
Hybrid Composite Membrane of Phosphorylated Chitosan/Poly (Vinyl Alcohol)/Silica as a Proton Exchange Membrane

 ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of N-Methylene Phosphonic Chitosan (NMPC)
2.3. Preparation of the N-Methylene Phosphonic Chitosan/Poly (Vinyl Alcohol) Composite Membrane
2.4. Preparation of the N-Methylene Phosphonic Chitosan/Poly (Vinyl Alcohol) Composite Membrane Modified with Silicon Dioxide Filler (NMPC/PVA-SiO2)
2.5. Characterization
2.5.1. Fourier Transform Infrared (FTIR) Spectroscopy
2.5.2. X-ray Diffraction (XRD)
2.5.3. Field-Emission Scanning Electron Microscopy (FESEM)
2.5.4. Thermogravimetric Analysis (TGA)
2.5.5. Dynamic Mechanical Analysis (DMA)
2.5.6. Water Uptake and Swelling Ratio
2.5.7. Ion-Exchange Capacity (IEC)
2.5.8. Proton Conductivity
3. Results and Discussion
3.1. Chitosan and N-Methylene Phosphonic Chitosan (NMPC)
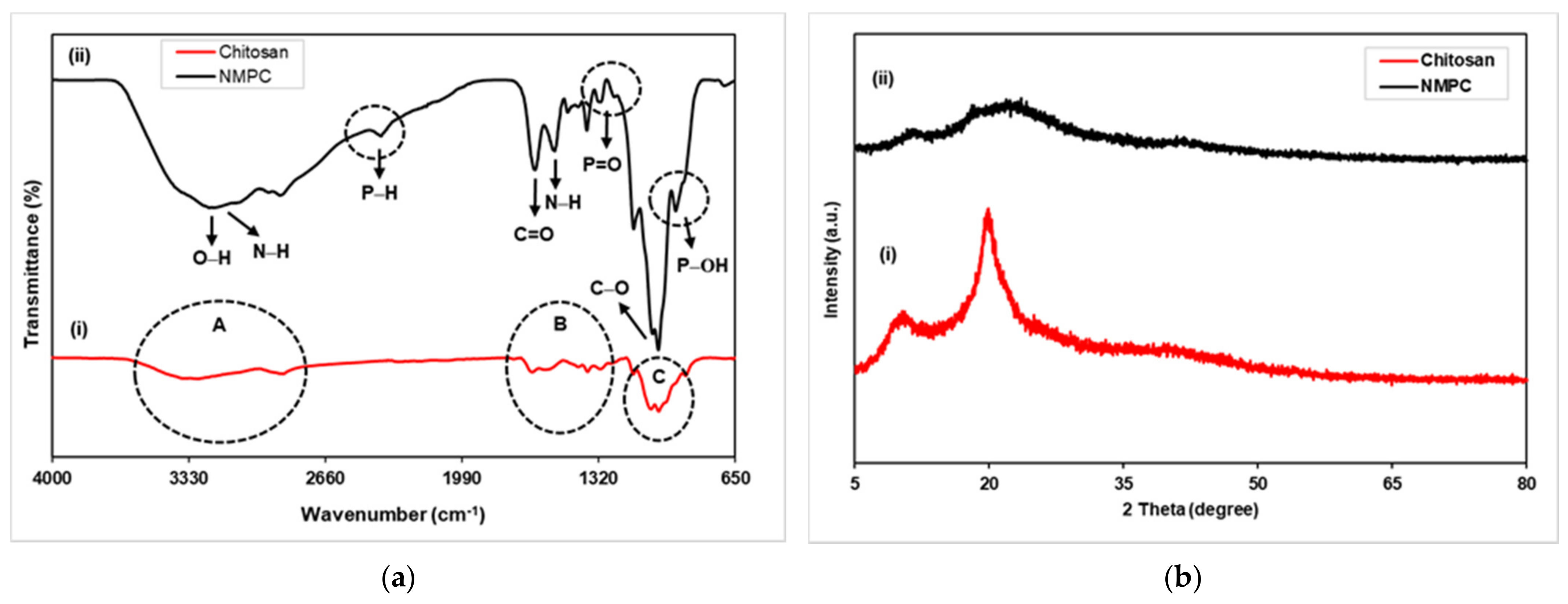
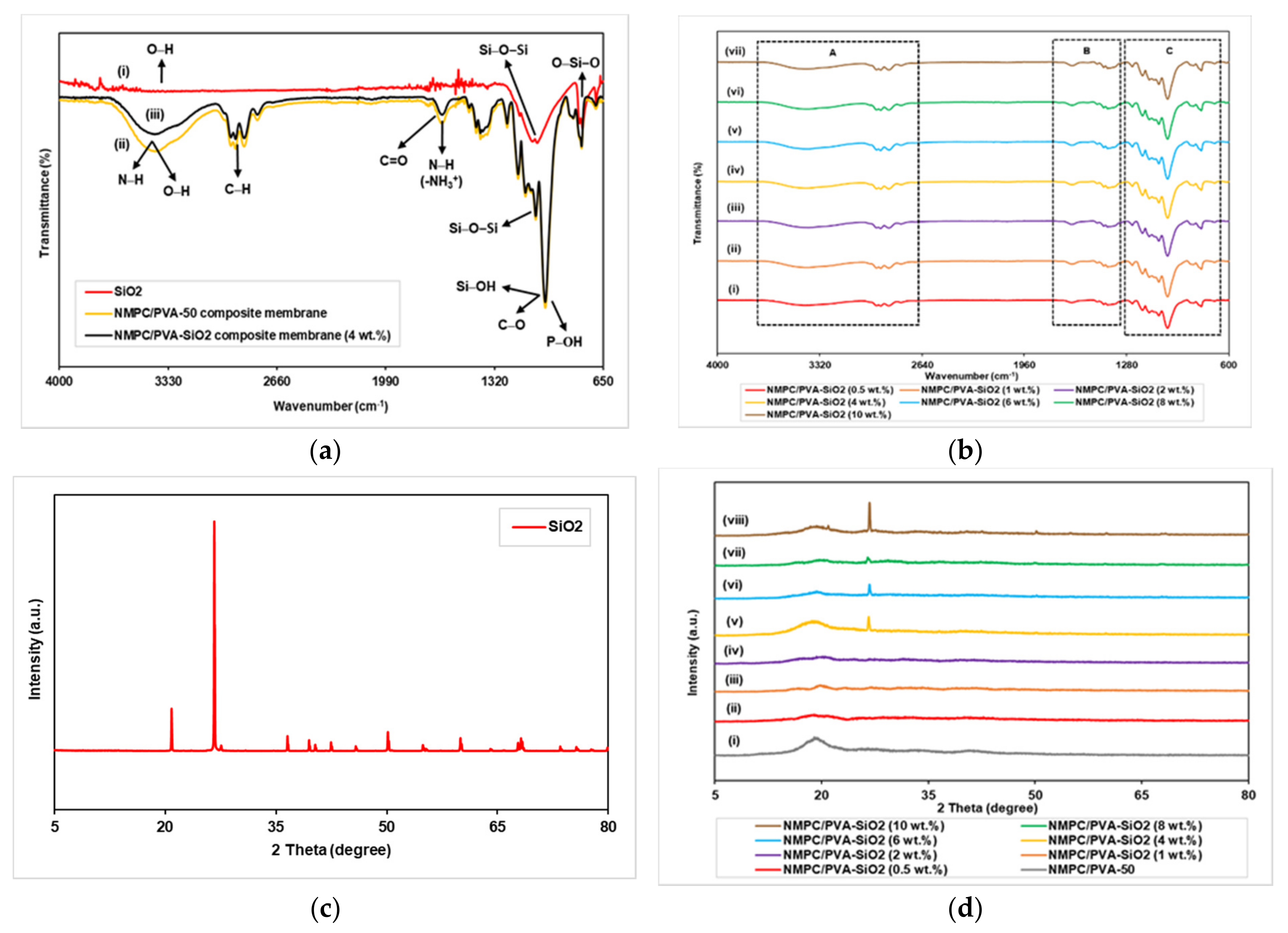
3.1.1. FTIR and XRD Analysis
3.2. N-Methylene Phosphonic Chitosan/Poly (Vinyl Alcohol) (NMPC/PVA) Composite Membranes
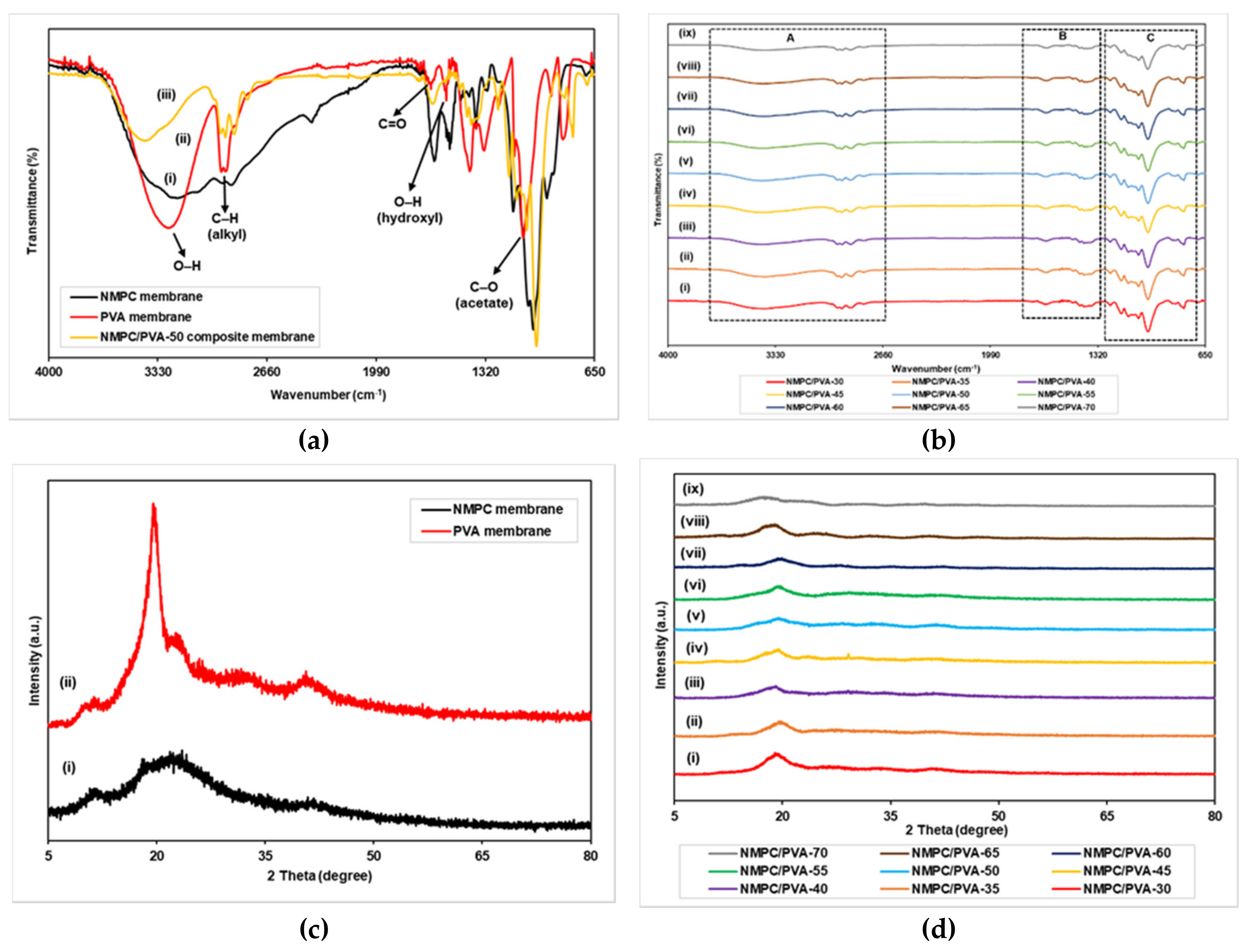
3.2.1. FTIR and XRD Analysis
3.2.2. Morphological Studies
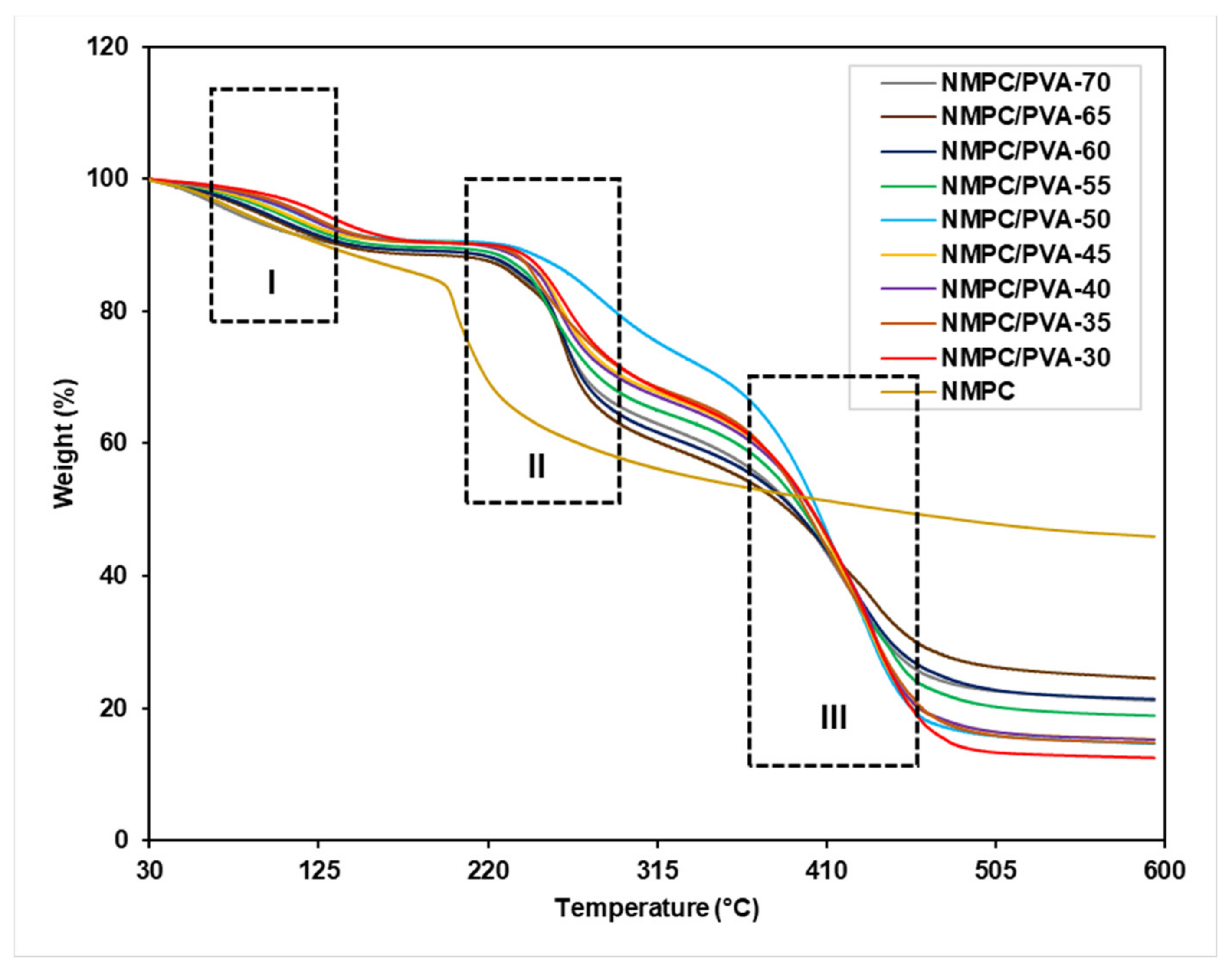
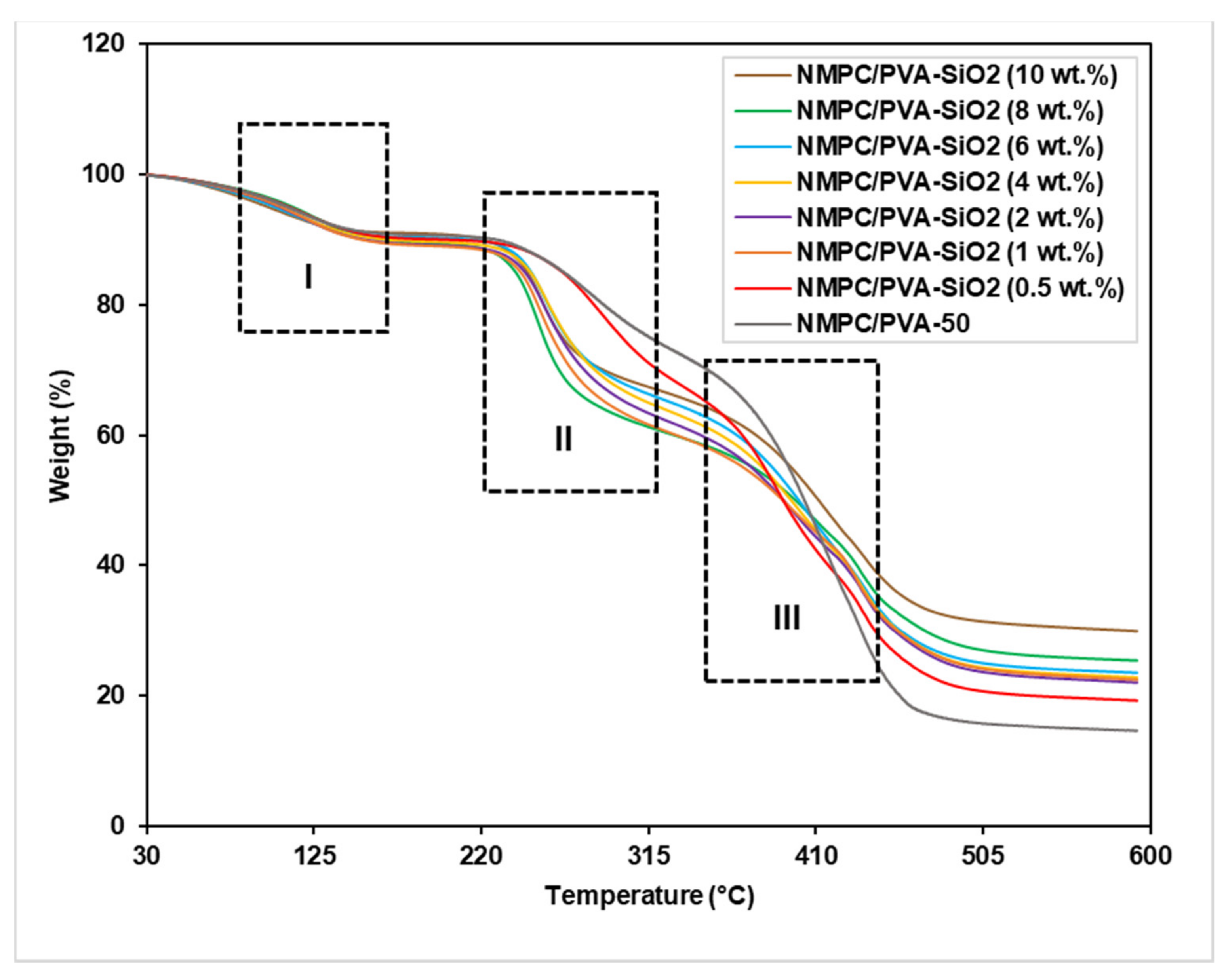
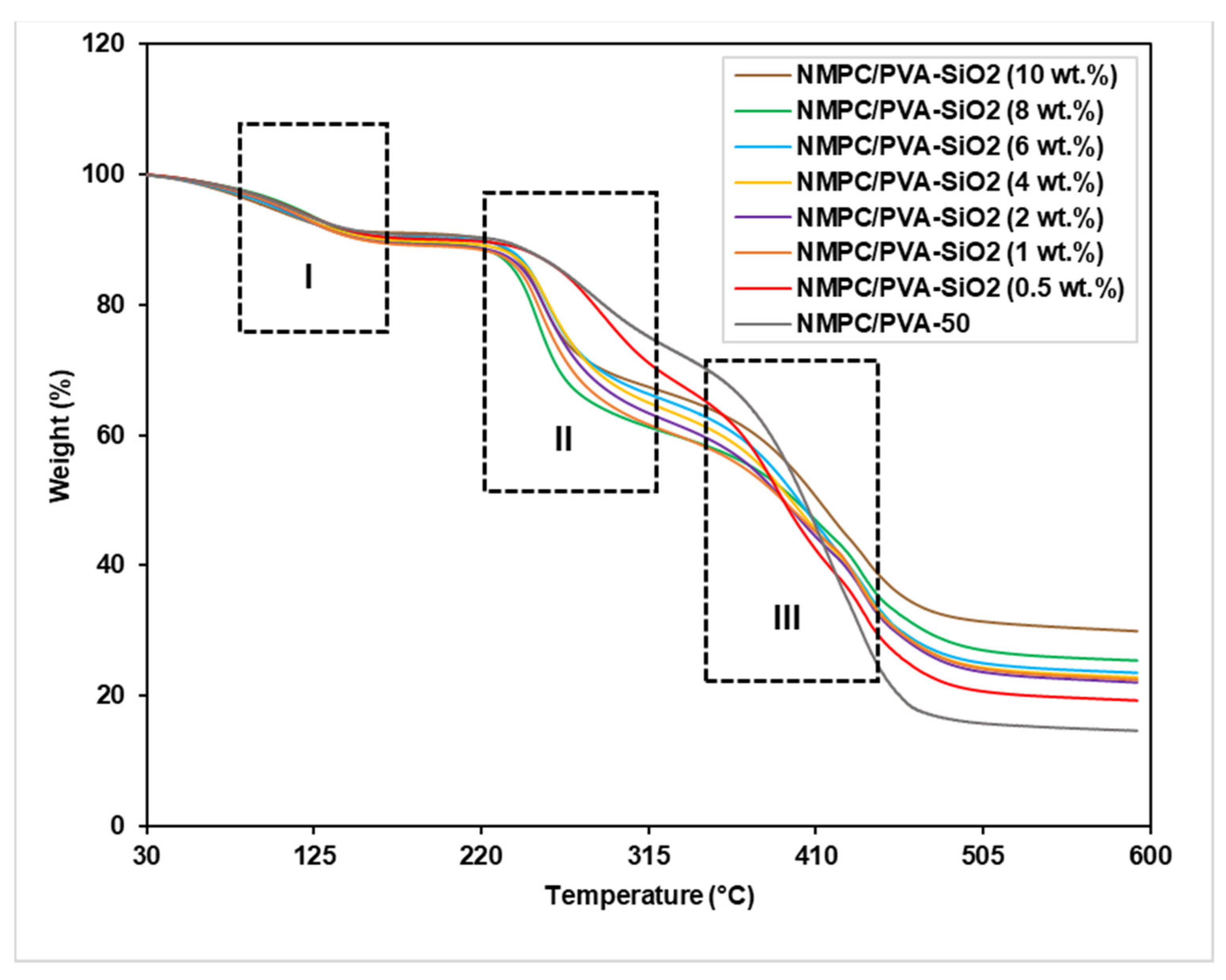
3.2.3. Thermal Stability
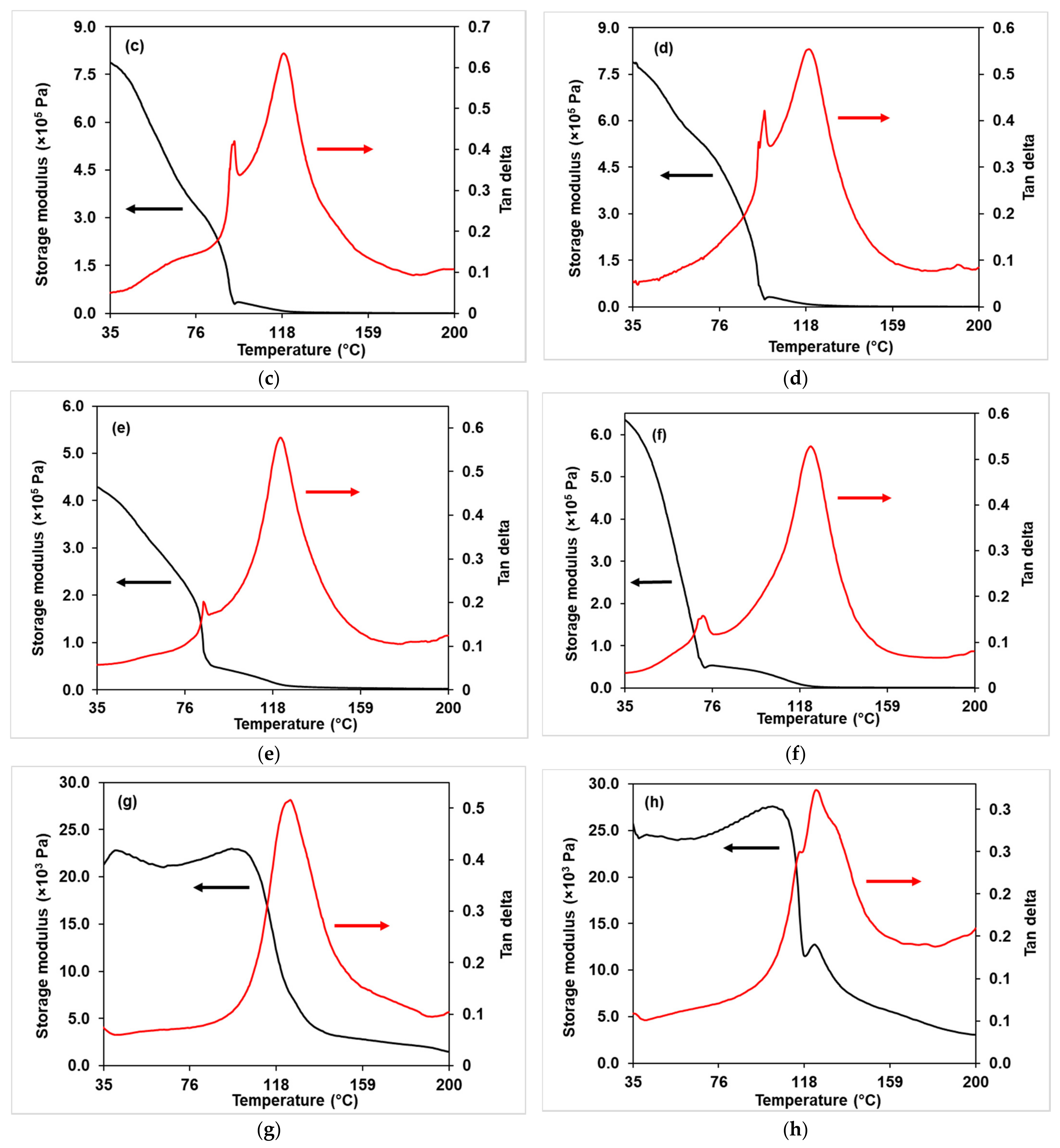
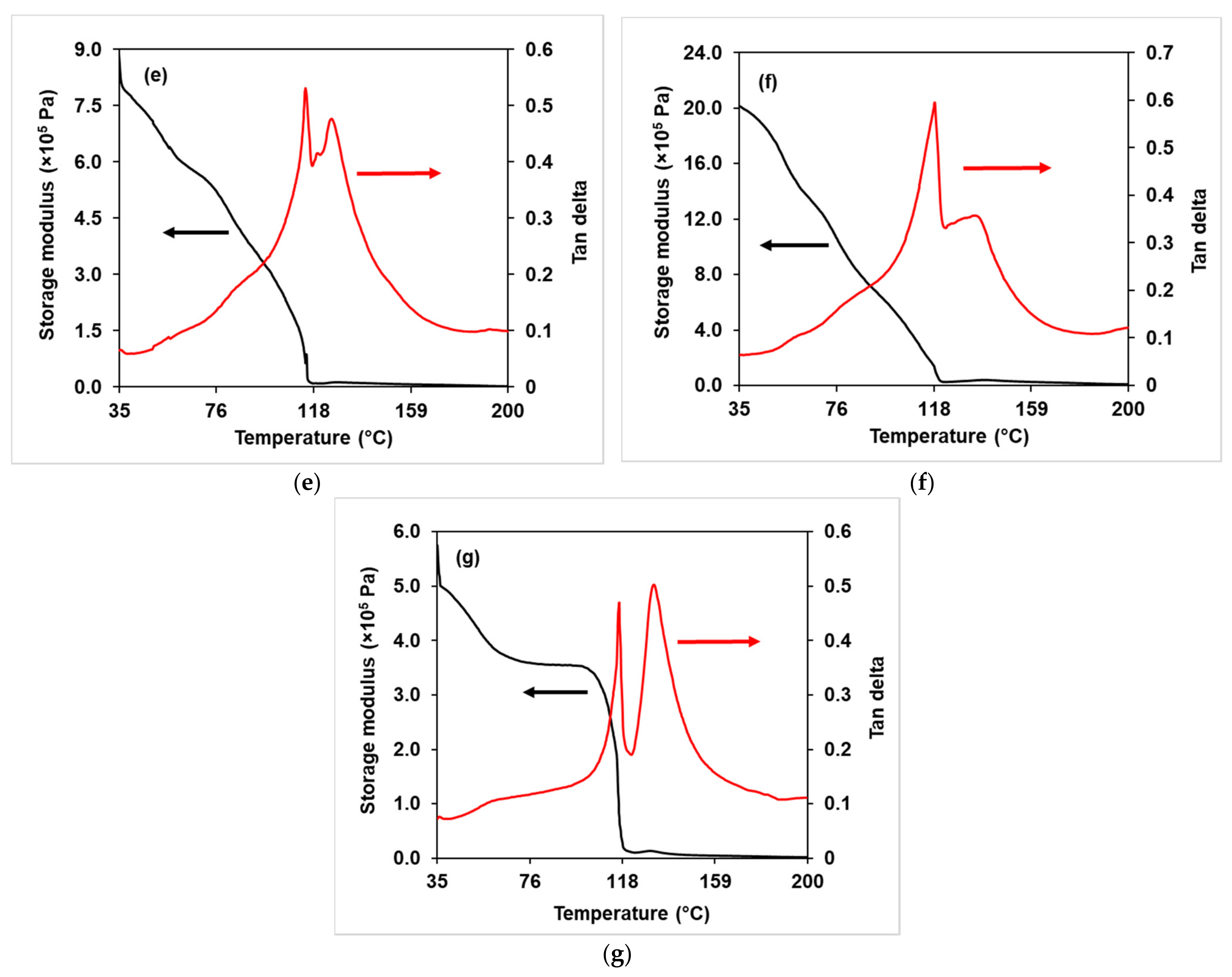
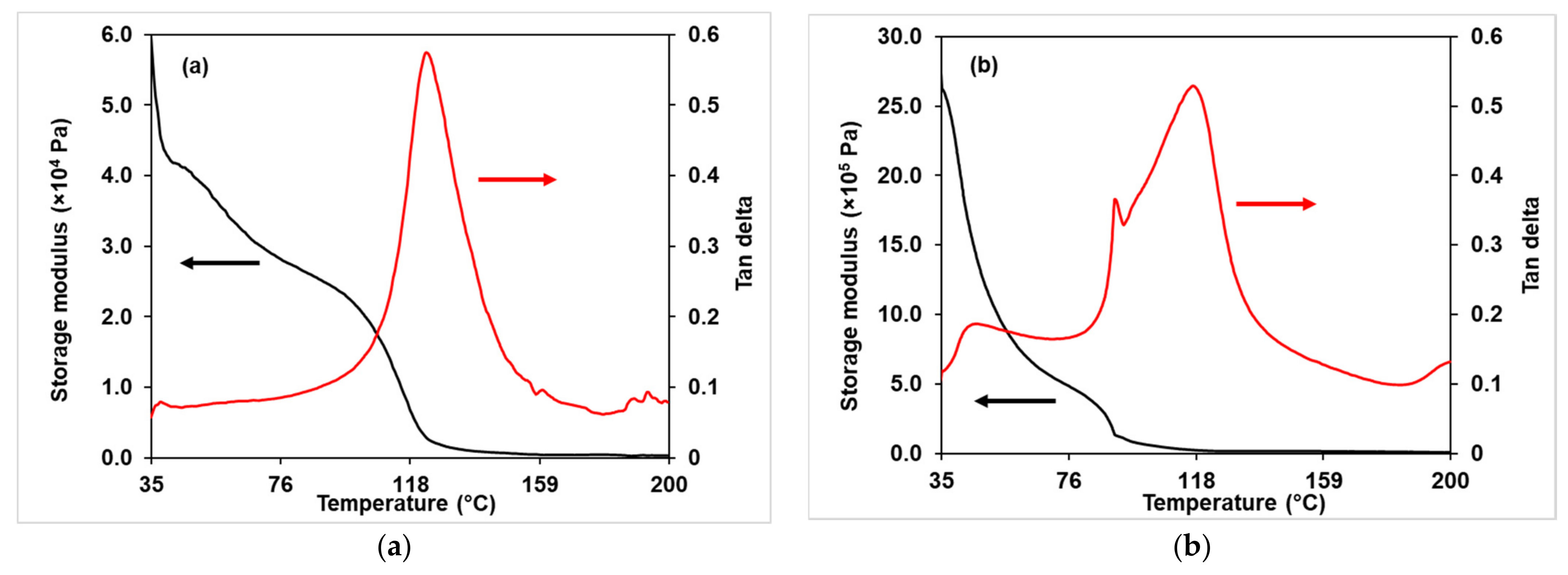
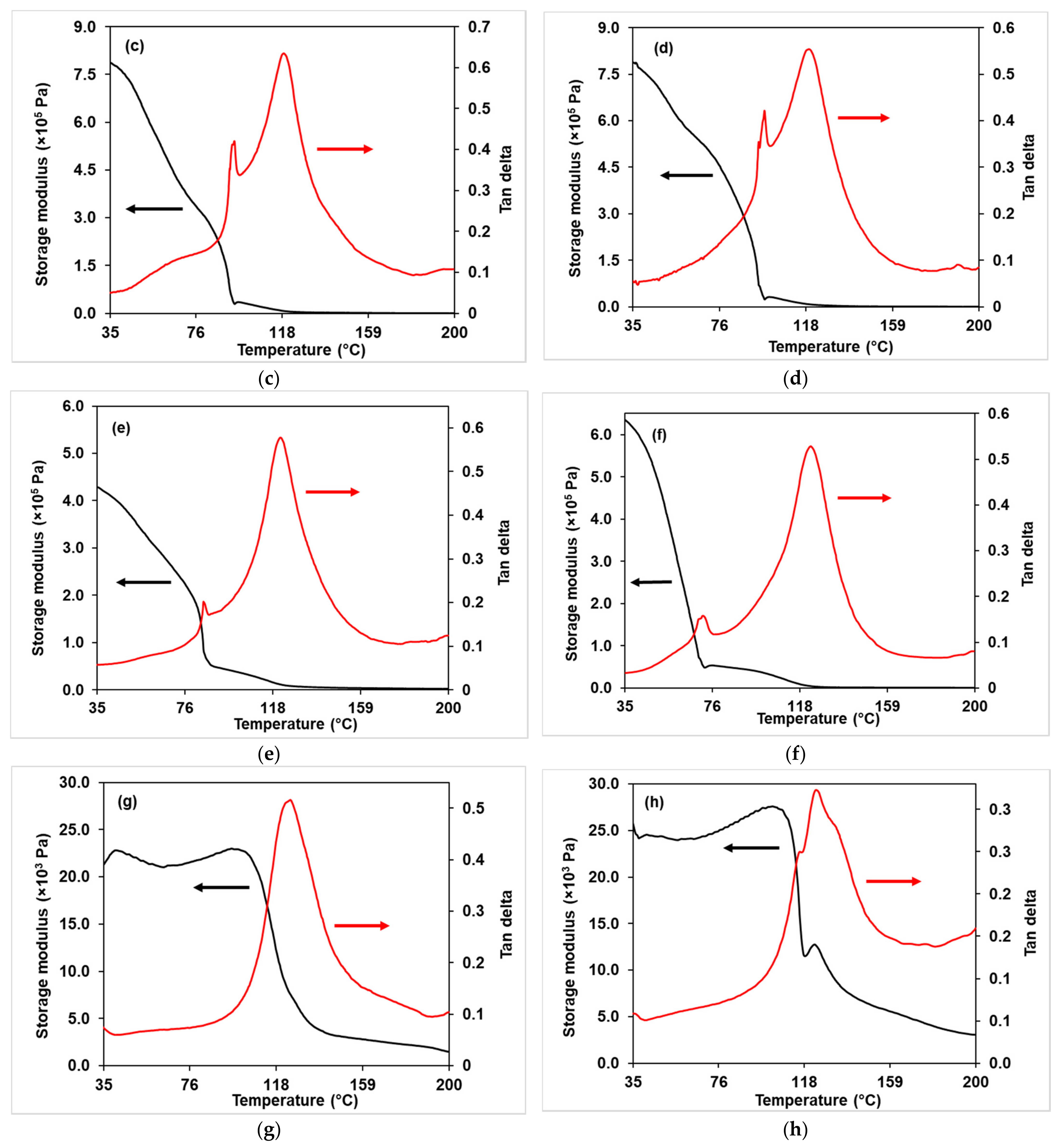
3.2.4. DMA Analysis
3.2.5. Water Uptake, Swelling Ratio, Ion Exchange Capacity, and Proton Conductivity
3.3. N-Methylene Phosphonic Chitosan/Poly (Vinyl Alcohol) Composite Membranes Modified with SiO2 Filler (NMPC/PVA-SiO2)
3.3.1. FTIR and XRD Analysis
3.3.2. Morphological Studies
3.3.3. Thermal Stability
3.3.4. Dynamic Mechanical Analysis (DMA)
3.3.5. Water Uptake, Swelling Ratio, Ion Exchange Capacity, and Proton Conductivity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Peighambardoust, S.J.; Rowshanzamir, S.; Amjadi, M. Review of the Proton Exchange Membranes for Fuel Cell Applications; Elsevier Ltd: Amsterdam, The Netherlands, 2010; Volume 35, ISBN 2177491223. [Google Scholar]
- Chen, Q.; Zhang, G.; Zhang, X.; Sun, C.; Jiao, K.; Wang, Y. Thermal management of polymer electrolyte membrane fuel cells: A review of cooling methods, material properties, and durability. Appl. Energy 2021, 286, 116496. [Google Scholar] [CrossRef]
- Adiera, N.; Rosli, H.; Loh, K.S.; Wong, W.Y.; Yunus, R.M.; Lee, T.K.; Ahmad, A.; Chong, S.T. Review of Chitosan-Based Polymers as Proton Exchange Membranes and Roles of Chitosan-Supported Ionic Liquids. Int. J. Mol. Sci. 2020, 21, 632. [Google Scholar]
- Alaswad, A.; Omran, A.; Sodre, J.R.; Wilberforce, T.; Pignatelli, G.; Dassisti, M.; Baroutaji, A.; Olabi, A.G. Technical and commercial challenges of proton-exchange membrane (Pem) fuel cells. Energies 2021, 14, 144. [Google Scholar] [CrossRef]
- Wan, Y.; Creber, K.A.M.; Peppley, B.; Bui, V.T. Ionic conductivity of chitosan membranes. Polymer 2003, 44, 1057–1065. [Google Scholar] [CrossRef]
- López-Chávez, E.; Martínez-Magadán, J.M.; Oviedo-Roa, R.; Guzmán, J.; Ramírez-Salgado, J.; Marín-Cruz, J. Molecular modeling and simulation of ion-conductivity in chitosan membranes. Polymer 2005, 46, 7519–7527. [Google Scholar] [CrossRef]
- Wan, Y.; Creber, K.A.M.; Peppley, B.; Bui, V.T. Synthesis, characterization and ionic conductive properties of phosphorylated chitosan membranes. Macromol. Chem. Phys. 2003, 204, 850–858. [Google Scholar] [CrossRef]
- Jayakumar, R.; Selvamurugan, N.; Nair, S.V.; Tokura, S.; Tamura, H. Preparative methods of phosphorylated chitin and chitosan-An overview. Int. J. Biol. Macromol. 2008, 43, 221–225. [Google Scholar] [CrossRef]
- Ramos, V.M.; Rodríguez, N.M.; Díaz, M.F.; Rodríguez, M.S.; Heras, A.; Agulló, E. N-methylene phosphonic chitosan. Effect of preparation methods on its properties. Carbohydr. Polym. 2003, 52, 39–46. [Google Scholar] [CrossRef]
- Binsu, V.V.; Nagarale, R.K.; Shahi, V.K.; Ghosh, P.K. Studies on N-methylene phosphonic chitosan/poly(vinyl alcohol) composite proton-exchange membrane. React. Funct. Polym. 2006, 66, 1619–1629. [Google Scholar] [CrossRef]
- Saxena, A.; Kumar, A.; Shahi, V.K. Preparation and characterization of N-methylene phosphonic and quaternized chitosan composite membranes for electrolyte separations. J. Colloid Interface Sci. 2006, 303, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Datta, P.; Dhara, S.; Chatterjee, J. Hydrogels and electrospun nanofibrous scaffolds of N-methylene phosphonic chitosan as bioinspired osteoconductive materials for bone grafting. Carbohydr. Polym. 2012, 87, 1354–1362. [Google Scholar] [CrossRef]
- Dadhich, P.; Das, B.; Dhara, S. Microwave assisted rapid synthesis of N-methylene phosphonic chitosan via Mannich-type reaction. Carbohydr. Polym. 2015, 133, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Ramos, V.M.; Rodríguez, M.S.; Agulló, E.; Rodríguez, N.M.; Heras, A. Chitosan with phosphonic and carboxylic group: New multidentate ligands. Int. J. Polym. Mater. Polym. Biomater. 2002, 51, 711–720. [Google Scholar] [CrossRef]
- Albu, A.M.; Maior, I.; Nicolae, C.A.; Bocăneală, F.L. Novel Pva Proton Conducting Membranes Doped With Polyaniline Generated By in-Situ Polymerization. Electrochim. Acta 2016, 211, 911–917. [Google Scholar] [CrossRef]
- Di Noto, V.; Boaretto, N.; Negro, E.; Pace, G. New inorganic-organic proton conducting membranes based on Nafion and hydrophobic fluoroalkylated silica nanoparticles. J. Power Sources 2010, 195, 7734–7742. [Google Scholar] [CrossRef]
- Dupuis, A.C. Proton exchange membranes for fuel cells operated at medium temperatures: Materials and experimental techniques. Prog. Mater. Sci. 2011, 56, 289–327. [Google Scholar] [CrossRef]
- Sun, C.; Negro, E.; Vezzù, K.; Pagot, G.; Cavinato, G.; Nale, A.; Herve Bang, Y.; Di Noto, V. Hybrid inorganic-organic proton-conducting membranes based on SPEEK doped with WO3 nanoparticles for application in vanadium redox flow batteries. Electrochim. Acta 2019, 309, 311–325. [Google Scholar] [CrossRef]
- Sun, C.; Negro, E.; Nale, A.; Pagot, G.; Vezzù, K.; Zawodzinski, T.A.; Meda, L.; Gambaro, C.; Di Noto, V. An efficient barrier toward vanadium crossover in redox flow batteries: The bilayer [Nafion/(WO3) x] hybrid inorganic-organic membrane. Electrochim. Acta 2021, 378, 138133. [Google Scholar] [CrossRef]
- Vijayakumar, V.; Khastgir, D. Hybrid composite membranes of chitosan/sulfonated polyaniline/silica as polymer electrolyte membrane for fuel cells. Carbohydr. Polym. 2018, 179, 152–163. [Google Scholar] [CrossRef]
- Dreyer, D.R.; Park, S.; Bielawski, C.W.; Ruoff, R.S. The chemistry of graphene oxide. Chem. Soc. Rev. 2010, 39, 228–240. [Google Scholar] [CrossRef]
- Hatakeyama, K.; Karim, M.R.; Ogata, C.; Tateishi, H.; Funatsu, A.; Taniguchi, T.; Koinuma, M.; Hayami, S.; Matsumoto, Y. Proton conductivities of graphene oxide nanosheets: Single, multilayer, and modified nanosheets. Angew. Chemie-Int. Ed. 2014, 53, 6997–7000. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.G.; Hong, J.; Park, Y.C.; Jung, D.H.; Hong, W.H.; Hammond, P.T.; Park, H. Innovative polymer nanocomposite electrolytes: Nanoscale manipulation of ion channels by functionalized graphenes. ACS Nano 2011, 5, 5167–5174. [Google Scholar] [CrossRef]
- Ou, X.; Jiang, L.; Chen, P.; Zhu, M.; Hu, W.; Liu, M.; Zhu, J.; Ju, H. Highly stable graphene-based multilayer films immobilized via covalent bonds and their applications in organic field-effect transistors. Adv. Funct. Mater. 2013, 23, 2422–2435. [Google Scholar] [CrossRef]
- Tseng, C.Y.; Ye, Y.S.; Cheng, M.Y.; Kao, K.Y.; Shen, W.C.; Rick, J.; Chen, J.C.; Hwang, B.J. Sulfonated polyimide proton exchange membranes with graphene oxide show improved proton conductivity, methanol crossover impedance, and mechanical properties. Adv. Energy Mater. 2011, 1, 1220–1224. [Google Scholar] [CrossRef]
- Zarrin, H.; Higgins, D.; Jun, Y.; Chen, Z.; Fowler, M. Functionalized graphene oxide nanocomposite membrane for low humidity and high temperature proton exchange membrane fuel cells. J. Phys. Chem. C 2011, 115, 20774–20781. [Google Scholar] [CrossRef]
- Bai, H.; Li, Y.; Zhang, H.; Chen, H.; Wu, W.; Wang, J.; Liu, J. Anhydrous proton exchange membranes comprising of chitosan and phosphorylated graphene oxide for elevated temperature fuel cells. J. Memb. Sci. 2015, 495, 48–60. [Google Scholar] [CrossRef]
- Mohd Nasir, N.F.; Zain, N.M.; Raha, M.G.; Kadri, N.A. Characterization of Chitosan-poly (Ethylene Oxide) Blends as Haemodialysis Membrane. Am. J. Appl. Sci. 2005, 2, 1578–1583. [Google Scholar] [CrossRef]
- Mansur, H.S.; de S. Costa, E., Jr.; Mansur, A.A.P.; Barbosa-Stancioli, E.F. Cytocompatibility evaluation in cell-culture systems of chemically crosslinked chitosan/PVA hydrogels. Mater. Sci. Eng. C 2009, 29, 1574–1583. [Google Scholar] [CrossRef]
- Wang, J.; Wang, X.; Xu, C.; Zhang, M.; Shang, X. Preparation of graphene/poly(vinyl alcohol) nanocomposites with enhanced mechanical properties and water resistance. Polym. Int. 2011, 60, 816–822. [Google Scholar] [CrossRef]
- Santos, C.; Silva, C.J.; Büttel, Z.; Guimarães, R.; Pereira, S.B.; Tamagnini, P.; Zille, A. Preparation and characterization of polysaccharides/PVA blend nanofibrous membranes by electrospinning method. Carbohydr. Polym. 2014, 99, 584–592. [Google Scholar] [CrossRef] [Green Version]
- Smitha, B.; Sridhar, S.; Khan, A.A. Synthesis and characterization of poly(vinyl alcohol)-based membranes for direct methanol fuel cell. J. Appl. Polym. Sci. 2005, 95, 1154–1163. [Google Scholar] [CrossRef]
- Xiong, Y.; Liu, Q.L.; Zhang, Q.G.; Zhu, A.M. Synthesis and characterization of cross-linked quaternized poly(vinyl alcohol)/chitosan composite anion exchange membranes for fuel cells. J. Power Sources 2008, 183, 447–453. [Google Scholar] [CrossRef]
- Ming Yang, J.; Chih Chiu, H. Preparation and characterization of polyvinyl alcohol/chitosan blended membrane for alkaline direct methanol fuel cells. J. Memb. Sci. 2012, 419–420, 65–71. [Google Scholar] [CrossRef]
- El Miri, N.; Abdelouahdi, K.; Zahouily, M.; Fihri, A.; Barakat, A.; Solhy, A.; El Achaby, M. Bio-nanocomposite films based on cellulose nanocrystals filled polyvinyl alcohol/chitosan polymer blend. J. Appl. Polym. Sci. 2015, 132, 42004. [Google Scholar] [CrossRef]
- Yusoff, Y.N.; Loh, K.S.; Wong, W.Y.; Daud, W.R.W.; Lee, T.K. Sulfonated graphene oxide as an inorganic filler in promoting the properties of a polybenzimidazole membrane as a high temperature proton exchange membrane. Int. J. Hydrogen Energy 2020, 45, 27510–27526. [Google Scholar] [CrossRef]
- Staiti, P.; Lufrano, F.; Aricò, A.S.; Passalacqua, E.; Antonucci, V. Sulfonated polybenzimidazole membranes—Preparation and physico-chemical characterization. J. Memb. Sci. 2001, 188, 71–78. [Google Scholar] [CrossRef]
- Fadzallah, I.A.; Majid, S.R.; Careem, M.A.; Arof, A.K. A study on ionic interactions in chitosan-oxalic acid polymer electrolyte membranes. J. Memb. Sci. 2014, 463, 65–72. [Google Scholar] [CrossRef]
- Amaral, I.F.; Granja, P.L.; Barbosa, M.A. Chemical modification of chitosan by phosphorylation: An XPS, FT-IR and SEM study. J. Biomater. Sci. Polym. Ed. 2005, 16, 1575–1593. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, J.; Huang, K.; Zhang, X.; Xu, L.; Shi, Z. guo Preparation, characterization and application of N-methylene phosphonic acid chitosan grafted magnesia-zirconia stationary phase. Anal. Chim. Acta 2015, 854, 191–201. [Google Scholar] [CrossRef]
- Liew, J.W.Y.; Loh, K.S.; Ahmad, A.; Lim, K.L.; Daud, W.R. Effect of modified natural filler o-methylene phosphonic κ-carrageenan on chitosan-based polymer electrolytes. Energies 2018, 11, 1910. [Google Scholar] [CrossRef] [Green Version]
- Ma, G.; Qian, B.; Yang, J.; Hu, C.; Nie, J. Synthesis and properties of photosensitive chitosan derivatives(1). Int. J. Biol. Macromol. 2010, 46, 558–561. [Google Scholar] [CrossRef]
- Mobarak, N.N.; Ahmad, A.; Abdullah, M.P.; Ramli, N.; Rahman, M.Y.A. Conductivity enhancement via chemical modification of chitosan based green polymer electrolyte. Electrochim. Acta 2013, 92, 161–167. [Google Scholar] [CrossRef]
- Coates, J. Interpretation of Infrared Spectra, A Practical Approach. Encycl. Anal. Chem. 2006, 1–23. [Google Scholar] [CrossRef]
- Andrade, G.; Barbosa-Stancioli, E.F.; Piscitelli Mansur, A.A.; Vasconcelos, W.L.; Mansur, H.S. Design of novel hybrid organic-inorganic nanostructured biomaterials for immunoassay applications. Biomed. Mater. 2006, 1, 221–234. [Google Scholar] [CrossRef]
- Andrade, G.I.; Barbosa-Stancioli, E.F.; Mansur, A.A.P.; Vasconcelos, W.L.; Mansur, H.S. Small-angle X-ray scattering and FTIR characterization of nanostructured poly (vinyl alcohol)/silicate hybrids for immunoassay applications. J. Mater. Sci. 2008, 43, 450–463. [Google Scholar] [CrossRef]
- Wang, T.; Turhan, M.; Gunasekaran, S. Selected properties of pH-sensitive, biodegradable chitosan-poly(vinyl alcohol) hydrogel. Polym. Int. 2004, 53, 911–918. [Google Scholar] [CrossRef]
- Mansur, H.S.; Sadahira, C.M.; Souza, A.N.; Mansur, A.A.P. FTIR spectroscopy characterization of poly (vinyl alcohol) hydrogel with different hydrolysis degree and chemically crosslinked with glutaraldehyde. Mater. Sci. Eng. C 2008, 28, 539–548. [Google Scholar] [CrossRef]
- Shao, C.; Kim, H.Y.; Gong, J.; Ding, B.; Lee, D.R.; Park, S.J. Fiber mats of poly(vinyl alcohol)/silica composite via electrospinning. Mater. Lett. 2003, 57, 1579–1584. [Google Scholar] [CrossRef]
- Lue, S.J.; Chen, J.Y.; Yang, J.M. Crystallinity and stability of poly(vinyl alcohol)-fumed silica mixed matrix membranes. J. Macromol. Sci. Part B Phys. 2008, 47, 39–51. [Google Scholar] [CrossRef]
- Sapalidis, A.A. Porous Polyvinyl alcohol membranes: Preparation methods and applications. Symmetry 2020, 12, 960. [Google Scholar] [CrossRef]
- Menard, K.P.; Bilyeu, B.W. Dynamic Mechanical Analysis of Polymers and Rubbers. Encycl. Anal. Chem. 2008. [Google Scholar] [CrossRef]
- Turi, E.A. Thermal Characterization of Polymeric Materials; Academic Press: New York, NY, USA, 1997. [Google Scholar]
- Ferry, J.D. Viscoelastic Properties of Polymers; John Wiley & Sons: Hoboken, NJ, USA, 1980. [Google Scholar]
- Boyer, R.F. Dependence of mechanical properties on molecular motion in polymers. Polym. Eng. Sci. 1968, 8, 161–185. [Google Scholar] [CrossRef]
- Rohn, C.L. Analytical Polymer Rheology: Structure-Processing-Property Relationships; Hanser: Liberty Township, OH, USA, 1995. [Google Scholar]
- Menard, K.P. Dynamic Mechanical Analysis. Encycl. Anal. Chem. 2017, 1–22. [Google Scholar] [CrossRef]
- Du, L.; Yan, X.; He, G.; Wu, X.; Hu, Z.; Wang, Y. SPEEK proton exchange membranes modified with silica sulfuric acid nanoparticles. Int. J. Hydrogen Energy 2012, 37, 11853–11861. [Google Scholar] [CrossRef]
- Kannan, R.; Kakade, B.A.; Pillai, V.K. Polymer electrolyte fuel cells using nafion-based composite membranes with functionalized carbon nanotubes. Angew. Chemie-Int. Ed. 2008, 47, 2653–2656. [Google Scholar] [CrossRef]
- Kalaiselvimary, J.; Sundararajan, M.; Prabhu, M.R. Preparation and characterization of chitosan-based nanocomposite hybrid polymer electrolyte membranes for fuel cell application. Ionics 2018, 24, 3555–3571. [Google Scholar] [CrossRef]
- Isahak, W.N.R.W.; Ismail, M.; Nordin, N.M.; Jahim, J.M.; Yarmo, M.A. Synthesis, characterization and catalytic performance of H 3SiW 12O 40/SiO 2 prepared by sol-gel technique. J. Nanotechnol. 2011, 2011, 13194–13203. [Google Scholar] [CrossRef] [Green Version]
- Lakshminarayana, G.; Nogami, M. Synthesis and characterization of proton conducting inorganic-organic hybrid nanocomposite membranes based on tetraethoxysilane/trimethylphosphate/3-glycidoxypropyltrimethoxysilane/heteropoly acids. Electrochim. Acta 2009, 54, 4731–4740. [Google Scholar] [CrossRef]
- Shanmugam, S.; Viswanathan, B.; Varadarajan, T.K. Synthesis and characterization of silicotungstic acid based organic-inorganic nanocomposite membrane. J. Memb. Sci. 2006, 275, 105–109. [Google Scholar] [CrossRef]
- Joni, I.M.; Nulhakim, L.; Vanitha, M.; Panatarani, C. Characteristics of crystalline silica (SiO2) particles prepared by simple solution method using sodium silicate (Na2SiO3) precursor. J. Phys. Conf. Ser. 2018, 1080. [Google Scholar] [CrossRef]
- Liu, H.; Gong, C.; Wang, J.; Liu, X.; Liu, H.; Cheng, F.; Wang, G.; Zheng, G.; Qin, C.; Wen, S. Chitosan/silica coated carbon nanotubes composite proton exchange membranes for fuel cell applications. Carbohydr. Polym. 2016, 136, 1379–1385. [Google Scholar] [CrossRef]
- Ou, Y.; Tsen, W.C.; Gong, C.; Wang, J.; Liu, H.; Zheng, G.; Qin, C.; Wen, S. Chitosan-based composite membranes containing chitosan-coated carbon nanotubes for polymer electrolyte membranes. Polym. Adv. Technol. 2018, 29, 612–622. [Google Scholar] [CrossRef]
- Ou, Y.; Tsen, W.C.; Jang, S.C.; Chuang, F.S.; Wang, J.; Liu, H.; Wen, S.; Gong, C. Novel composite polymer electrolyte membrane using solid superacidic sulfated zirconia—Functionalized carbon nanotube modified chitosan. Electrochim. Acta 2018, 264, 251–259. [Google Scholar] [CrossRef]
- Ciu, W.; Du, F.; Zhao, J.; Zhang, W.; Yang, Y.; Xie, X.; Mai, Y.W. Improving thermal conductivity while retaining high electrical resistivity of epoxy composites by incorporating silica-coated multi-walled carbon nanotubes. Carbon 2011, 49, 495–500. [Google Scholar] [CrossRef]
- Gong, C.; Zheng, X.; Liu, H.; Wang, G.; Cheng, F.; Zheng, G.; Wen, S.; Law, W.C.; Tsui, C.P.; Tang, C.Y. A new strategy for designing high-performance sulfonated poly(ether ether ketone) polymer electrolyte membranes using inorganic proton conductor-functionalized carbon nanotubes. J. Power Sources 2016, 325, 453–464. [Google Scholar] [CrossRef]
- Wong, C.Y.; Wong, W.Y.; Walvekar, R.; Loh, K.S.; Khalid, M.; Lim, K.L. Effect of deep eutectic solvent in proton conduction and thermal behaviour of chitosan-based membrane. J. Mol. Liq. 2018, 269, 675–683. [Google Scholar] [CrossRef]
- Xie, Z.; Wu, H.; Wu, Q.; Ai, L. Synthesis and performance of solid proton conductor molybdovanadosilicic acid. RSC Adv. 2018, 8, 13984–13988. [Google Scholar] [CrossRef] [Green Version]
- Vijayalekshmi, V.; Khastgir, D. Fabrication and comprehensive investigation of physicochemical and electrochemical properties of chitosan-silica supported silicotungstic acid nanocomposite membranes for fuel cell applications. Energy 2018, 142, 313–330. [Google Scholar] [CrossRef]
- Gnana Kumar, G.; Lee, D.N.; Kim, P.; Nahm, K.S.; Nimma Elizabeth, R. Characterization of PVdF-HFP/Nafion/AlO[OH]n composite membranes for direct methanol fuel cell (DMFC). Eur. Polym. J. 2008, 44, 2225–2230. [Google Scholar] [CrossRef]
- Kumar, P.; Kundu, P.P. Coating and lamination of Nafion117 with partially sulfonated PVdF for low methanol crossover in DMFC applications. Electrochim. Acta 2015, 173, 124–130. [Google Scholar] [CrossRef]
- Xue, Y.; Chan, S. Layer-by-layer self-assembly of CHI/PVS-Nafion composite membrane for reduced methanol crossover and enhanced DMFC performance. Int. J. Hydrogen Energy 2015, 40, 1877–1885. [Google Scholar] [CrossRef]
- Hasani-Sadrabadi, M.M.; Dashtimoghadam, E.; Majedi, F.S.; Wu, S.; Bertsch, A.; Moaddel, H.; Renaud, P. Nafion/chitosan-wrapped CNT nanocomposite membrane for high-performance direct methanol fuel cells. RSC Adv. 2013, 3, 7337–7346. [Google Scholar] [CrossRef]
- Wong, C.Y.; Wong, W.Y.; Loh, K.S.; Khalid, M.; Wan Daud, W.R.; Lim, K.L.; Walvekar, R. Influences of crosslinked carboxylic acid monomers on the proton conduction characteristics of chitosan/SPVA composite membranes. Polymer 2020, 203, 122782. [Google Scholar] [CrossRef]
- Gaur, S.S.; Dhar, P.; Sonowal, A.; Sharma, A.; Kumar, A.; Katiyar, V. Thermo-mechanically stable sustainable polymer based solid electrolyte membranes for direct methanol fuel cell applications. J. Memb. Sci. 2017, 526, 348–354. [Google Scholar] [CrossRef]
- Aziz, N.A.; Majid, S.R.; Arof, A.K. Synthesis and characterizations of phthaloyl chitosan-based polymer electrolytes. J. Non-Cryst. Solids 2012, 358, 1581–1590. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Membrane Sample | Thickness (mm) | Water Uptake (%) | Swelling Area (%) | Swelling Thickness (%) | IEC (mequiv g−1) | Proton Conductivity (10−5 S cm−1) |
|---|---|---|---|---|---|---|
| NMPC/PVA-30 | 0.06 ± 0.02 | 32.1 ± 5.2 | 15.1 ± 3.6 | 46.5 ± 5.9 | 0.24 ± 0.08 | 2.61 ± 0.29 |
| NMPC/PVA-35 | 0.07 ± 0.03 | 37.3 ± 5.1 | 25.4 ± 7.1 | 51.2 ± 5.3 | 0.39 ± 0.02 | 3.45 ± 0.38 |
| NMPC/PVA-40 | 0.08 ± 0.01 | 44.4 ± 2.3 | 29.6 ± 16.3 | 58.6 ± 5.9 | 0.40 ± 0.03 | 4.96 ± 0.33 |
| NMPC/PVA-45 | 0.08 ± 0.02 | 48.4 ± 3.5 | 33.4 ± 7.1 | 61.3 ± 1.6 | 0.42 ± 0.06 | 6.59 ± 0.19 |
| NMPC/PVA-50 | 0.09 ± 0.01 | 51.9 ± 2.3 | 35.1 ± 16.3 | 62.2 ± 3.3 | 0.45 ± 0.02 | 8.76 ± 0.16 |
| NMPC/PVA-55 | 0.10 ± 0.02 | 49.5 ± 5.5 | 33.3 ± 12.9 | 54.3 ± 2.3 | 0.38 ± 0.04 | 7.47 ± 0.25 |
| NMPC/PVA-60 | 0.10 ± 0.04 | 48.3 ± 6.5 | 30.7 ± 7.8 | 51.2 ± 2.8 | 0.36 ± 0.03 | 5.38 ± 0.41 |
| NMPC/PVA-65 | 0.11 ± 0.01 | 42.5 ± 6.2 | 24.1 ± 5.1 | 45.1 ± 2.2 | 0.33 ± 0.02 | 4.26 ± 0.34 |
| NMPC/PVA-70 | 0.12 ± 0.03 | 39.1 ± 2.9 | 17.8 ± 9.8 | 39.2 ± 1.9 | 0.29 ± 0.07 | 3.59 ± 0.46 |
| Membrane Sample | Thickness (mm) | Water Uptake (%) | Swelling Area (%) | Swelling Thickness (%) | IEC (mequiv g−1) | Proton Conductivity (10−4 S cm−1) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| 25 °C | 40 °C | 60 °C | 80 °C | 100 °C | ||||||
| NMPC/PVA-50 | 0.09 ± 0.01 | 51.9 ± 2.3 | 35.1 ± 16.3 | 62.2 ± 3.3 | 0.45 ± 0.02 | 0.87 ± 0.02 | 1.10 ± 0.03 | 1.69 ± 0.01 | 2.03 ± 0.02 | 2.22 ± 0.01 |
| NMPC/PVA-SiO2 (0.5 wt.%) | 0.10 ± 0.01 | 45.7 ± 2.0 | 28.4 ± 5.9 | 44.6 ± 8.8 | 0.43 ± 0.02 | 1.14 ± 0.04 | 1.78 ± 0.08 | 2.22 ± 0.01 | 2.87 ± 0.01 | 3.20 ± 0.06 |
| NMPC/PVA-SiO2 (1 wt.%) | 0.10 ± 0.02 | 47.1 ± 1.8 | 33.1 ± 10.8 | 48.3 ± 4.9 | 0.45 ± 0.02 | 1.36 ± 0.07 | 1.93 ± 0.09 | 2.47 ± 0.10 | 3.25 ± 0.02 | 3.81 ± 0.05 |
| NMPC/PVA-SiO2 (2 wt.%) | 0.10 ± 0.01 | 47.4 ± 2.8 | 37.6 ± 11.3 | 52.9 ± 3.6 | 0.49 ± 0.03 | 1.63 ± 0.06 | 2.28 ± 0.04 | 3.42 ± 0.14 | 3.94 ± 0.13 | 4.41 ± 0.14 |
| NMPC/PVA-SiO2 (4 wt.%) | 0.11 ± 0.01 | 55.7 ± 1.9 | 43.9 ± 5.5 | 63.1 ± 1.0 | 0.56 ± 0.02 | 1.90 ± 0.09 | 2.38 ± 0.09 | 3.51 ± 0.07 | 4.65 ± 0.06 | 5.08 ± 0.05 |
| NMPC/PVA-SiO2 (6 wt.%) | 0.11 ± 0.02 | 50.6 ± 1.9 | 35.3 ± 6.6 | 55.8 ± 4.8 | 0.44 ± 0.03 | 1.82 ± 0.07 | 2.22 ± 0.17 | 3.42 ± 0.11 | 4.16 ± 0.10 | 4.67 ± 0.09 |
| NMPC/PVA-SiO2 (8 wt.%) | 0.12 ± 0.03 | 46.5 ± 1.2 | 27.6 ± 6.1 | 51.4 ± 5.1 | 0.37 ± 0.03 | 1.56 ± 0.08 | 1.96 ± 0.09 | 2.79 ± 0.10 | 3.53 ± 0.05 | 4.38 ± 0.08 |
| NMPC/PVA-SiO2 (10 wt.%) | 0.13 ± 0.02 | 48.8 ± 1.2 | 32.8 ± 6.1 | 54.1 ± 8.2 | 0.41 ± 0.02 | 1.71 ± 0.16 | 2.22 ± 0.08 | 3.05 ± 0.09 | 3.97 ± 0.08 | 4.53 ± 0.05 |
| Nafion 212 | 0.02 ± 0.01 | - | - | - | - | * 1.25 ± 0.03 | * 1.58 ± 0.05 | * 1.80 ± 0.04 | * 2.06 ± 0.03 | * 1.87 ± 0.06 |
| Membrane | Water Uptake (%) | IEC (mequiv g−1) | Proton Conductivity (S cm−1) | Applications | References |
|---|---|---|---|---|---|
| PVdF-coHFP/Nafion | 33.8 | - | 1.00 × 10−3 | DMFC | [73] |
| Nafion-sulfonated PVdF coated | 13.0 | - | 5.91 × 10−3 | DMFC | [74] |
| CS/PVS-Nafion | 29.1 | - | 7.01 × 10−2 | DMFC | [75] |
| Nafion/CNT | 29.5 | 0.90 | 7.35 × 10−2 | DMFC | [76] |
| Nafion 212 | - | - | 2.06 × 10−2 | PEM | This study |
| SPVA-SPTA | 150.4 | 0.45 | 8.80 × 10−4 | PEM | [77] |
| PVA-CS-CNC | 78 | - | 6.42 × 10−4 | DMFC | [78] |
| CS/SPVA-SSA | 220 | 2.60 | 2.58 × 10−4 | PEM | [77] |
| Ph/CS-NH4SCN | - | - | 2.42 × 10−5 | PEM | [79] |
| NMPC | - | - | 2.74 × 10−6 | PEM | This study |
| NMPC-OMPk | - | - | 1.43 × 10−5 | PEM | [41] |
| NMPC/PVA | 51.9 | 0.45 | 2.22 × 10−4 | PEM | This study |
| NMPC/PVA-SiO2 | 55.7 | 0.56 | 5.08 × 10−4 | PEM | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosli, N.A.H.; Loh, K.S.; Wong, W.Y.; Lee, T.K.; Ahmad, A. Hybrid Composite Membrane of Phosphorylated Chitosan/Poly (Vinyl Alcohol)/Silica as a Proton Exchange Membrane. Membranes 2021, 11, 675. https://doi.org/10.3390/membranes11090675
Rosli NAH, Loh KS, Wong WY, Lee TK, Ahmad A. Hybrid Composite Membrane of Phosphorylated Chitosan/Poly (Vinyl Alcohol)/Silica as a Proton Exchange Membrane. Membranes. 2021; 11(9):675. https://doi.org/10.3390/membranes11090675
Chicago/Turabian StyleRosli, Nur Adiera Hanna, Kee Shyuan Loh, Wai Yin Wong, Tian Khoon Lee, and Azizan Ahmad. 2021. "Hybrid Composite Membrane of Phosphorylated Chitosan/Poly (Vinyl Alcohol)/Silica as a Proton Exchange Membrane" Membranes 11, no. 9: 675. https://doi.org/10.3390/membranes11090675
APA StyleRosli, N. A. H., Loh, K. S., Wong, W. Y., Lee, T. K., & Ahmad, A. (2021). Hybrid Composite Membrane of Phosphorylated Chitosan/Poly (Vinyl Alcohol)/Silica as a Proton Exchange Membrane. Membranes, 11(9), 675. https://doi.org/10.3390/membranes11090675