
Cell Surface Binding and Lipid Interactions behind Chemotherapy-Drug-Induced Ion Pore Formation in Membranes

 ,
,  ,
, 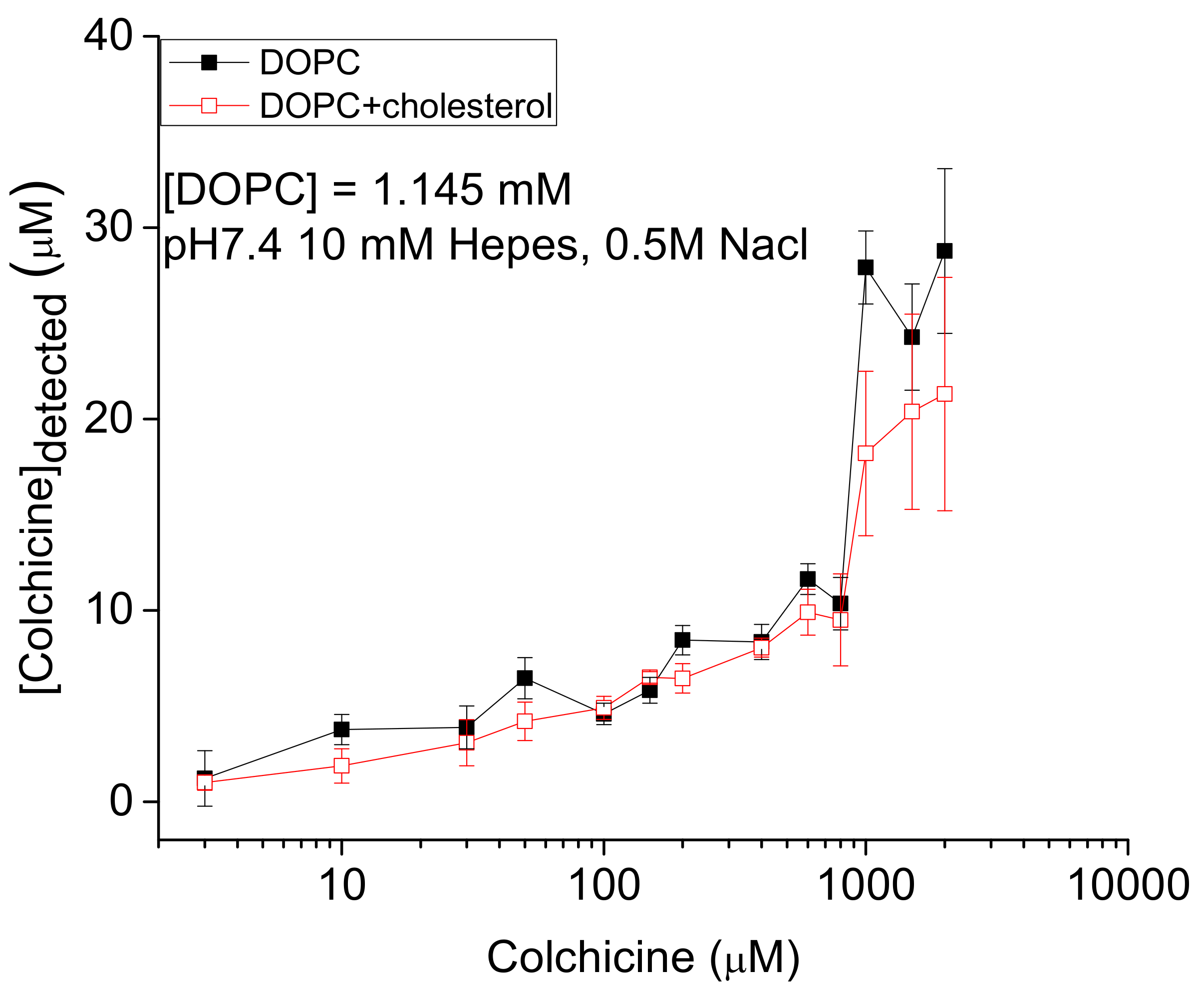{kind=link}
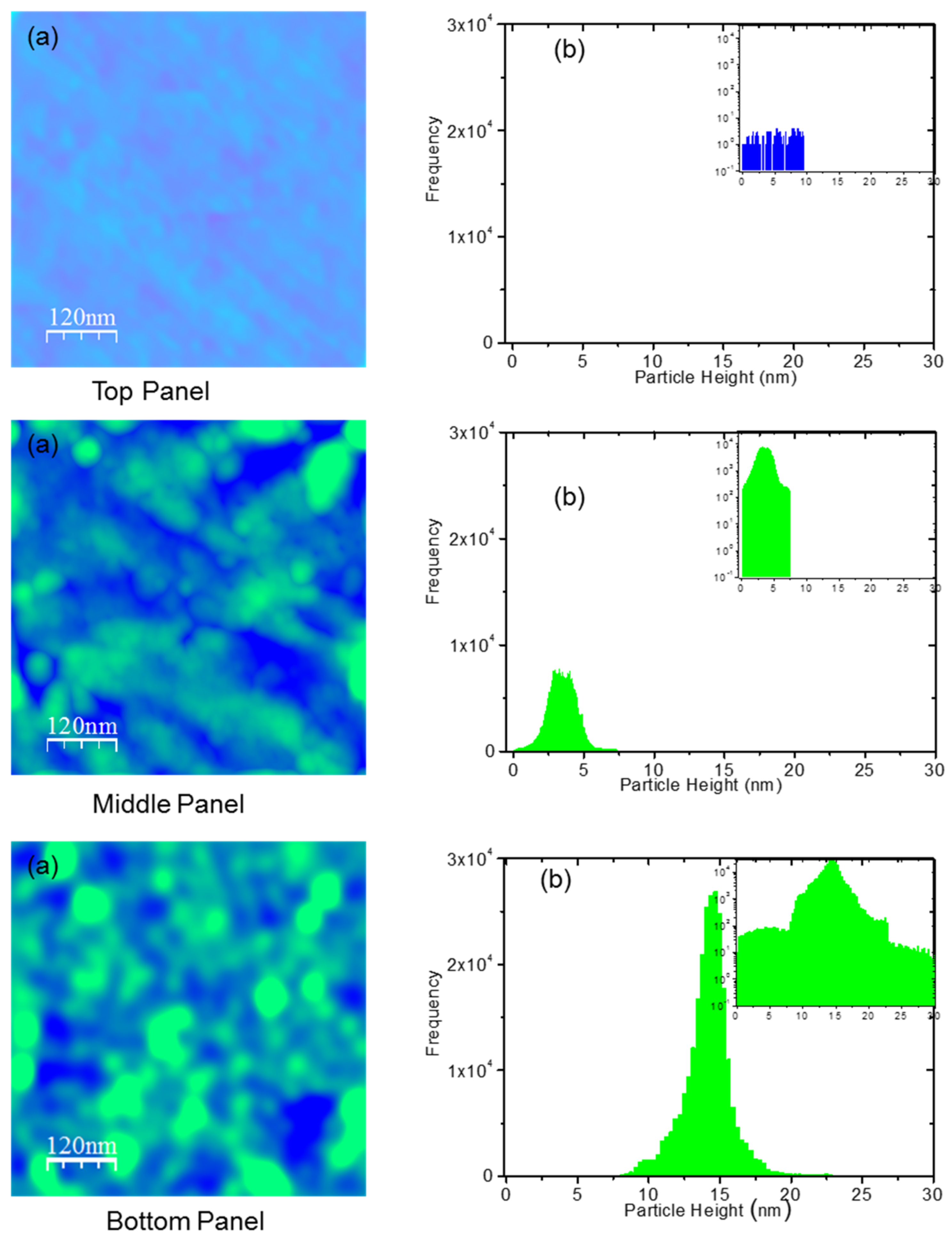{kind=link}
{kind=link}
{kind=link}
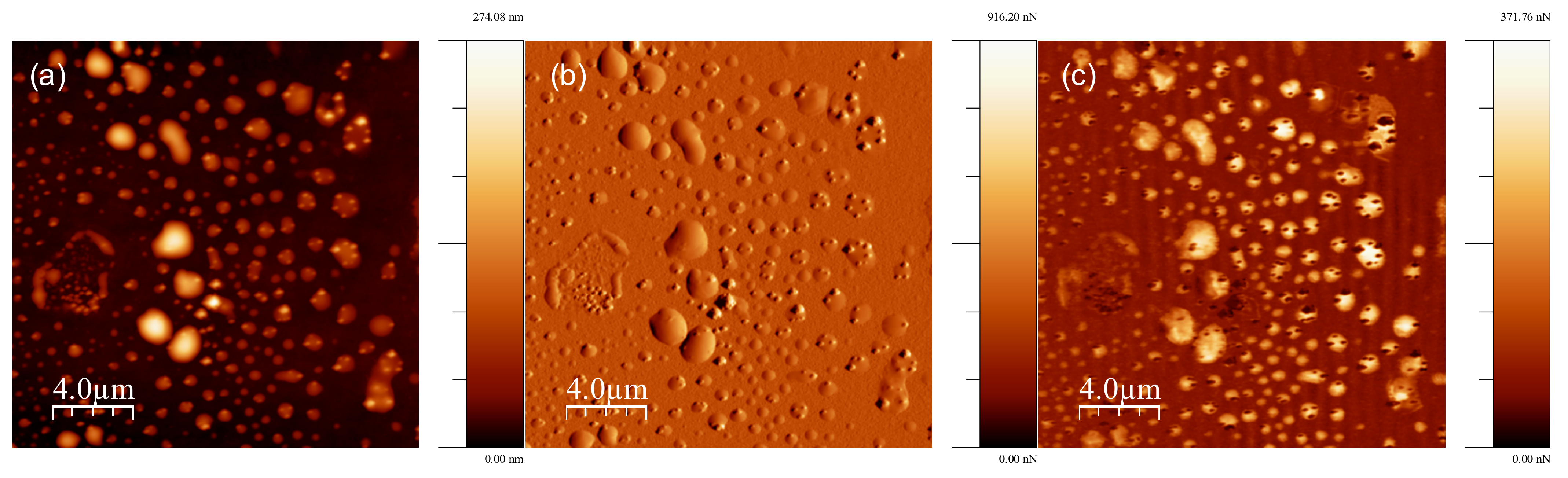{kind=link}
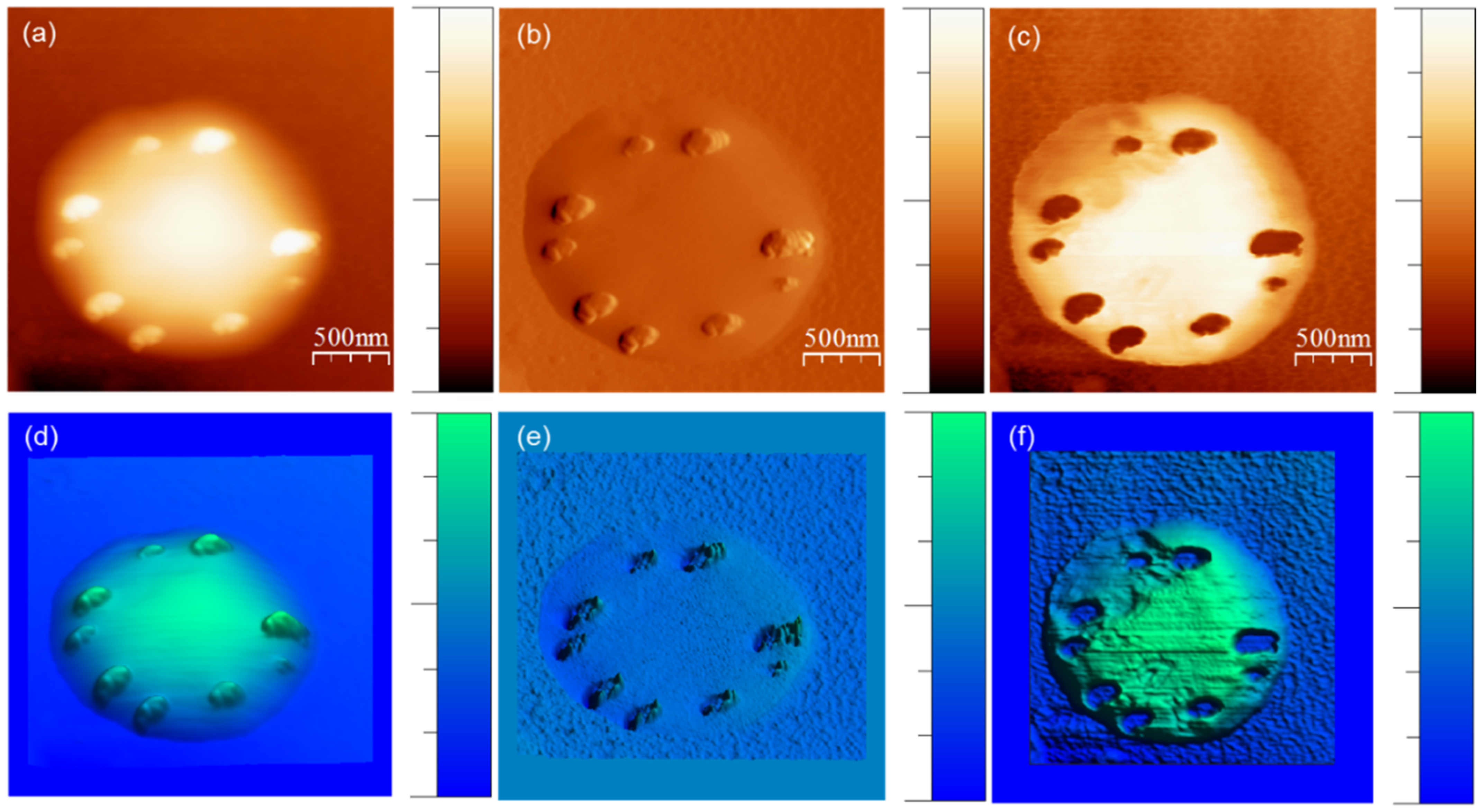{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- (i)
- address the statistical significance of the cell surface binding of CDs;
- (ii)
- understand the nature of the surface distribution (random or cluster creation) of drug molecules and propose drug distribution models; and
- (iii)
- finally, measure the energetics of cell surface adsorption of drugs.
2. Materials and Methods
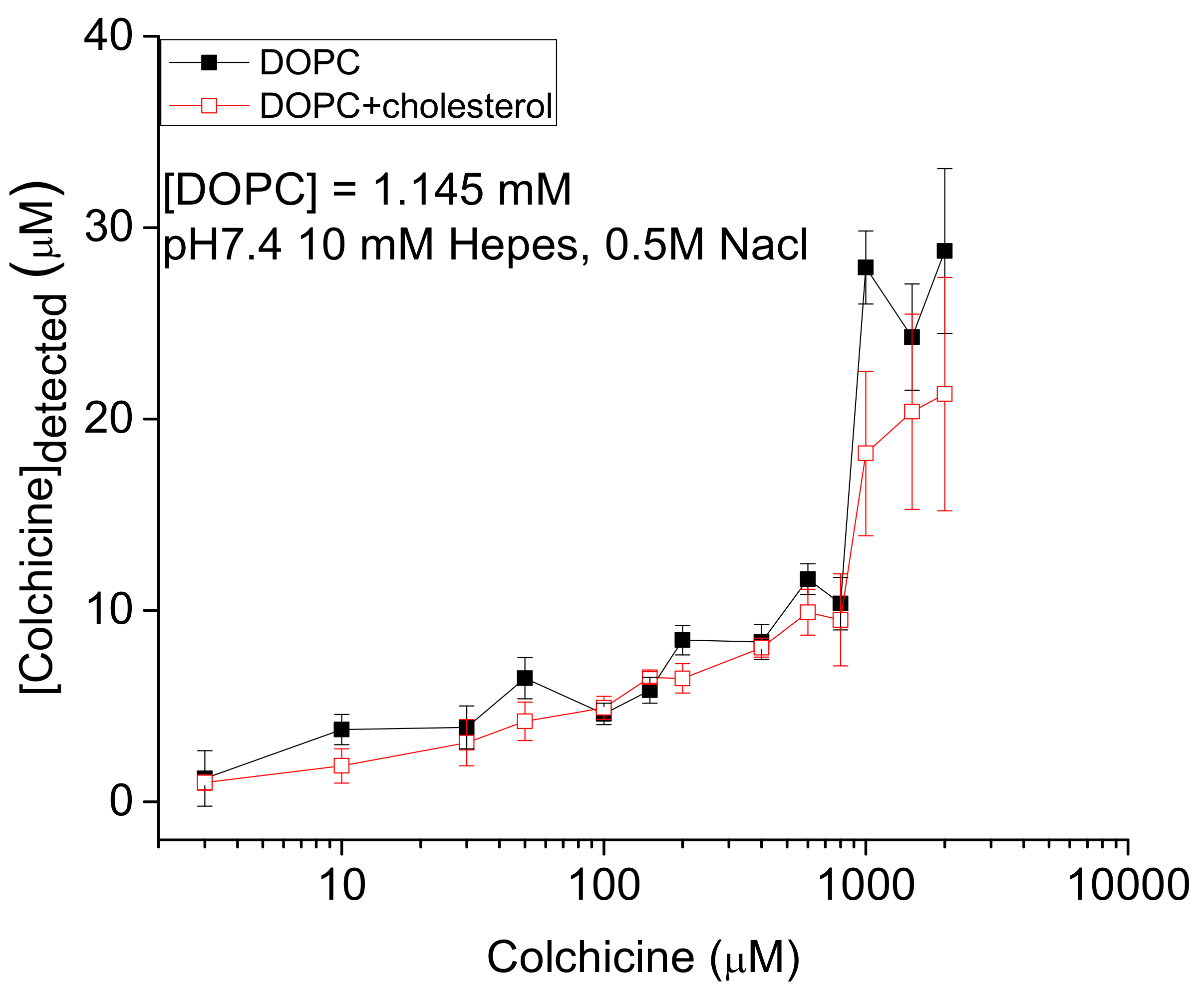
2.1. Detection of In Vitro Liposome Bound Colchicine
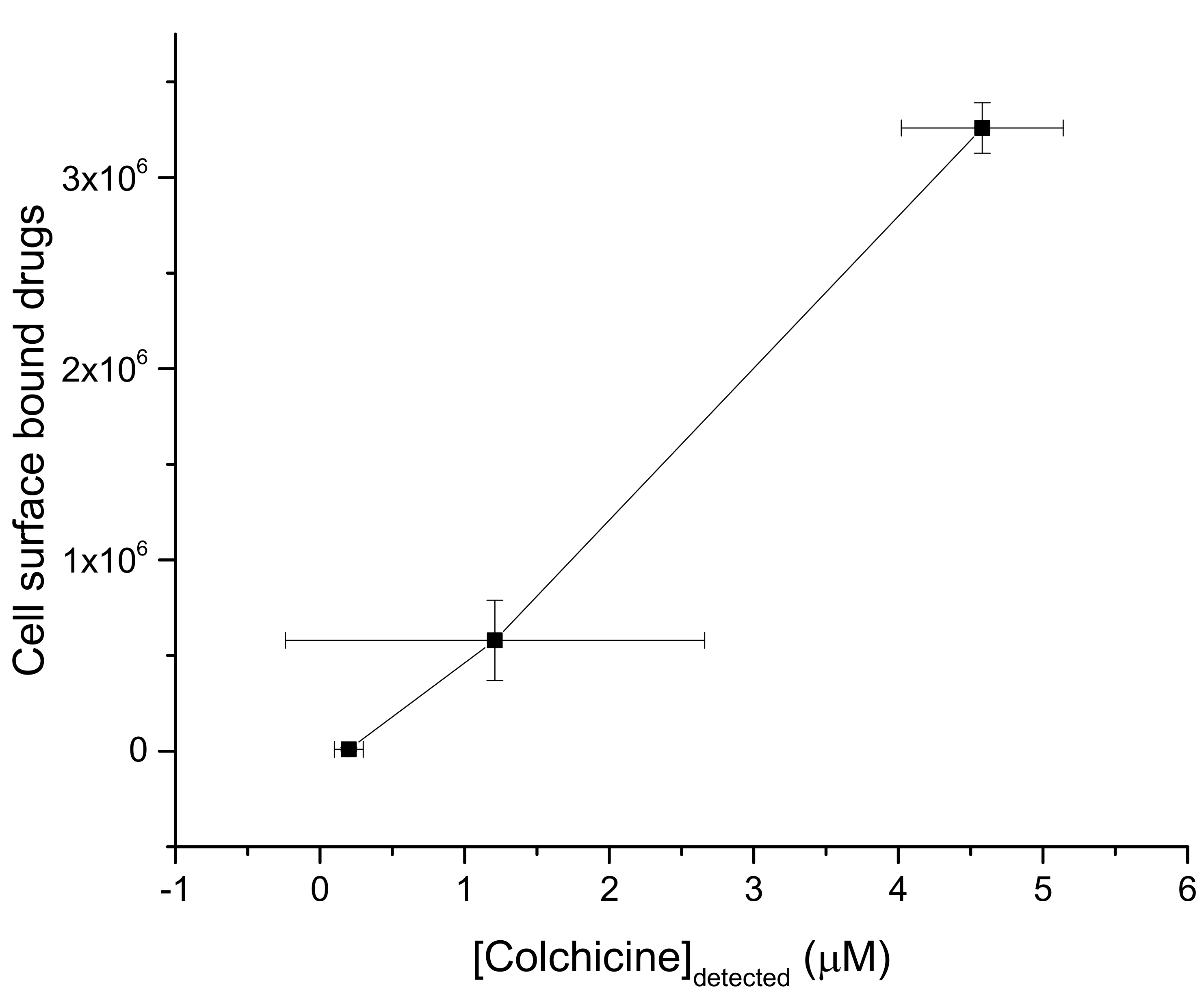
2.2. Cell Culture and Treatment of Cells with Colchicine
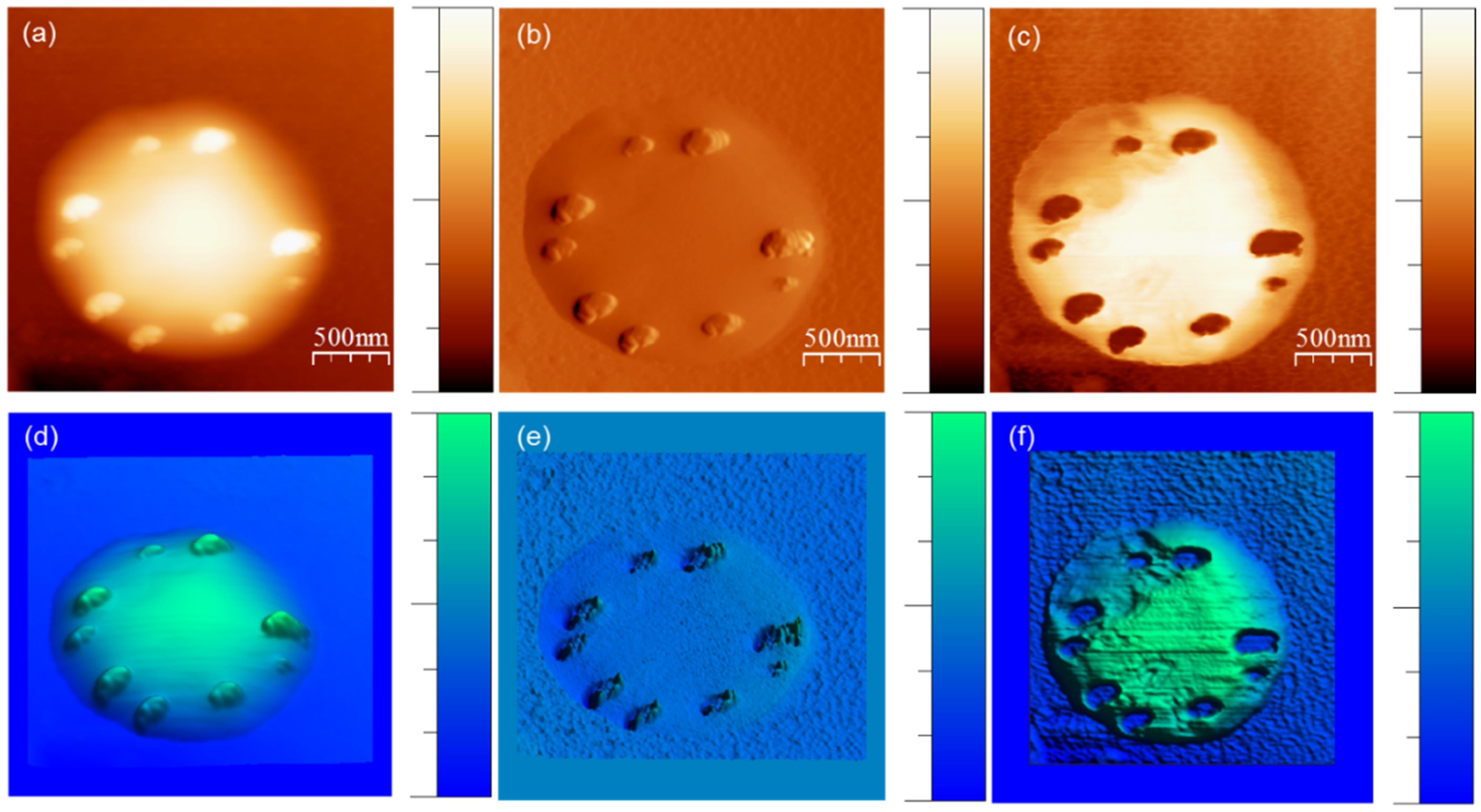
2.3. AFM Experimental Methods
2.4. NCs Considering Relevant Physiological Parameters
3. Results
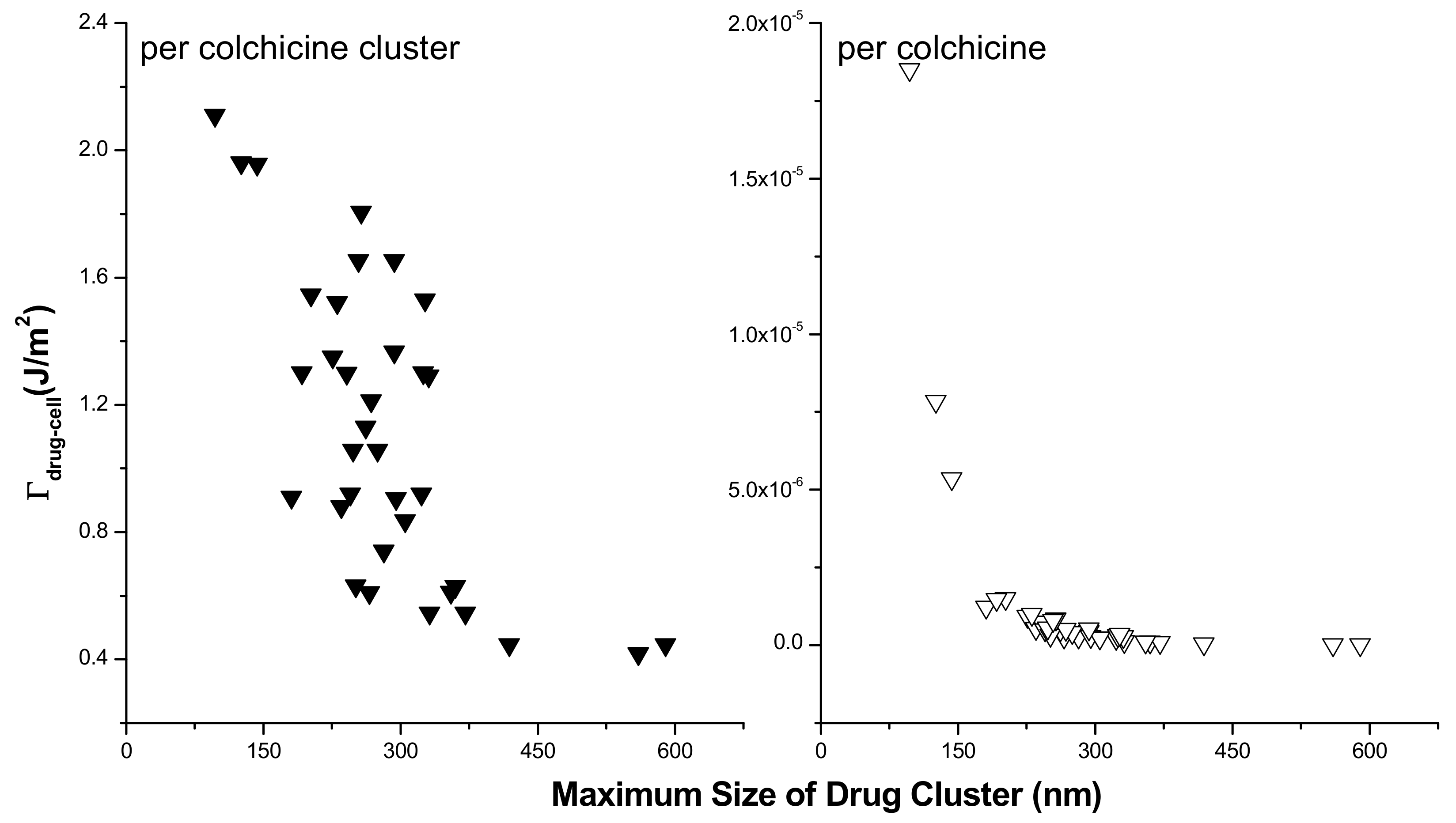
3.1. Strong Adhesion between Colchicine Cluster and Cell Membrane
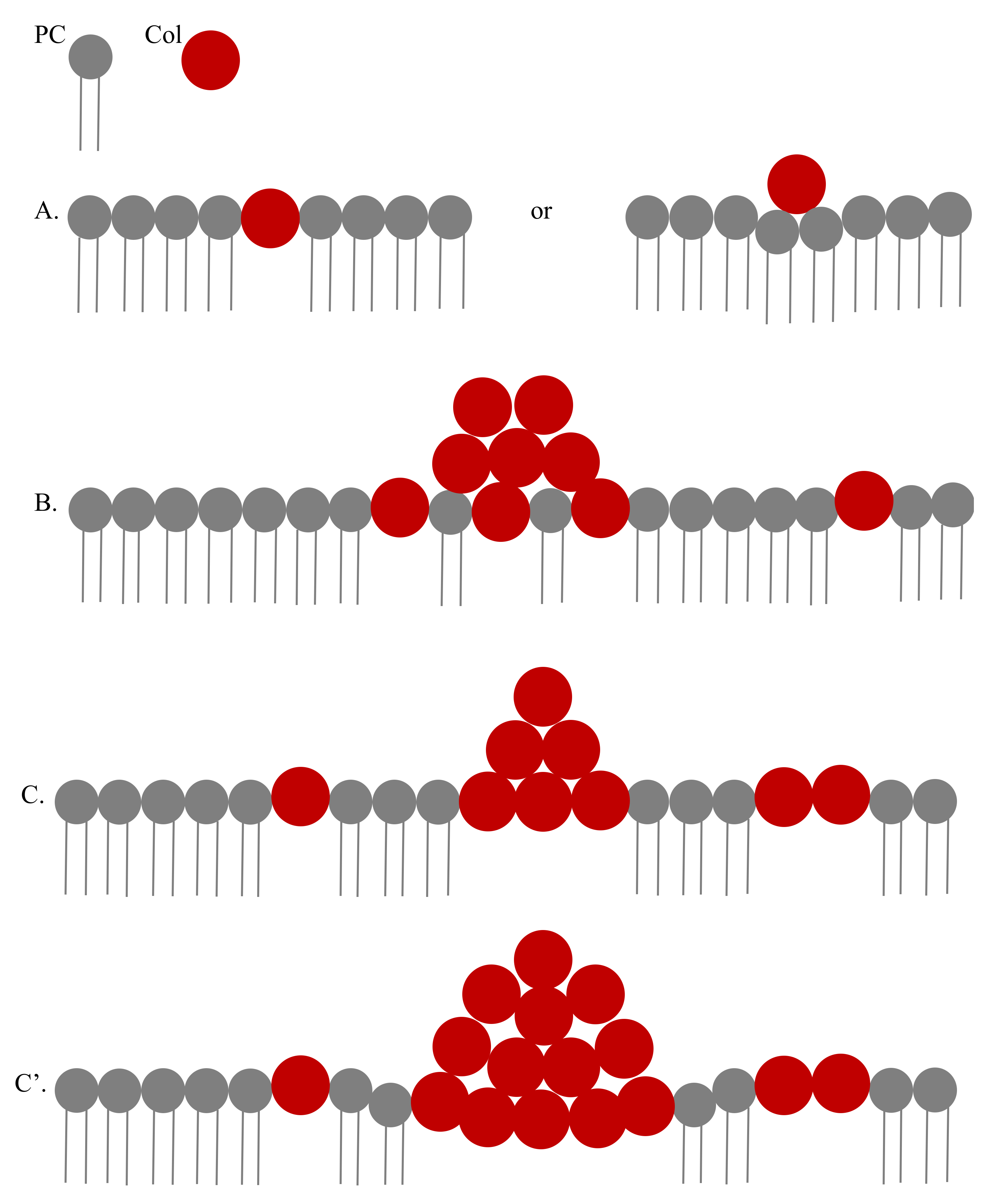
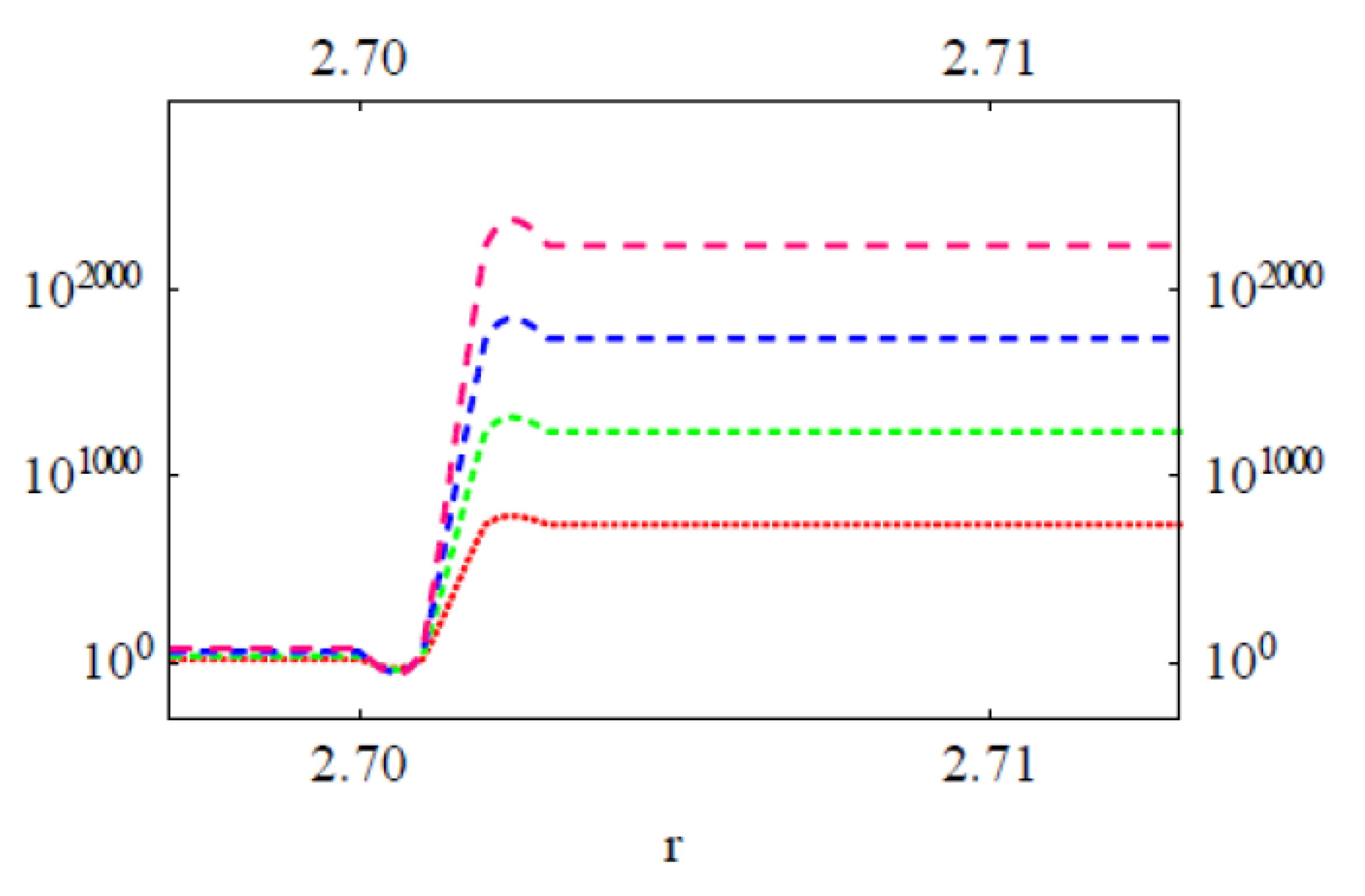
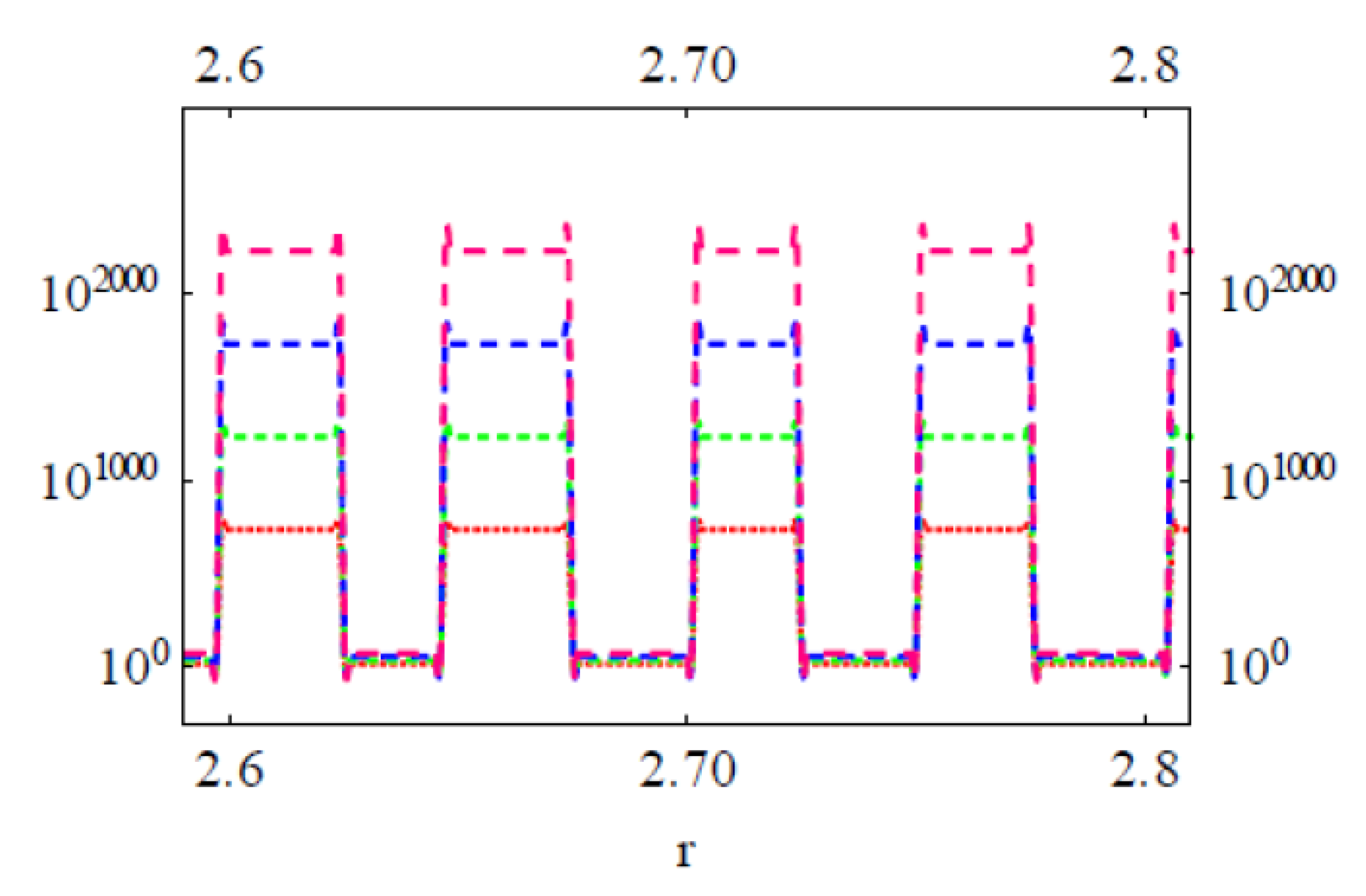
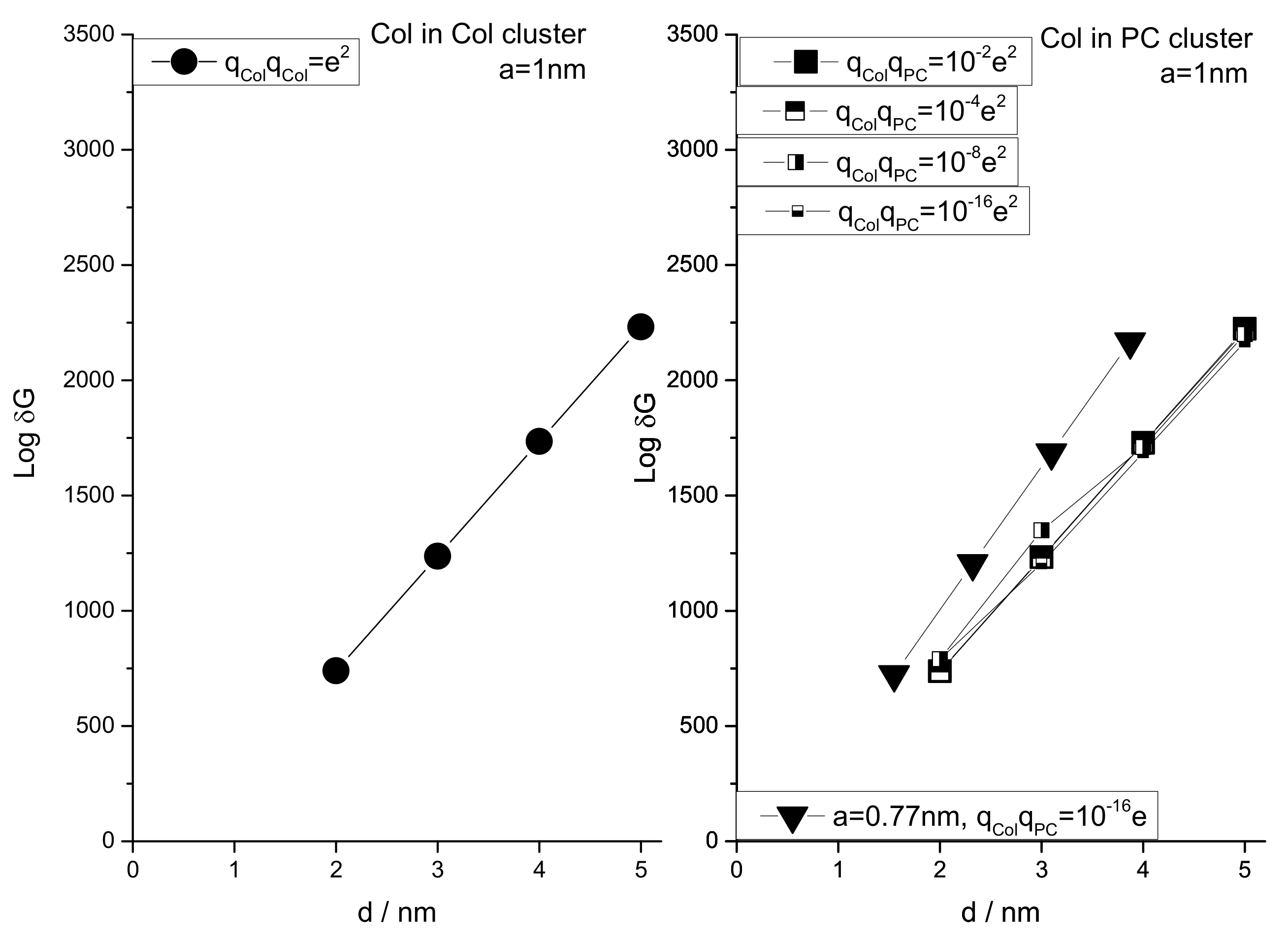
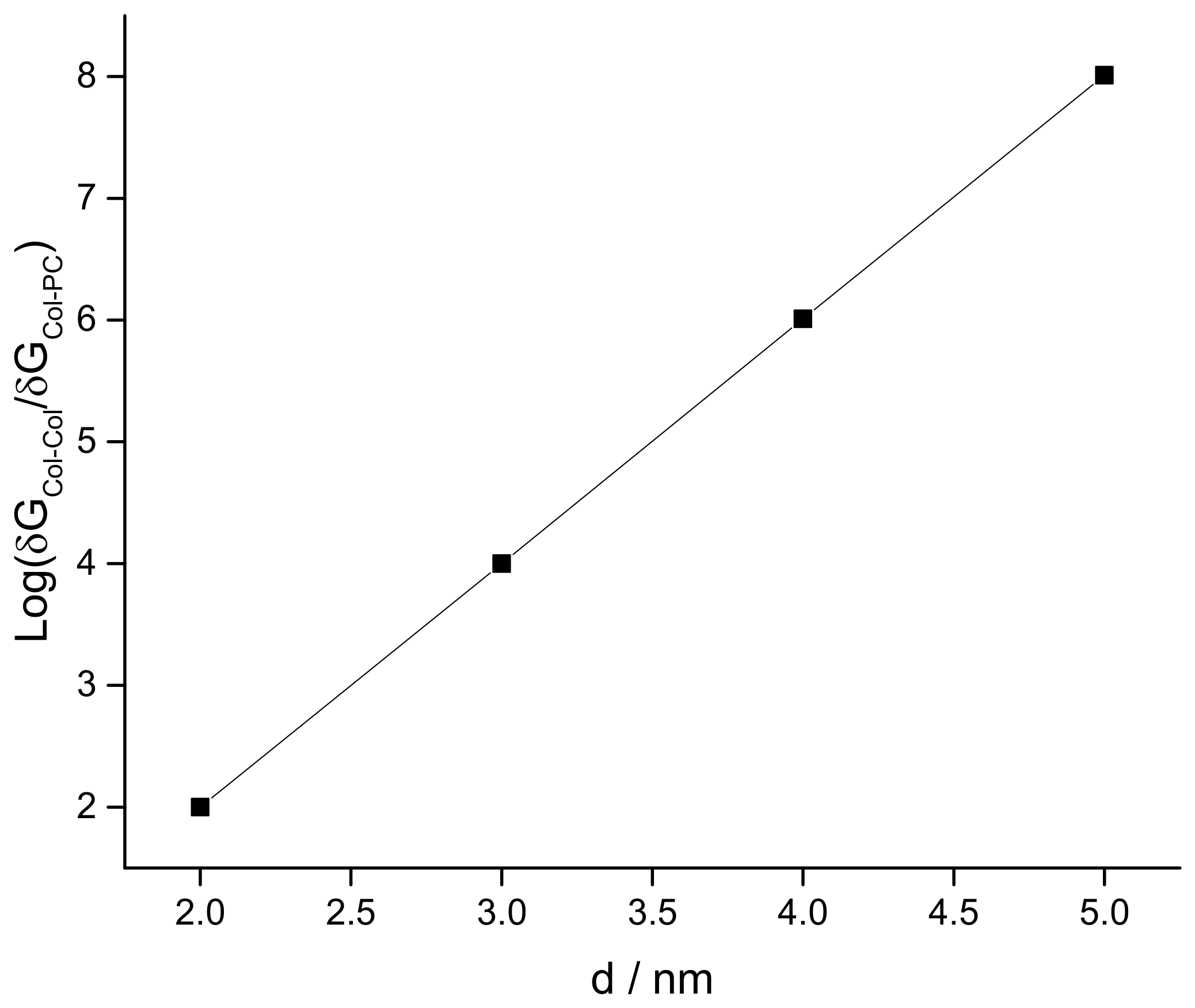
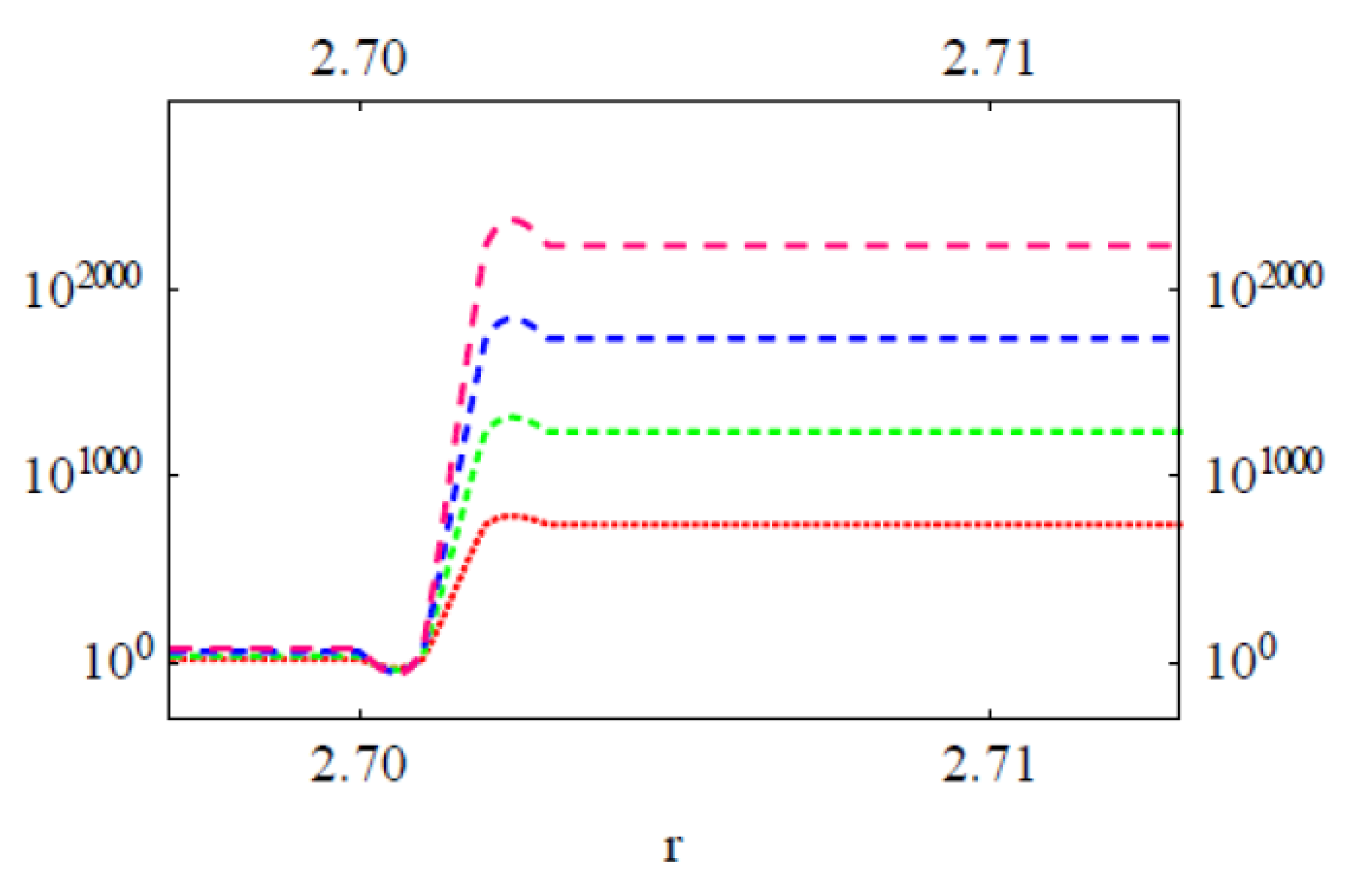
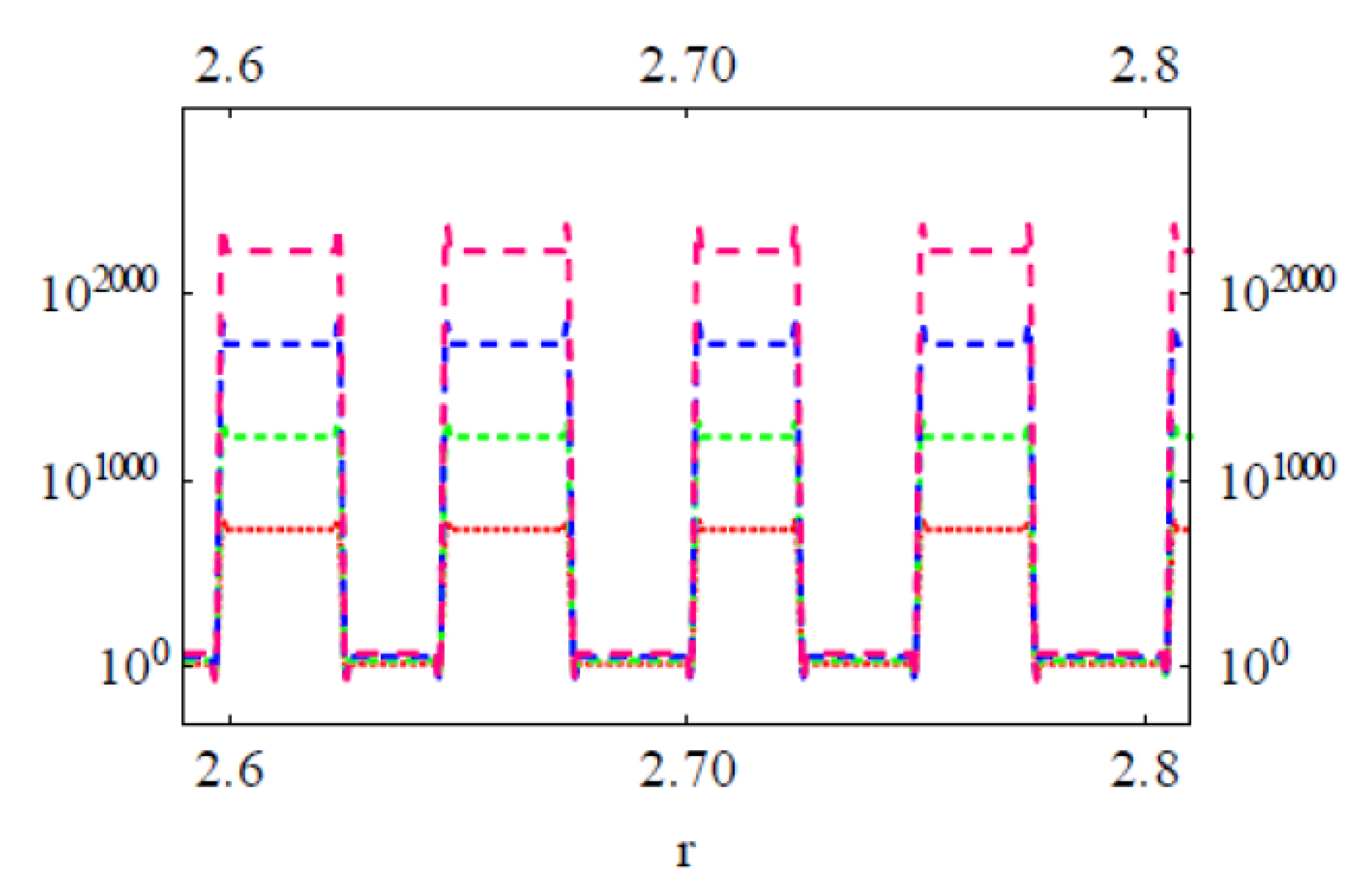
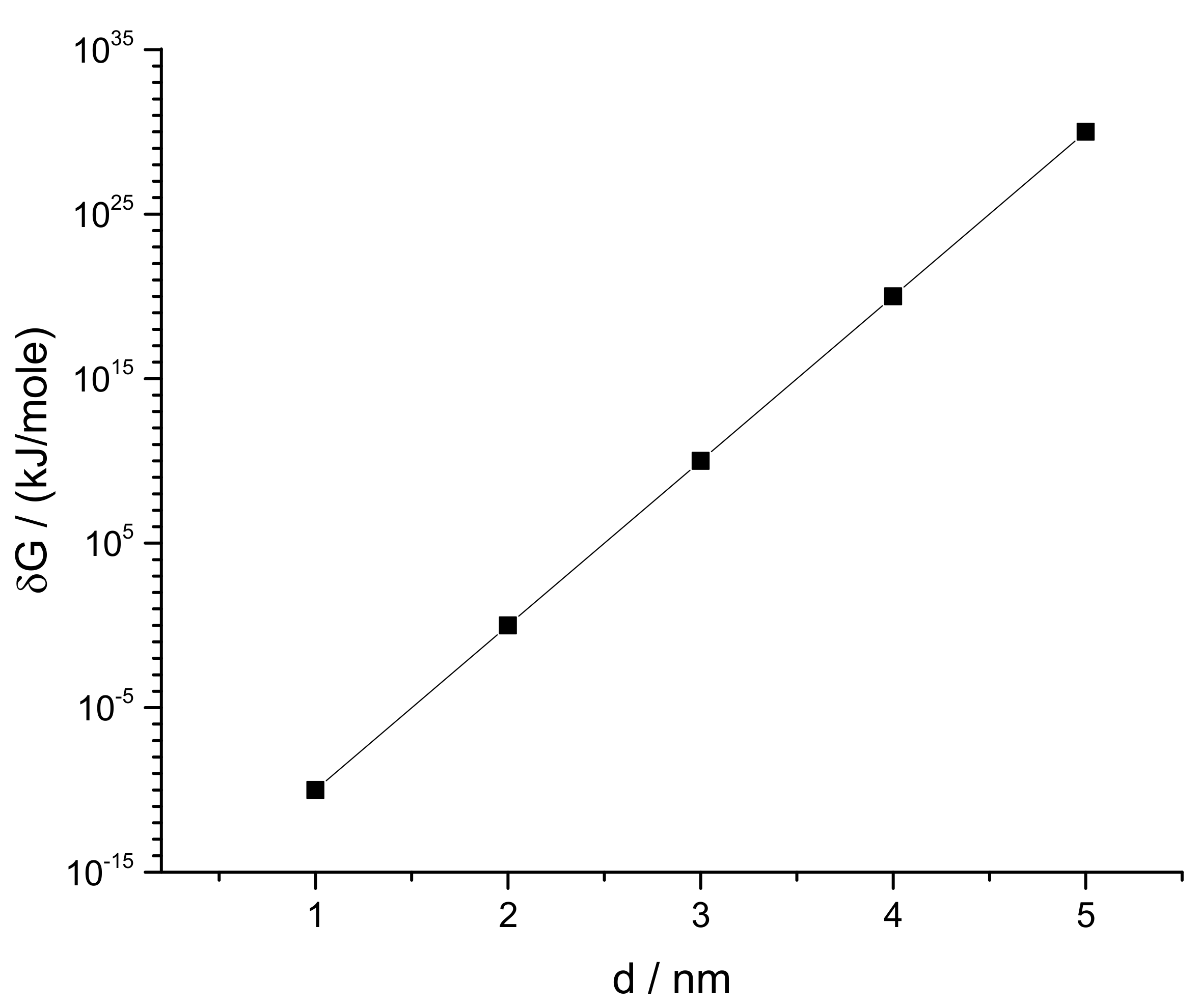
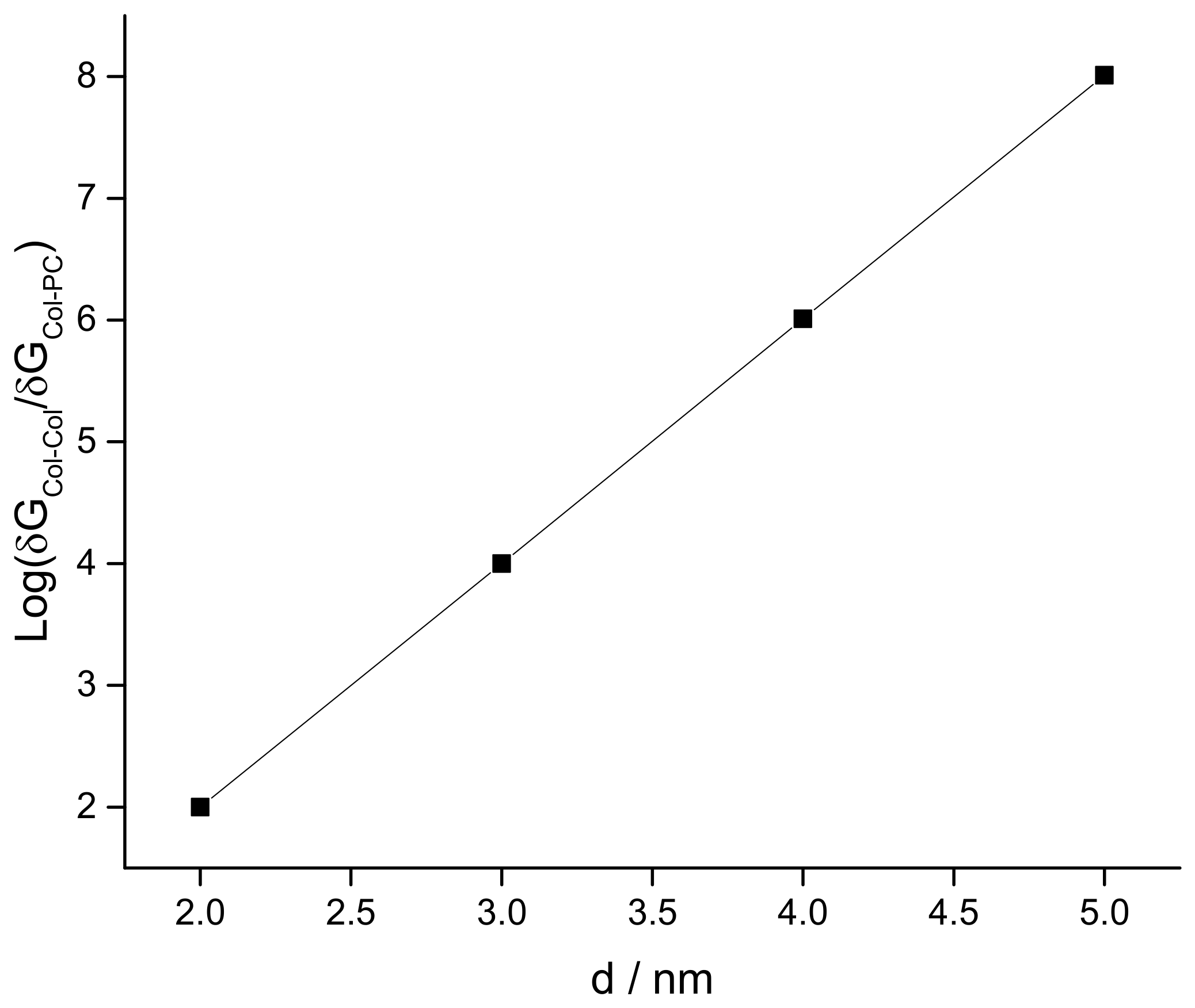
3.2. Modeling the Distribution and Computing the Energetics of Drugs on the Cell Surface
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ashrafuzzaman, M.; Duszyk, M.; Tuszynski, J. Chemotherapy drug molecules thiocochicoside and taxol permeabilize lipid bilayer membranes by forming ion channels. J. Phys. Conf. Ser. 2011, 329, 012029. [Google Scholar] [CrossRef]
- Ashrafuzzaman, M.; Tseng, C.-Y.; Duszyk, M.; Tuszynski, J.A. Chemotherapy Drugs form Ion Pores in Membranes Due to Physical Interactions with Lipids. Chem. Biol. Drug Des. 2012, 80, 992–1002. [Google Scholar] [CrossRef] [PubMed]
- Andersen, O. Ion movement through gramicidin A channels. Studies on the diffusion-controlled association step. Biophys. J. 1983, 41, 147–165. [Google Scholar] [CrossRef] [Green Version]
- Huang, H. Deformation free energy of bilayer membrane and its effect on gramicidin channel lifetime. Biophys. J. 1986, 50, 1061–1070. [Google Scholar] [CrossRef] [Green Version]
- Latorre, M.; Alvarez, O. Voltage-dependent channels in planar lipid bilayer membranes. Physiol. Rev. 1981, 6, 77–150. [Google Scholar] [CrossRef]
- Ludtke, S.; He, K.; Heller, W.T.; Harroun, T.; Yang, L.; Huang, H.W. Membrane Pores Induced by Magainin. Biochemistry 1996, 35, 13723–13728. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Murase, O.; Fujii, N.; Miyajima, K. An Antimicrobial Peptide, Magainin 2, Induced Rapid Flip-Flop of Phospholipids Coupled with Pore Formation and Peptide Translocation. Biochemistry 1996, 35, 11361–11368. [Google Scholar] [CrossRef]
- Ashrafuzzaman, M.; Tuszynski, J.A. Regulation of Channel Function Due to Coupling with a Lipid Bilayer. J. Comput. Theor. Nanosci. 2012, 9, 564–570. [Google Scholar] [CrossRef]
- Ashrafuzzaman, M.; Tseng, C.-Y.; Tuszynski, J.A. Regulation of channel function due to physical energetic coupling with a lipid bilayer. Biochem. Biophys. Res. Commun. 2014, 445, 463–468. [Google Scholar] [CrossRef] [Green Version]
- Ashrafuzzaman, M.; Tseng, C.-Y. Method for Direct Detection of Lipid Binding Agents in Membrane. U.S. Patent US9529006B1, 27 December 2016. [Google Scholar]
- Binning, G.; Quate, C.F.; Gerber, C.; Weibel, E. Atomic Force Microscope. Phys. Rev. Lett. 1986, 56, 930–933. [Google Scholar] [CrossRef] [Green Version]
- Schneider, S.W.; Sritharan, K.C.; Geibel, J.P.; Oberleithner, H.; Jena, B.P. Surface dynamics in living acinar cells imaged by atomic force microscopy: Identification of plasma membrane structures involved in exocytosis. Proc. Natl. Acad. Sci. USA 1997, 94, 316–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhushan, B. (Ed.) Springer Handbook of Nanotechnology; Springer: Berlin/Heidelberg, Germany, 2004. [Google Scholar]
- Franz, C.M.; Puech, P.-H. Atomic Force Microscopy: A Versatile Tool for Studying Cell Morphology, Adhesion and Mechanics. Cell. Mol. Bioeng. 2008, 1, 289–300. [Google Scholar] [CrossRef]
- Kuznetsova, T.G.; Starodubtseva, M.N.; Yegorenkov, N.I.; Chizhik, S.A.; Zhdanov, R.I. Atomic force microscopy probing of cell elasticity. Micron 2007, 38, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Caille, N.; Thoumine, O.; Tardy, Y.; Meister, J.-J. Contribution of the nucleus to the mechanical properties of endothelial cells. J. Biomech. 2002, 35, 177–187. [Google Scholar] [CrossRef]
- Laurent, V.M.; Kasas, S.; Yersin, A.; Schäffer, T.; Catsicas, S.; Dietler, G.; Verkhovsky, A.; Meister, J.-J. Gradient of Rigidity in the Lamellipodia of Migrating Cells Revealed by Atomic Force Microscopy. Biophys. J. 2005, 89, 667–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preiner, J.; Horner, A.; Karner, A.; Ollinger, N.; Siligan, C.; Pohl, P.; Hinterdorfer, P. High-Speed AFM Images of Thermal Motion Provide Stiffness Map of Interfacial Membrane Protein Moieties. Nano Lett. 2015, 15, 759–763. [Google Scholar] [CrossRef]
- Langer, M.; Koitschev, A.; Haase, H.; Rexhausen, U.; Hörber, J.; Ruppersberg, J. Mechanical stimulation of individual stereocilia of living cochlear hair cells by atomic force microscopy. Ultramicroscopy 2000, 82, 269–278. [Google Scholar] [CrossRef]
- Madl, J.; Rhode, S.; Stangl, H.; Stockinger, H.; Hinterdorfer, P.; Schütz, G.J.; Kada, G. A combined optical and atomic force microscope for live cell investigations. Ultramicroscopy 2006, 106, 645–651. [Google Scholar] [CrossRef]
- Heinisch, J.J.; Lipke, P.N.; Beaussart, A.; El-Kirat-Chatel, S.; Dupres, V.; Alsteens, D.; Dufrêne, Y.F. Atomic force microscopy—Looking at mechanosensors on the cell surface. J. Cell Sci. 2012, 125, 4189–4195. [Google Scholar] [CrossRef] [Green Version]
- Ashrafuzzaman, M.; Lampson, M.A.; Greathouse, D.V.; Koeppe, R.E.; Andersen, O.S. Manipulating lipid bilayer material properties using biologically active amphipathic molecules. J. Phys. Condens. Matter 2006, 18, S1235–S1255. [Google Scholar] [CrossRef]
- Ashrafuzzaman, M.; Andersen, O.S.; McElhaney, R.N. The antimicrobial peptide gramicidin S permeabilizes phospholipid bilayer membranes without forming discrete ion channels. Biochim. Biophys. Acta 2008, 1778, 2814–2822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashrafuzzaman, M.; Tuszynski, J. Membrane Biophysics; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Schiff, P.B.; Fant, J.; Horwitz, S.B. Promotion of microtubule assembly in vitro by taxol. Nature 1979, 277, 665–667. [Google Scholar] [CrossRef] [PubMed]
- Callen, J.P. Colchicine is effective in controlling chronic cutaneous vasculitis in lupus erythematosus. J. Am. Acad. Dermatol. 1985, 13, 193–200. [Google Scholar] [CrossRef]
- Seideman, P.; Fjellner, B.; Johannesson, A. Psoriatic arthritis treated with oral colchicine. J. Rheumatol. 1987, 14, 777–779. [Google Scholar]
- Fisherman, J.; McCabe, M.; Hillig, M. Phase I study of taxol and doxorubucin (Dox) with G-CSF in previously untreated metastatic breast cancer. Proc. Am. Soc. Clin. Oncol. 1992, 11, 57. [Google Scholar]
- Holmes, F.A.; Kudelka, A.P.; Kavanagh, J.J.; Huber, M.H.; Ajani, J.A.; Valero, V. Taxane Anticancer Agents: Basic Science and Current Status. In ACS Symposium Series 583; Georg, G.I., Chen, T.T., Ojima, I., Vyas, D.M., Eds.; American Chemical Society: Washington, DC, USA, 1994; pp. 31–57. [Google Scholar]
- Ashrafuzzaman, M. Diffusion across cell phase states. Biomed. Sci. Today 2015, 1, e4. [Google Scholar]
- Ashrafuzzaman, M. Phenomenology and energetics of diffusion across cell phase states. Saudi J. Biol. Sci. 2015, 22, 666–673. [Google Scholar] [CrossRef] [Green Version]
- Rosenman, S.J.; Ganji, A.A.; Gallatin, W.M. Contact dependent redistribution of cell surface adhesion and activation molecules reorganization. FASEB J. 1991, 5, 1603. [Google Scholar]
- Mekory, Y.A.; Baram, D.; Goldberg, A.; Klajman, A. Inhibition of delayed hypersensitivity in mice by colchicines: Mechanism of inhibition of contact sensibility in vivo. Cell. Immunol. 1989, 120, 330–340. [Google Scholar] [CrossRef]
- Borisy, G.O.; Taylor, E.W. The mechanism of action of colchicine: Colchicine binding to seaurchin eggs and the mitotic apparatus. J. Cell. Biol. 1967, 34, 533–548. [Google Scholar] [CrossRef]
- Agutter, P.S.; Suckling, K.E. Effect of colchicine on mammalian liver nuclear envelope and on nucleo-cytoplasmic RNA transport. Biochim. Biophys. Acta 1982, 698, 223–229. [Google Scholar] [CrossRef]
- Balasubramanian, S.V.; Straubinger, R.M. Taxol-lipid interactions: Taxol-dependent effects on the physical properties of model membranes. Biochemistry 1994, 33, 8941–8947. [Google Scholar] [CrossRef]
- Matsumoto, G.; Sakai, H. Microtubules inside the plasma membrane of squid giant axons and their possible physiological function. J. Membr. Biol. 1979, 50, 1–14. [Google Scholar] [CrossRef]
- Sonee, M.; Barrón, E.; Yarber, F.A.; Hamm-Alvarez, S.F. Taxol inhibits endosomal-lysosomal membrane trafficking at two distinct steps in CV-1 cells. Am. J. Physiol. Cell Physiol. 1998, 275, C1630–C1639. [Google Scholar] [CrossRef]
- Shiba, M.; Watanabe, E.; Sasakawa, S.; Ikeda, Y. Effects of taxol and colchicine on platelet membrane properties. Thromb. Res. 1988, 52, 313–323. [Google Scholar] [CrossRef]
- Mons, S.; Veretout, F.; Carlier, M.-F.; Erk, I.; Lepault, J.; Trudel, E.; Salesse, C.; Ducray, P.; Mioskowski, C.; Lebeau, L. The interaction between lipid derivatives of colchicine and tubulin: Consequences of the interaction of the alkaloid with lipid membranes. Biochim. Biophys. Acta 2000, 1468, 381–395. [Google Scholar] [CrossRef] [Green Version]
- Parsegian, A. Energy of an Ion crossing a Low Dielectric Membrane: Solutions to Four Relevant Electrostatic Problems. Nature 1969, 221, 844–846. [Google Scholar] [CrossRef]
- Horcas, I.; Fernández, R.; Gómez-Rodríguez, J.M.; Colchero, J.; Gómez-Herrero, J.; Baro, A.M. WSXM: A software for scanning probe microscopy and a tool for nanotechnology. Rev. Sci. Instrum. 2007, 78, 013705. [Google Scholar] [CrossRef] [PubMed]
- Grierson, D.S.; Flater, E.E.; Carpick, R.W. Accounting for the JKR-DMT transition in adhesion and friction measurements with atomic force microscopy. J. Adhes. Sci. Technol. 2005, 19, 291–311. [Google Scholar] [CrossRef] [Green Version]
- Ashrafuzzaman, M.; Tseng, C.-Y.; Tuszynski, J.A. Charge-based interactions of antimicrobial peptides and general drugs with lipid bilayers. J. Mol. Graph. Model. 2020, 95, 107502. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.Y.; Ashrafuzzaman, M.; Mane, J.Y.; Kapty, J.; Mercer, J.R.; Tuszynski, J.A. Entropic fragment based approach to aptamer design. Chem. Biol. Drug Des. 2011, 78, 1–13. [Google Scholar] [CrossRef]
- Ashrafuzzaman, M.; Tseng, C.-Y.; Kapty, J.A.; Mercer, J.R.; Tuszynski, J.A. A Computationally Designed DNA Aptamer Template with Specific Binding to Phosphatidylserine. Nucleic Acid Ther. 2013, 23, 418–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prenner, E.J.; Lewis, R.N.; Jelokhani-Niaraki, M.; Hodges, R.S.; McElhaney, R.N. Cholesterol attenuates the interaction of the antimicrobial peptide gramicidin S with phospholipid bilayer membranes. Biochim. Biophys. Acta 2001, 1510, 83–92. [Google Scholar] [CrossRef] [Green Version]
- Abraham, T.; Lewis, R.N.A.H.; Hodges, R.S.; McElhaney, R.N. Isothermal Titration Calorimetry Studies of the Binding of the Antimicrobial Peptide Gramicidin S to Phospholipid Bilayer Membranes. Biochemistry 2005, 44, 11279–11285. [Google Scholar] [CrossRef]
- Alves, A.C.; Magarkar, A.; Horta, M.; Lima, J.L.F.C.; Bunker, A.; Nunes, C.; Reis, S. Influence of doxorubicin on model cell membrane properties: Insights from in vitro and in silico studies. Sci. Rep. 2017, 7, 6343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zalba, S.; Hagen, T.L.T. Cell membrane modulation as adjuvant in cancer therapy. Cancer Treat. Rev. 2017, 52, 48–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashrafuzzaman, M. Amphiphiles capsaicin and triton X-100 regulate the chemotherapy drug colchicine’s membrane adsorption and ion pore formation potency. Saudi J. Biol. Sci. 2021, 28, 3100–3109. [Google Scholar] [CrossRef]
- Ashrafuzzaman, M. The Antimicrobial Peptide Gramicidin S Enhances Membrane Adsorption and Ion Pore Formation Potency of Chemotherapy Drugs in Lipid Bilayers. Membranes 2021, 11, 247. [Google Scholar] [CrossRef] [PubMed]
- Grage, S.L.; Afonin, S.; Kara, S.; Buth, G.; Ulrich, A.S. Membrane Thinning and Thickening Induced by Membrane-Active Amphipathic Peptides. Front. Cell Dev. Biol. 2016, 4, 65. [Google Scholar] [CrossRef] [Green Version]
- Kumariya, R.; Sood, S.K.; Rajput, Y.S.; Saini, N.; Garsa, A.K. Increased membrane surface positive charge and altered membrane fluidity leads to cationic antimicrobial peptide resistance in Enterococcus faecalis. Biochim. Biophys. Acta Biomembr. 2015, 1848, 1367–1375. [Google Scholar] [CrossRef] [Green Version]
- Henderson, J.; Iyengar, N.S.; Lam, K.L.H.; Maldonado, E.; Suwatthee, T.; Roy, I.; Waring, A.J.; Lee, K.Y.C. Beyond electrostatics: Antimicrobial peptide selectivity and the influence of cholesterol-mediated fluidity and lipid chain length on protegrin-1 activity. Biochim. Biophys. Acta Biomembr. 2019, 1861, 182977. [Google Scholar] [CrossRef] [PubMed]
- Prenner, E.J.; Lewis, R.N.; McElhaney, R.N. The interaction of the antimicrobial peptide gramicidin S with lipid bilayer model and biological membranes. Biochim. Biophys. Acta Biomembr. 1999, 1462, 201–221. [Google Scholar] [CrossRef] [Green Version]
- Bitler, A.; Dover, R.; Shai, Y. Anticancer Drug Colchicine Increases Disorder and Reduces Complexity in the Macrophage Membrane. Biophys. J. 2016, 110, 83A. [Google Scholar] [CrossRef]
- Ashrafuzzaman, M. Nanoscale Biophysics of the Cell; Springer International Publishing AG: Basel, Switzerland, 2018; ISBN 978-3-319-77463-3. [Google Scholar]
- Ashrafuzzaman, M. Energy-Based Method for Drug Design. US Patent US10916330B1, 9 February 2021. [Google Scholar]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ashrafuzzaman, M.; Khan, Z.; Alqarni, A.; Alanazi, M.; Alam, M.S. Cell Surface Binding and Lipid Interactions behind Chemotherapy-Drug-Induced Ion Pore Formation in Membranes. Membranes 2021, 11, 501. https://doi.org/10.3390/membranes11070501
Ashrafuzzaman M, Khan Z, Alqarni A, Alanazi M, Alam MS. Cell Surface Binding and Lipid Interactions behind Chemotherapy-Drug-Induced Ion Pore Formation in Membranes. Membranes. 2021; 11(7):501. https://doi.org/10.3390/membranes11070501
Chicago/Turabian StyleAshrafuzzaman, Md., Zahid Khan, Ashwaq Alqarni, Mohammad Alanazi, and Mohammad Shahabul Alam. 2021. "Cell Surface Binding and Lipid Interactions behind Chemotherapy-Drug-Induced Ion Pore Formation in Membranes" Membranes 11, no. 7: 501. https://doi.org/10.3390/membranes11070501
APA StyleAshrafuzzaman, M., Khan, Z., Alqarni, A., Alanazi, M., & Alam, M. S. (2021). Cell Surface Binding and Lipid Interactions behind Chemotherapy-Drug-Induced Ion Pore Formation in Membranes. Membranes, 11(7), 501. https://doi.org/10.3390/membranes11070501