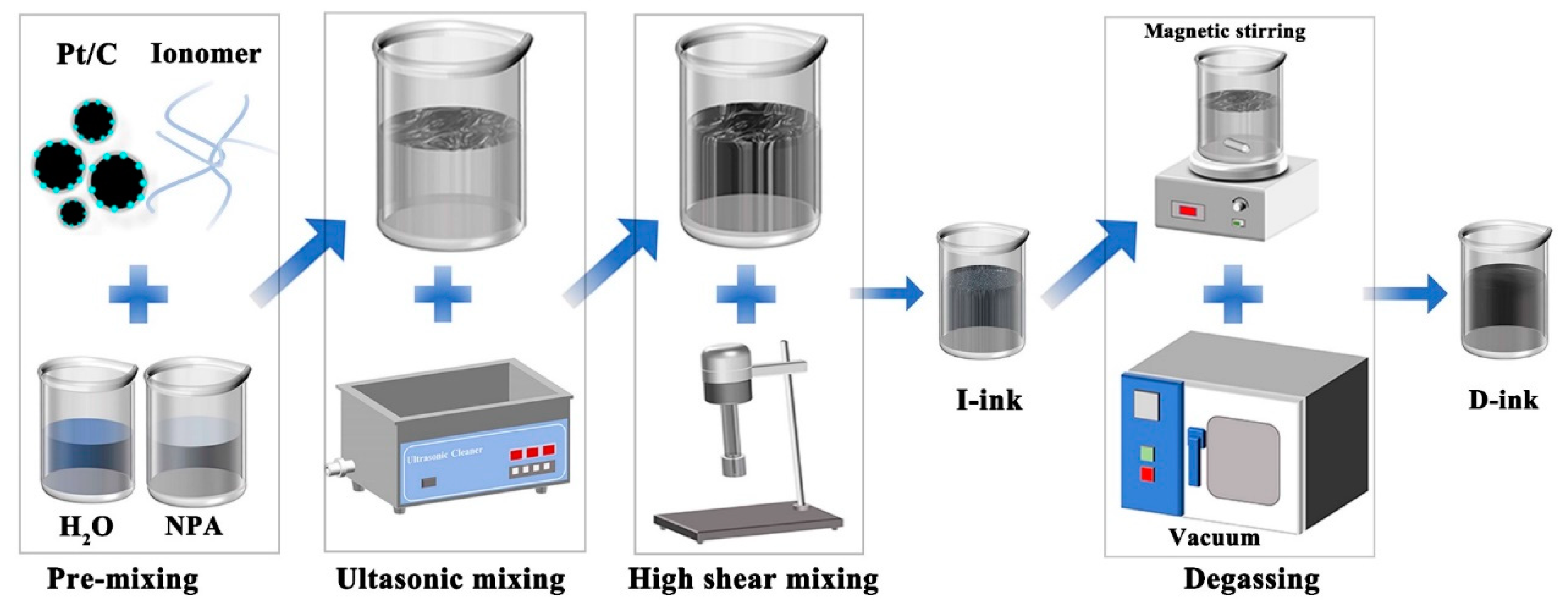
2.1. Preparation of Catalyst Ink
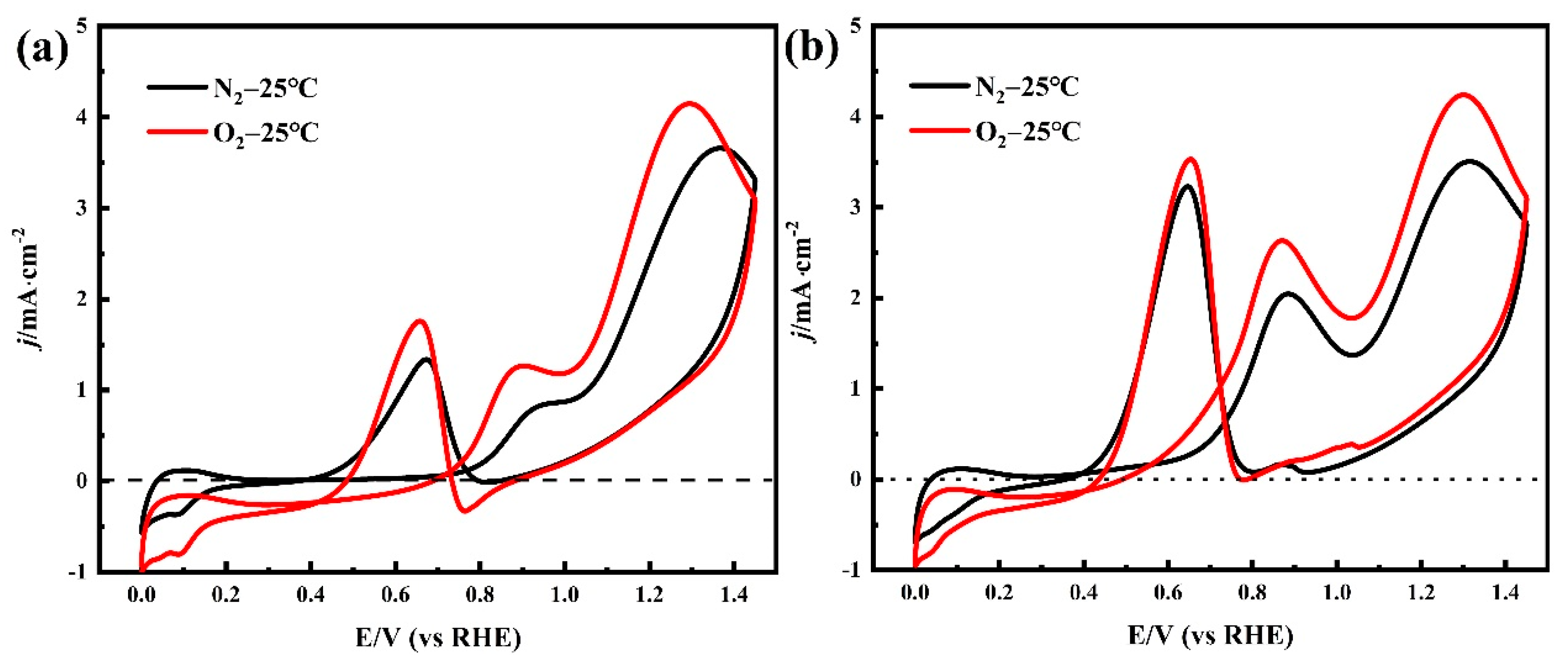
2.2. Electrochemical Evaluation
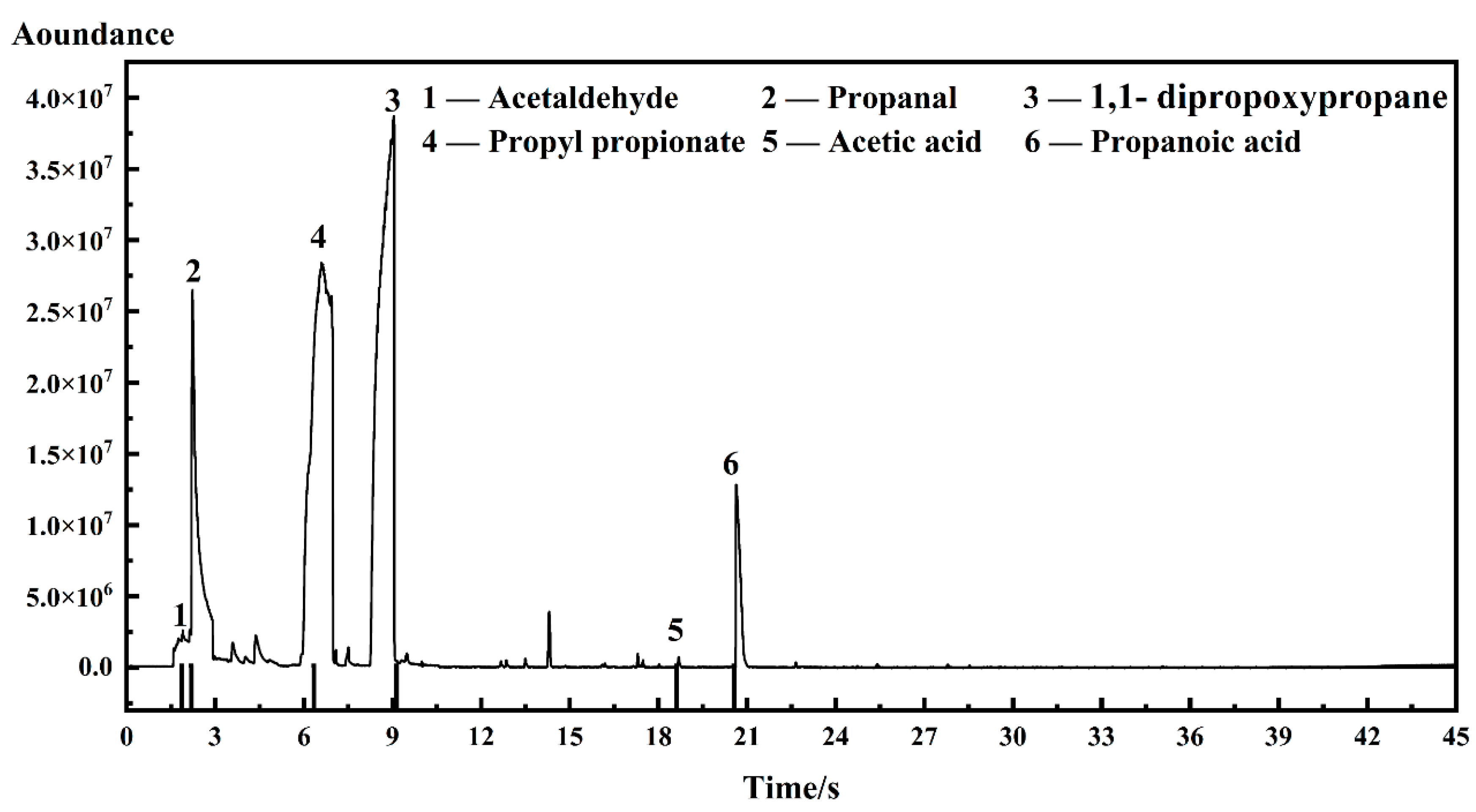
2.3. GC-MS Instrumentation
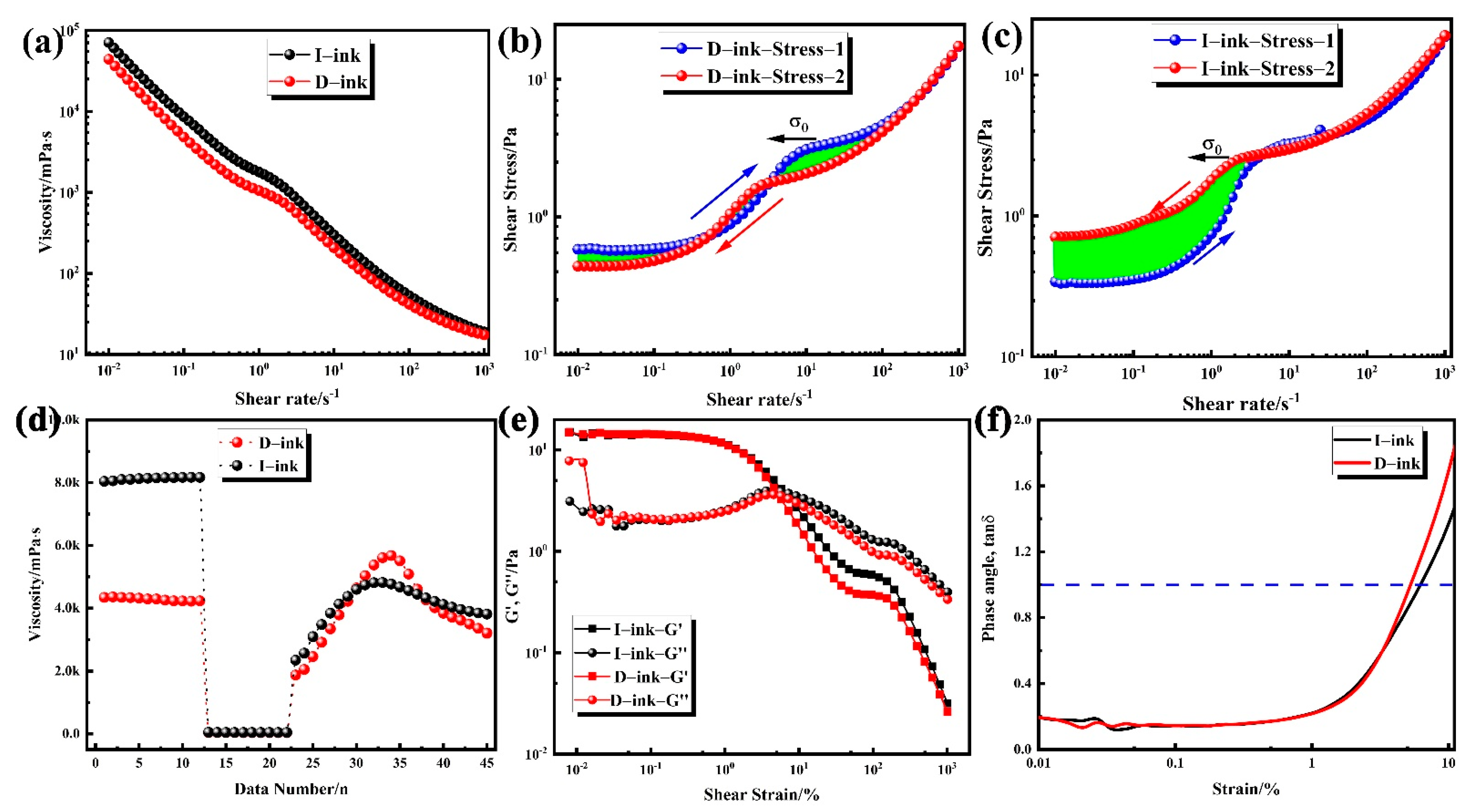
2.4. Rheological Measurements
2.5. Measurement of Ink Cluster Size and Zeta Potential
2.6. Determination of Contact Angle and Deposition of LSC Ink
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ioroi, T.; Siroma, Z.; Yamazaki, S.I.; Yasuda, K. Electrocatalysts for pem fuel cells. Adv. Energy Mater. 2018, 9, 1801284. [Google Scholar] [CrossRef]
- Wang, Y.; Ruiz Diaz, D.F.; Chen, K.S.; Wang, Z.; Adroher, X.C. Materials, technological status, and fundamentals of pem fuel cells—A review. Mater. Today 2020, 32, 178–203. [Google Scholar] [CrossRef]
- Son, T.Y.; Kim, T.H.; Nam, S.Y. Crosslinked pore-filling anion exchange membrane using the cylindrical centrifugal force for anion exchange membrane fuel cell system. Polymers 2020, 12, 2758. [Google Scholar] [CrossRef] [PubMed]
- Qaisrani, N.A.; Ma, Y.; Ma, L.; Liu, J.; Gao, L.; Li, L.; Gong, S.; Yan, X.; Zhang, F.; He, G. Facile and green fabrication of polybenzoxazine-based composite anion-exchange membranes with a self-cross-linked structure. Ionics 2018, 24, 3053–3063. [Google Scholar] [CrossRef]
- Guo, Y.; Yang, D.; Li, B.; Yang, D.; Ming, P.; Zhang, C. Effect of dispersion solvents and ionomers on the rheology of catalyst inks and catalyst layer structure for proton exchange membrane fuel cells. ACS Appl. Mater. Interfaces 2021, 13, 27119–27128. [Google Scholar] [CrossRef]
- Fouzaï, I.; Gentil, S.; Bassetto, V.C.; Silva, W.O.; Maher, R.; Girault, H.H. Catalytic layer-membrane electrode assembly methods for optimum triple phase boundaries and fuel cell performances. J. Mater. Chem. A 2021, 9, 11096–11123. [Google Scholar] [CrossRef]
- Chu, T.; Zhang, R.; Wang, Y.; Ou, M.; Xie, M.; Shao, H.; Yang, D.; Li, B.; Ming, P.; Zhang, C. Performance degradation and process engineering of the 10 kw proton exchange membrane fuel cell stack. Energy 2021, 219, 119623. [Google Scholar] [CrossRef]
- Yoshino, S.; Shinohara, A.; Kodama, K.; Morimoto, Y. Fabrication of catalyst layer with ionomer nanofiber scaffolding for polymer electrolyte fuel cells. J. Power Sources 2020, 476, 228584. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, R.; Kang, Q.; Tao, W.-Q. Pore-scale study of pore-ionomer interfacial reactive transport processes in proton exchange membrane fuel cell catalyst layer. Chem. Eng. J. 2020, 391, 123590. [Google Scholar] [CrossRef]
- Stähler, M.; Stähler, A.; Scheepers, F.; Carmo, M.; Stolten, D. A completely slot die coated membrane electrode assembly. Int. J. Hydrogen Energy 2019, 44, 7053–7058. [Google Scholar] [CrossRef]
- Li, B.; Liu, Y.; Guo, Y.; Yang, D.; Yang, D.; Ming, P.; Zhang, C. Controlling the microscopic morphology and permeability of catalyst layers in proton exchange membrane fuel cells by adjusting catalyst ink agglomerates. Int. J. Hydrogen Energy 2021, 46, 32215–32225. [Google Scholar] [CrossRef]
- Gong, Q.; Li, C.; Liu, Y.; Ilavsky, J.; Guo, F.; Cheng, X.; Xie, J. Effects of ink formulation on construction of catalyst layers for high-performance polymer electrolyte membrane fuel cells. ACS Appl. Mater. Interfaces 2021, 13, 37004–37013. [Google Scholar] [CrossRef] [PubMed]
- Kumano, N.; Kudo, K.; Akimoto, Y.; Ishii, M.; Nakamura, H. Influence of ionomer adsorption on agglomerate structures in high-solid catalyst inks. Carbon 2020, 169, 429–439. [Google Scholar] [CrossRef]
- Uemura, S.; Yoshida, T.; Koga, M.; Matsumoto, H.; Yang, X.; Shinohara, K.; Sasabe, T.; Hirai, S. Ink degradation and its effects on the crack formation of fuel cell catalyst layers. J. Electrochem. Soc. 2019, 166, F89–F92. [Google Scholar] [CrossRef]
- Balu, R.; Choudhury, N.R.; Mata, J.P.; de Campo, L.; Rehm, C.; Hill, A.J.; Dutta, N.K. Evolution of the interfacial structure of a catalyst ink with the quality of the dispersing solvent: A contrast variation small-angle and ultrasmall-angle neutron scattering investigation. ACS Appl. Mater. Interfaces 2019, 11, 9934–9946. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Chen, Z.; Yao, D.; Chen, X.; Wang, F.; Dai, G. Microstructure and macroscopic rheology of microporous layer nanoinks for pem fuel cells. Chem. Eng. Sci. 2021, 246, 117001. [Google Scholar] [CrossRef]
- Uemura, S.; Yoshida, T.; Koga, M.; Matsumoto, H.; Shinohara, K.; Hirai, S. Ink degradation phenomena and its impact on crack formation of fuel cell catalyst. ECS Trans. 2018, 86, 151–156. [Google Scholar] [CrossRef]
- Uemura, S.; Sakai, K.; Sasabe, T.; Matsumoto, H.; Sugimori, H.; Shinohara, K.; Hirai, S. Effect of reaction products on the pefc catalyst ink property and catalyst layer quality. ECS Trans. 2020, 98, 61–65. [Google Scholar] [CrossRef]
- Uemura, S.; Kameya, Y.; Iriguchi, N.; Yoshida, T.; Shinohara, K.; Hirai, S. Communication—investigation of catalyst ink degradation by x-ray ct. J. Electrochem. Soc. 2018, 165, F142–F144. [Google Scholar] [CrossRef]
- Uemura, S.; Sasabe, T.; Sakai, K.; Shinohara, K.; Hirai, S. Relation between degradation reaction and mixing at fuel cell catalyst ink fabrication process. ECS Trans. 2019, 92, 183–187. [Google Scholar] [CrossRef]
- Kameya, Y.; Iriguchi, N.; Ohki, M.; Yokoyama, K.; Sugawara, S.; Sugimori, H.; Uemura, S.; Sasabe, T.; Yoshida, T.; Hirai1, T. Mri and 1h/19f nmr investigation of dispersion state of pefc catalyst ink. ECS Trans. 2017, 80, 819. [Google Scholar] [CrossRef]
- Pastor, E.; Wasmus, S.; Iwasita, T.; Arévalo, M.C.; González, S.; Arvia, A.J. Spectroscopic investigations of c3 primary alcohols on platinum electrodes in acid solutions. J. Electroanal. Chem. 1993, 350, 97–116. [Google Scholar] [CrossRef] [Green Version]
- Pastor, E.; González, S.; Arvia, A.J. Electroreactivity of isopropanol on platinum in acids studied by dems and ftirs. J. Electroanal. Chem. 1995, 395, 233–242. [Google Scholar] [CrossRef] [Green Version]
- Garsany, Y.; Baturina, O.A.; Swider-Lyons, K.E.; Kocha, S.S. Experimental methods for quantifying the activity of platinum electrocatalysts for the oxygen reduction reaction. Anal. Chem. 2010, 82, 6321–6328. [Google Scholar] [CrossRef]
- Tang, X.; Zeng, Y.; Cao, L.; Yang, L.; Wang, Z.; Fang, D.; Gao, Y.; Shao, Z.; Yi, B. Anchoring ultrafine pt nanoparticles on the 3d hierarchical self-assembly of graphene/functionalized carbon black as a highly efficient oxygen reduction catalyst for pemfcs. J. Mater. Chem. A 2018, 6, 15074–15082. [Google Scholar] [CrossRef]
- Luo, L.; Zhu, F.; Tian, R.; Li, L.; Shen, S.; Yan, X.; Zhang, J. Composition-graded pdxni1–x nanospheres with pt monolayer shells as high-performance electrocatalysts for oxygen reduction reaction. ACS Catal. 2017, 7, 5420–5430. [Google Scholar] [CrossRef]
- Mayrhofer, K.J.J.; Strmcnik, D.; Blizanac, B.B.; Stamenkovic, V.; Arenz, M.; Markovic, N.M. Measurement of oxygen reduction activities via the rotating disc electrode method: From pt model surfaces to carbon-supported high surface area catalysts. Electrochim. Acta 2008, 53, 3181–3188. [Google Scholar] [CrossRef]
- Umeda, M.; Sugii, H.; Uchida, I. Alcohol electrooxidation at pt and pt–ru sputtered electrodes under elevated temperature and pressurized conditions. J. Power Sources 2008, 179, 489–496. [Google Scholar] [CrossRef]
- Tu, K.; Li, G.; Jiang, Y. Effect of temperature on the electrocatalytic oxidation of ethanol. Acta Phys.-Chim. Sin 2020, 36, 1906026. [Google Scholar] [CrossRef]
- Kusoglu, A.; Weber, A.Z. New insights into perfluorinated sulfonic-acid ionomers. Chem. Rev. 2017, 117, 987–1104. [Google Scholar] [CrossRef]
- Woo, S.; Lee, S.; Taning, A.Z.; Yang, T.-H.; Park, S.-H.; Yim, S.-D. Current understanding of catalyst/ionomer interfacial structure and phenomena affecting the oxygen reduction reaction in cathode catalyst layers of proton exchange membrane fuel cells. Curr. Opin. Electrochem. 2020, 21, 289–296. [Google Scholar] [CrossRef]
- Khandavalli, S.; Iyer, R.; Park, J.H.; Myers, D.J.; Neyerlin, K.C.; Ulsh, M.; Mauger, S.A. Effect of dispersion medium composition and ionomer concentration on the microstructure and rheology of fe-n-c platinum group metal-free catalyst inks for polymer electrolyte membrane fuel cells. Langmuir 2020, 36, 12247–12260. [Google Scholar] [CrossRef] [PubMed]
- Berlinger, S.A.; Garg, S.; Weber, A.Z. Multicomponent, multiphase interactions in fuel-cell inks. Curr. Opin. Electrochem. 2021, 29, 100744. [Google Scholar] [CrossRef]
- Hatzell, K.B.; Dixit, M.B.; Berlinger, S.A.; Weber, A.Z. Understanding inks for porous-electrode formation. J. Mater. Chem. A 2017, 5, 20527–20533. [Google Scholar] [CrossRef] [Green Version]
- Dixit, M.B.; Harkey, B.A.; Shen, F.; Hatzell, K.B. Catalyst layer ink interactions that affect coatability. J. Electrochem. Soc. 2018, 165, F264–F271. [Google Scholar] [CrossRef]
- Lee, G.-W.; Ryu, J.H.; Han, W.; Ahn, K.H.; Oh, S.M. Effect of slurry preparation process on electrochemical performances of licoo2 composite electrode. J. Power Sources 2010, 195, 6049–6054. [Google Scholar] [CrossRef]
- Mizukawa, H.; Kawaguchi, M. Effects of perfluorosulfonic acid adsorption on the stability of carbon black suspensions. Langmuir 2009, 25, 11984–11987. [Google Scholar] [CrossRef]
- Lee, J.-H.; Paik, U.; Choi, J.-Y.; Kim, K.K.; Yoon, S.-M.; Lee, J.; Kim, B.-K.; Kim, J.M.; Park, M.H.; Yang, C.W.; et al. Dispersion stability of single-walled carbon nanotubes using nafion in bisolvent. J. Phys. Chem. C 2007, 111, 2477–2483. [Google Scholar] [CrossRef]
- Cho, K.Y.; Kwon, Y.I.; Youn, J.R.; Song, Y.S. Evaluation of slurry characteristics for rechargeable lithium-ion batteries. Mater. Res. Bull. 2013, 48, 2922–2926. [Google Scholar] [CrossRef]
- Kim, T.H.; Jang, L.W.; Lee, D.C.; Choi, H.J.; Jhon, M.S. Synthesis and rheology of intercalated polystyrene/na+-montmorillonite nanocomposites. Macromol. Rapid Commun. 2002, 23, 191–195. [Google Scholar] [CrossRef]
- Du, S.; Li, W.; Wu, H.; Abel Chuang, P.-Y.; Pan, M.; Sui, P.-C. Effects of ionomer and dispersion methods on rheological behavior of proton exchange membrane fuel cell catalyst layer ink. Int. J. Hydrogen Energy 2020, 45, 29430–29441. [Google Scholar] [CrossRef]
- Hawley, W.B.; Li, J. Beneficial rheological properties of lithium-ion battery cathode slurries from elevated mixing and coating temperatures. J. Energy Storage 2019, 26, 100994. [Google Scholar] [CrossRef]
- Zhao, J.; He, N. A mini-review of embedded 3d printing: Supporting media and strategies. J. Mater. Chem. B 2020, 8, 10474–10486. [Google Scholar] [CrossRef] [PubMed]
- Björn, A.; de La Monja, P.S.; Karlsson, A.; Ejlertsson, J.; Svensson, B.H. Rheological Characterization; InTech: London, UK, 2012. [Google Scholar]
- Bitsch, B.; Dittmann, J.; Schmitt, M.; Scharfer, P.; Schabel, W.; Willenbacher, N. A novel slurry concept for the fabrication of lithium-ion battery electrodes with beneficial properties. J. Power Sources 2014, 265, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Kumano, N.; Kudo, K.; Suda, A.; Akimoto, Y.; Ishii, M.; Nakamura, H. Controlling cracking formation in fuel cell catalyst layers. J. Power Sources 2019, 419, 219–228. [Google Scholar] [CrossRef]
- Su, F.-Y.; Dai, L.-Q.; Guo, X.-Q.; Xie, L.-J.; Sun, G.-H.; Chen, C.-M. Micro-structure evolution and control of lithium-ion battery electrode laminate. J. Energy Storage 2017, 14, 82–93. [Google Scholar] [CrossRef]
- Bauer, W.; Nötzel, D. Rheological properties and stability of nmp based cathode slurries for lithium ion batteries. Ceram. Int. 2014, 40, 4591–4598. [Google Scholar] [CrossRef]
- Lanceros-Méndez, S.; Costa, C.M. Printed Batteries: Materials, Technologies and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2018. [Google Scholar]
- Kiennemann, J.; Chartier, T.; Pagnoux, C.; Baumard, J.F.; Huger, M.; Lamérant, J.M. Drying mechanisms and stress development in aqueous alumina tape casting. J. Eur. Ceram. Soc. 2005, 25, 1551–1564. [Google Scholar] [CrossRef]
- Koos, E. Capillary suspensions: Particle networks formed through the capillary force. Curr. Opin. Colloid Interface Sci. 2014, 19, 575–584. [Google Scholar] [CrossRef] [Green Version]
- Velankar, S.S. A non-equilibrium state diagram for liquid/fluid/particle mixtures. Soft Matter 2015, 11, 8393–8403. [Google Scholar] [CrossRef]
- Kralchevsky, P.A.; Denkov, N.D. Capillary forces and structuring in layers of colloid particles. Curr. Opin. Colloid 2001, 6, 383–401. [Google Scholar] [CrossRef]
- Garsany, Y.; Atkinson, R.W.; Sassin, M.B.; Hjelm, R.M.E.; Gould, B.D.; Swider-Lyons, K.E. Improving pemfc performance using short-side-chain low-equivalent-weight pfsa ionomer in the cathode catalyst layer. J. Electrochem. Soc. 2018, 165, F381–F391. [Google Scholar] [CrossRef]
- Shukla, S.; Bhattacharjee, S.; Weber, A.Z.; Secanell, M. Experimental and theoretical analysis of ink dispersion stability for polymer electrolyte fuel cell applications. J. Electrochem. Soc. 2017, 164, F600–F609. [Google Scholar] [CrossRef]
- Kocha, S.S.; Shinozaki, K.; Zack, J.W.; Myers, D.J.; Kariuki, N.N.; Nowicki, T.; Stamenkovic, V.; Kang, Y.; Li, D.; Papageorgopoulos, D. Best practices and testing protocols for benchmarking orr activities of fuel cell electrocatalysts using rotating disk electrode. Electrocatalysis 2017, 8, 366–374. [Google Scholar] [CrossRef]
- Garsany, Y.; Ge, J.J.; St-Pierre, J.; Rocheleau, R.; Swider-Lyons, K.E. Standardizing thin-film rotating disk electrode measurements of the oxygen reduction activity of pt/c. ECS Trans. 2013, 58, 3–14. [Google Scholar] [CrossRef]
- Garsany, Y.; Singer, I.L.; Swider-Lyons, K.E. Impact of film drying procedures on rde characterization of pt/vc electrocatalysts. J. Electroanal. Chem. 2011, 662, 396–406. [Google Scholar] [CrossRef]
- Deegan, R.D.; Bakajin, O.; Dupont, T.F.; Huber, G.; Nagel, S.R.; Witten, T.A. Capillary flow as the cause of ring stains from dried liquid drops. Nature 1997, 389, 827–829. [Google Scholar] [CrossRef]
- Zhao, J.; Li, X.; Liu, Z.S. The effect of ink dilution and evaporation on the microstructures of catalyst layers in polymer electrolyte membrane fuel cells. Int. J. Energy Res. 2019, 43, 6799–6811. [Google Scholar] [CrossRef]
- Deegan, R.D. Pattern formation in drying drops. Phys. Rev. E 2000, 61, 475–485. [Google Scholar] [CrossRef] [Green Version]
- Deegan, R.D.; Bakajin, O.; Dupont, T.F.; Huber, G.; Nagel, S.R.; Witten, T.A. Contact line deposits in an evaporating drop. Phys. Rev. E 2000, 62, 756–765. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Larson, R.G. Marangoni effect reverses coffee-ring depositions. J. Phys. Chem. B 2006, 110, 7090–7094. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Lane, A.M. Resolving the hupd and hopd by dems to determine the ecsa of pt electrodes in pem fuel cells. Electrochem. Commun. 2011, 13, 913–916. [Google Scholar] [CrossRef]
- Garsany, Y.; Ge, J.; St-Pierre, J.; Rocheleau, R.; Swider-Lyons, K.E. Analytical procedure for accurate comparison of rotating disk electrode results for the oxygen reduction activity of pt/c. J. Electrochem. Soc. 2014, 161, F628–F640. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ink Type | Pt/C (g) | Deionized Water (g) | 1-Propanol (g) | Nafion® Solution (g) | Solid Content (%) |
|---|---|---|---|---|---|
| HSC ink | 5.7 | 14.082 | 13.705 | 20.517 | 12.45 |
| LSC ink | 0.002 | 0.000 | 0.967 | 0.0323 | 0.20 |
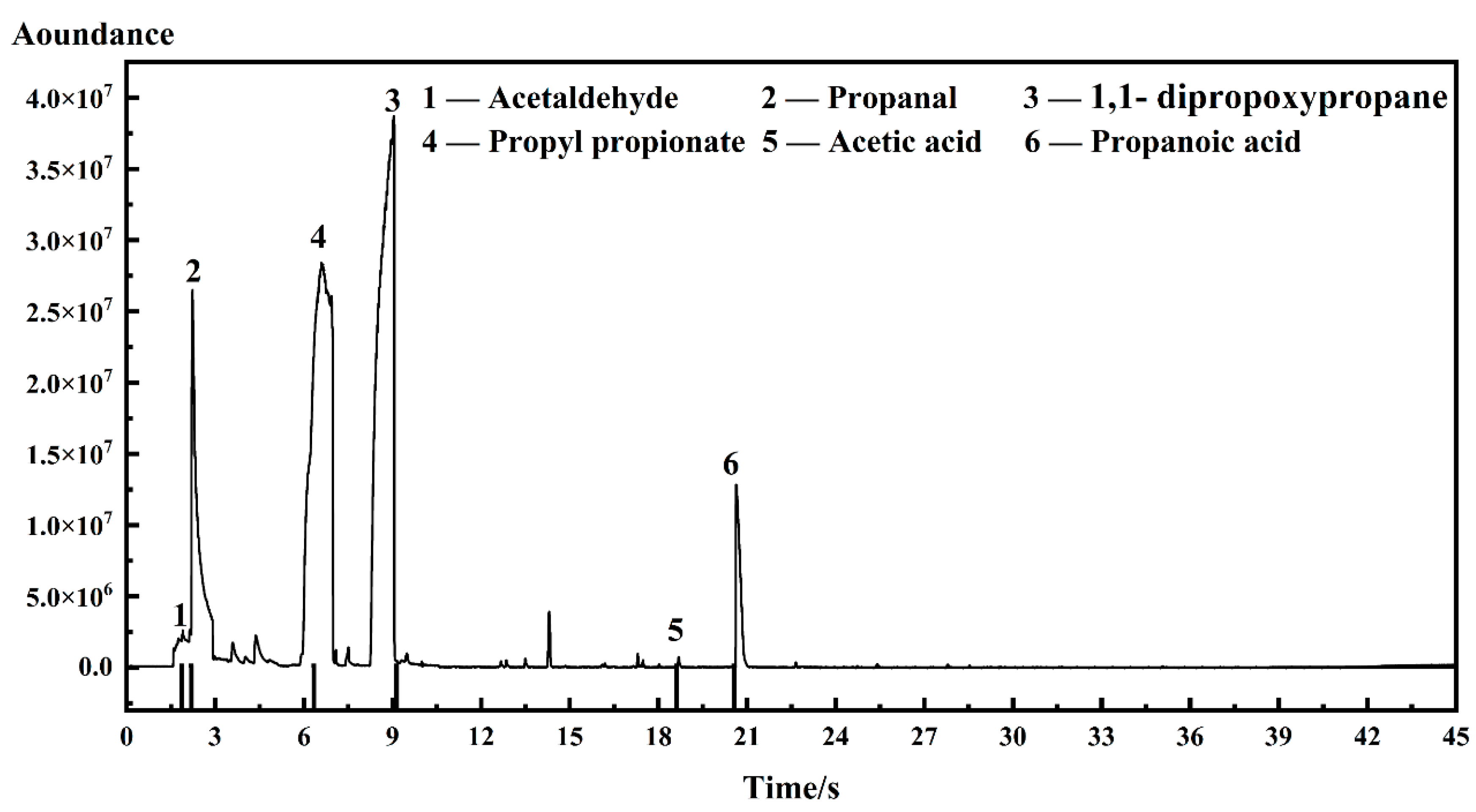
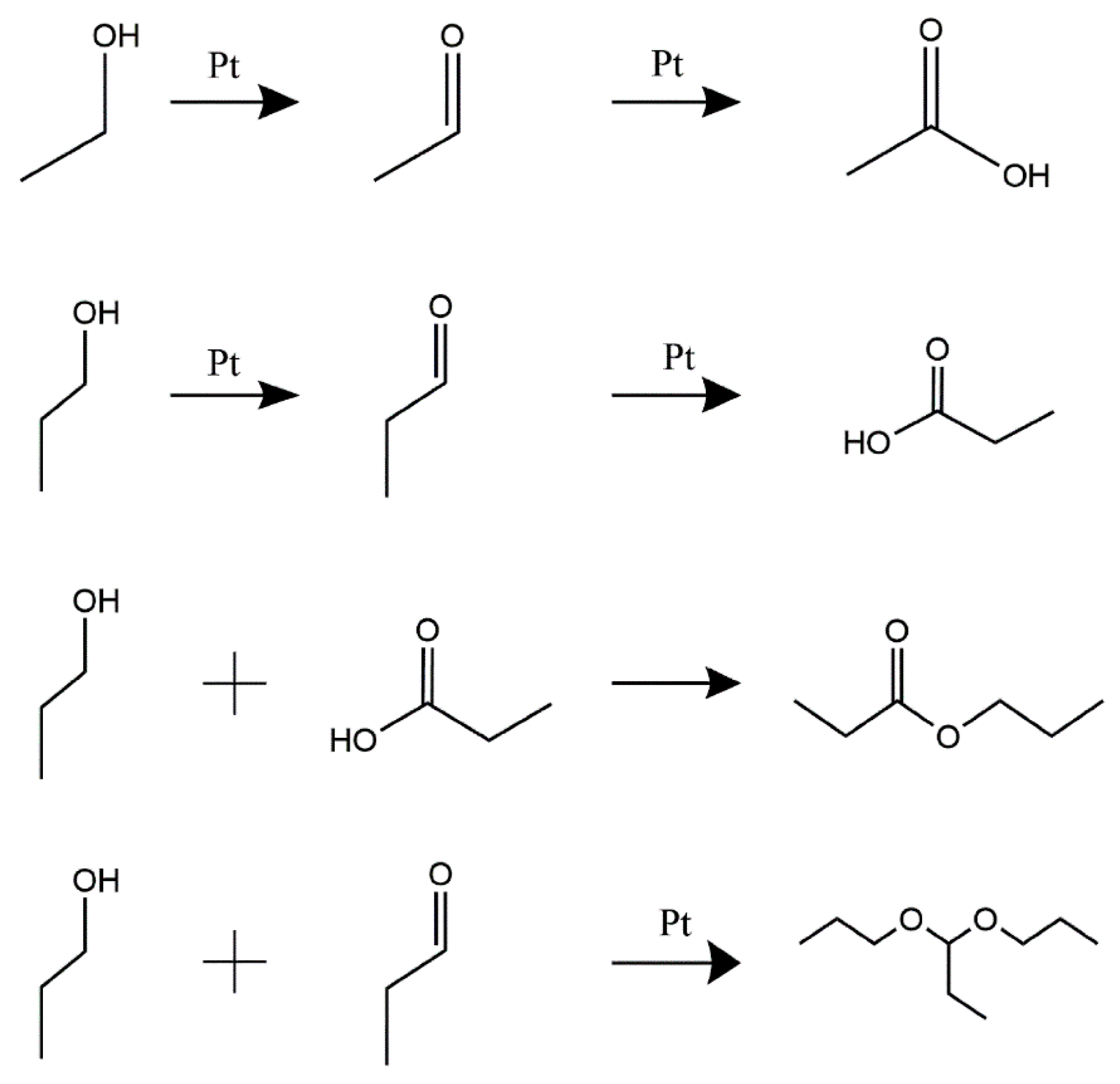
| Sample | Acetic Acid | Propionic Acid | Propyl Propionate |
|---|---|---|---|
| I-ink | 0.0898 wt.% | 0.0224 wt.% | 0.00046 wt.% |
| D-ink | 0.0025 wt.% | 0.0126 wt.% | 0.0003 wt.% |
| Condition | NPA’s Oxidation Current Density/mA cm−2 | EA’s Oxidation Current Density/mA cm−2 | ||||
|---|---|---|---|---|---|---|
| Peak I | Peak II | Peak III | Peak I | Peak II | Peak III | |
| N2-25 °C | 0.855 | 3.66 | 1.338 | 2.046 | 3.506 | 3.231 |
| O2-25 °C | 1.266 | 4.148 | 1.758 | 2.634 | 4.241 | 3.532 |
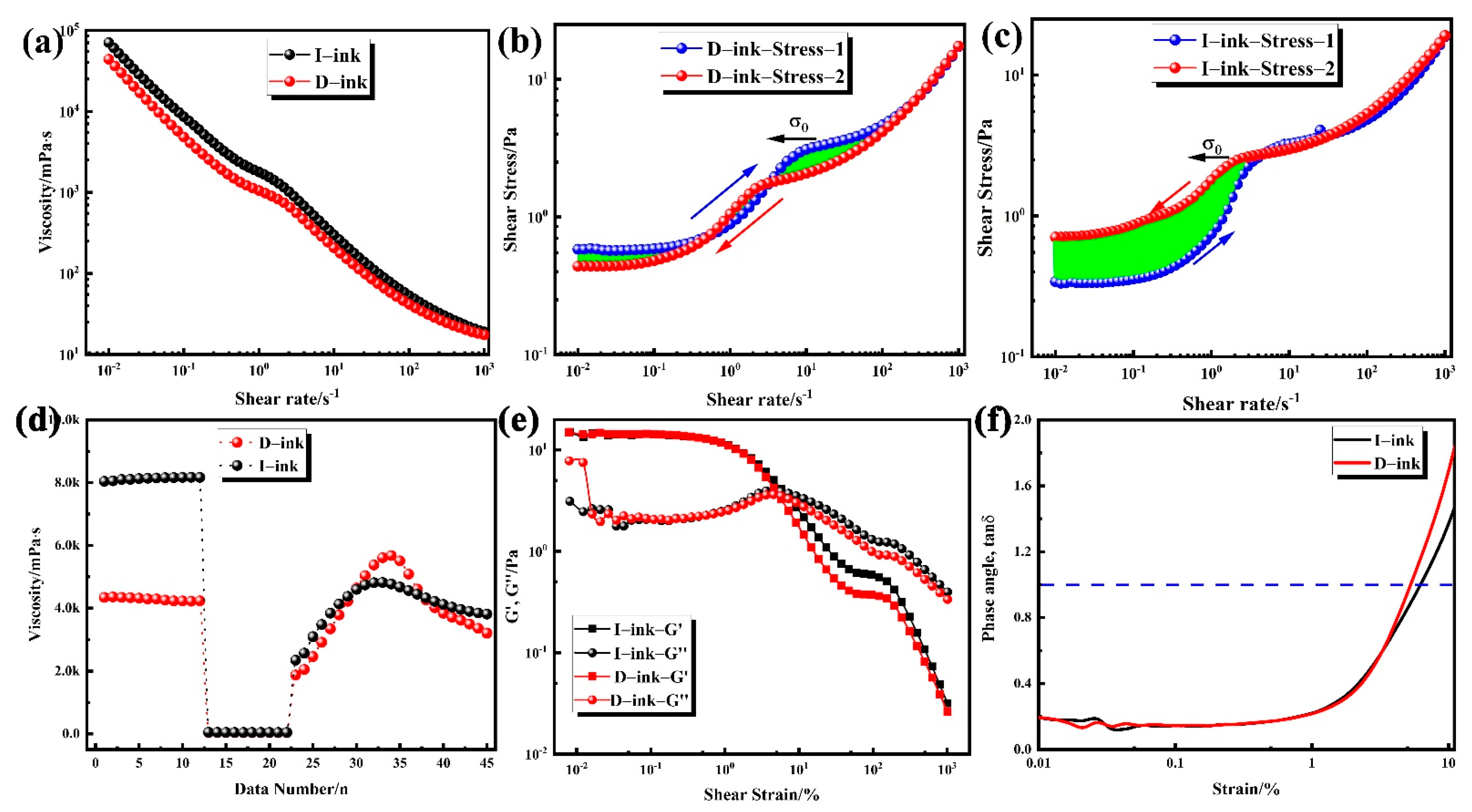
| Ink | R2 | |||
|---|---|---|---|---|
| I-ink | 2.49505 | 0.08131 | 0.76772 | 0.99982 |
| D-ink | 1.6832 | 0.05865 | 0.80801 | 0.99971 |
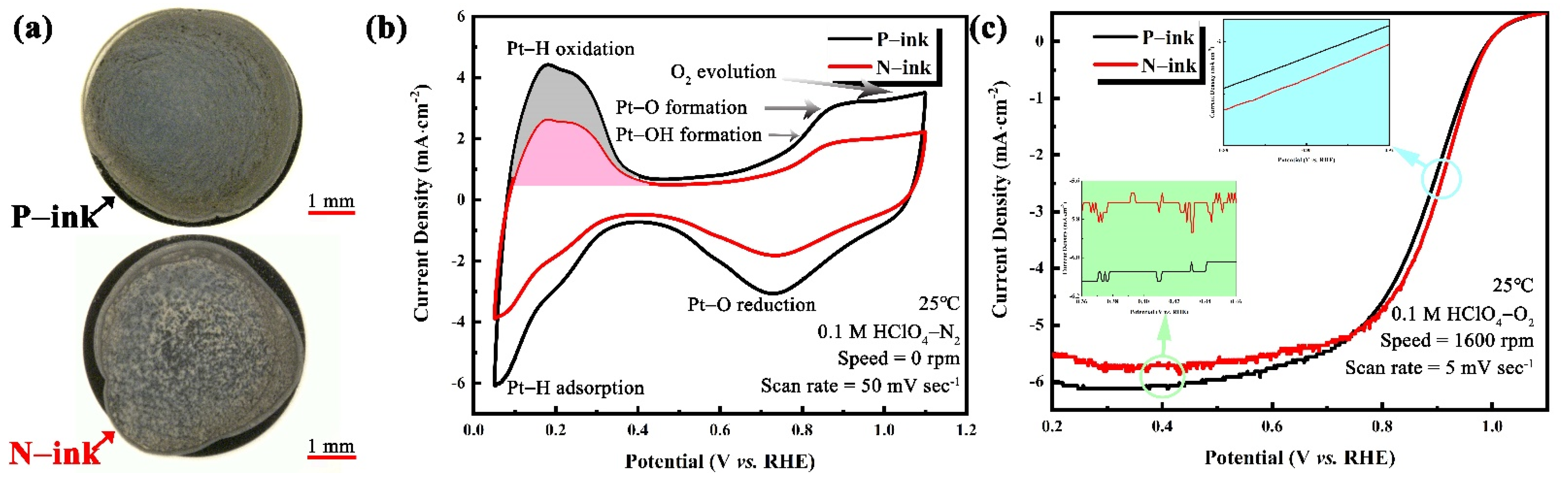
| Sample | ECSA (m2∙gPt−1) | MA (mA∙mgPt−1) |
|---|---|---|
| P-ink | 112.2 | 84.4 |
| N-ink | 62.7 | 60.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, P.; Yang, D.; Li, B.; Zhang, C.; Ming, P. Influence of Degassing Treatment on the Ink Properties and Performance of Proton Exchange Membrane Fuel Cells. Membranes 2022, 12, 541. https://doi.org/10.3390/membranes12050541
Liu P, Yang D, Li B, Zhang C, Ming P. Influence of Degassing Treatment on the Ink Properties and Performance of Proton Exchange Membrane Fuel Cells. Membranes. 2022; 12(5):541. https://doi.org/10.3390/membranes12050541
Chicago/Turabian StyleLiu, Pengcheng, Daijun Yang, Bing Li, Cunman Zhang, and Pingwen Ming. 2022. "Influence of Degassing Treatment on the Ink Properties and Performance of Proton Exchange Membrane Fuel Cells" Membranes 12, no. 5: 541. https://doi.org/10.3390/membranes12050541
APA StyleLiu, P., Yang, D., Li, B., Zhang, C., & Ming, P. (2022). Influence of Degassing Treatment on the Ink Properties and Performance of Proton Exchange Membrane Fuel Cells. Membranes, 12(5), 541. https://doi.org/10.3390/membranes12050541